

Abstract
Mu-opioid receptor agonists represent mainstays of pain management. However, the therapeutic use of these agents is associated with serious side effects, including potentially lethal respiratory depression. Accordingly, there is a longstanding interest in the development of new opioid analgesics with improved therapeutic profiles. The alkaloids of the Southeast Asian plant Mitragyna speciosa, represented by the prototypical member mitragynine, are an unusual class of opioid receptor modulators with distinct pharmacological properties. Here we describe the first receptor-level functional characterization of mitragynine and related natural alkaloids at the mu-, kappa-, and delta-opioid receptors. These results show that mitragynine and the oxidized analog 7-hydroxymitragynine, are partial agonists of the human mu-opioid receptor and competitive antagonists at the kappa- and delta-opioid receptors. We also show that mitragynine and 7-hydroxymitragynine are G-protein-biased agonists of the mu-opioid receptor, which do not recruit β-arrestin following receptor activation. Therefore, the Mitragyna alkaloid scaffold represents a novel framework for the development of functionally biased opioid modulators, which may exhibit improved therapeutic profiles. Also presented is an enantioselective total synthesis of both (-)-mitragynine and its unnatural enantiomer, (+)-mitragynine, employing a proline-catalyzed Mannich-Michael reaction sequence as the key transformation. Pharmacological evaluation of (+)-mitragynine revealed its much weaker opioid activity. Likewise, the intermediates and chemical transformations developed in the total synthesis allowed the elucidation of previously unexplored structure-activity relationships (SAR) within the Mitragyna scaffold. Molecular docking studies, in combination with the observed chemical SAR, suggest that Mitragyna alkaloids adopt a binding pose at the mu-opioid receptor that is distinct from that of classical opioids.
Graphical Abstract

Introduction
The opioid receptors, and in particular, the mu-opioid receptor (MOR), are among the longest and most intensely studied molecular signaling systems in the central nervous system.1 Likewise, the prototypical small molecule agonist of these receptors, morphine, has been used by humans as an important analgesic and recreational euphoriant since ancient times. Indeed, MOR agonists, including not only morphine itself, but also a vast number of synthetic and semi-synthetic opioids, remain the gold standard of pain therapy. Unfortunately, acute MOR activation is also associated with serious side effects, including respiratory depression, constipation, sedation, nausea, and itching.1,2 At sufficiently high doses, the evoked respiratory depression can be severe enough to cause death. Further, the pronounced euphoria produced by MOR agonists makes them major drugs of abuse. These properties have made overdose from prescription opioid analgesics a leading cause of accidental death in the United States, killing more than 18,000 people in 2014.3,4 Another shortcoming of MOR agonists is the rapid development of tolerance to their analgesic effects. Thus, continuing escalation of dose is required to maintain an equivalent level of pain management. Similarly, when they are abused, tolerance to the euphoric effects of opioids is also rapidly developed. Thus in either case, chronic use often results in severe physical dependence on MOR agonists due to cellular- and circuit-level adaptations to continuous receptor stimulation. Accordingly, much effort has been dedicated to the development of new MOR agonists retaining potent analgesic effects, while mitigating or eliminating the deleterious side effects of the agents currently in use.1–8
Historically, MOR agonists have also been applied in the treatment of mood disorders, notably including major depressive disorder (MDD). Indeed, until the mid-20th century, low doses of opium itself were used to treat depression, and the so called “opium cure” was purportedly quite effective.9 With the advent of tricyclic antidepressants (TCAs) in the 1950s however, the psychiatric use of opioids rapidly fell out of favor and has been largely dormant since, likely due to negative medical and societal perceptions stemming from their abuse potential. However, there have been scattered clinical reports (both case studies and small controlled trials) since the 1970s indicating the effectiveness of MOR agonists in treating depression. The endogenous opioid peptide β-endorphin, as well as a number of small molecules, have all been reported to rapidly and robustly improve the symptoms of MDD and/or anxiety disorders in the clinical setting, even in treatment resistant patients.10–17 These results have been recapitulated in rodent models, where a variety of MOR agonists show antidepressant effects.18–21 Most recently, we have found that the atypical antidepressant tianeptine, which has been used clinically for several decades and extensively studied in rodents and other mammalian species, is an MOR agonist, suggesting that this agent exerts its antidepressant effects via direct MOR activation.22 When taken together, this body of work establishes a clear precedent for the effectiveness of MOR agonists in the treatment of depression and anxiety. Unfortunately, the treatment of mood disorders with conventional MOR agonists is expected to suffer from the same liabilities as their use in the context of pain management. Accordingly, our laboratories have been concerned with the exploration of structurally and pharmacologically distinct classes of MOR agonists, with the aim of developing new opioid-based treatments for mood disorders and pain, which lack the classical side effects of these agents.
It was in this context that we became interested in the psychoactive plant Mitragyna speciosa (Figure 1), known as “kratom” in Thailand, or “biak biak” in Malaysia, a substance which has been used by humans in Southeast Asia for centuries to treat a variety of ailments. The plant material is typically consumed as a tea or chewed directly. At low doses, kratom is primarily used for its stimulating effects. At higher doses, opioid-like effects predominate, and the plant has been used as a general analgesic and as a substitute for opium or to treat opium withdrawal symptoms. Other medicinal applications are also known, including use as a treatment for fever, cough, diarrhea, and depression. There is also a precedent for recreational use of the plant, a fact that has contributed to legal control of Mitragyna speciosa in both Thailand and Malaysia, but the plant remains uncontrolled outside its endemic regions.23–27
Figure 1.

Leaves and fruiting bodies of Mitragyna speciosa and chemical structures of notable alkaloids.
In light of its well documented medicinal properties, the molecular constituents of Mitragyna speciosa responsible for its psychoactive effects have been studied, with more than 20 unique indole alkaloids having been identified in the plant.23–25,28 The indole alkaloid mitragynine (Figure 1) has been universally cited as the primary alkaloid constituent of Mitragyna speciosa, accounting for up to 66% by mass of crude alkaloid extracts.23 The other major alkaloids in the plant have been found to include paynantheine, speciogynine, and speciociliatine (Figure 1).23 The quantities of these major alkaloids, along with a wide variety of minor alkaloids, are considerably varied amongst different regional varieties of the plant and also depend on plant age, facts that considerably complicate the interpretation of reported psychoactive effects from the raw plant material.23–25,28 Amongst the minor alkaloids, the oxidized derivative 7-hydroxymitragynine (7-OH)29 (Figure 1) is of particular interest, as it has been reported to induce analgesic effects mediated through agonist activity at the mu-opioid receptor (MOR), exceeding in potency those of the prototypical opioid agonist morphine.27,30
The mechanism of action of Mitragyna alkaloids has also been studied both in vitro and in vivo. In particular, Takayama and coworkers have accumulated a large body of evidence implicating the opioid receptor system as the primary mediator of the psychoactive effects of these alkaloids. Specifically, both mitragynine and 7-OH exhibit nanomolar binding affinities for the MOR and possess functional activity in tissue assays.27,30 Likewise, the antinociceptive effects of mitragynine and 7-OH in several rodent models are also inhibited by naloxone.27,30,31 Despite this evidence for the involvement of opioid receptor systems, and specifically MOR, in mediating the analgesic effects of Mitragyna alkaloids, there are conflicting reports in the literature. Most notably, an early study with mitragynine indicated that the behavioral effects in cats, and analgesic effects in rats, were not reversed by treatment with nalorphine, an opioid antagonist, while at the same time, mitragynine was found to produce markedly less respiratory depression than codeine.32 Mitragynine has been shown to bind in some degree to several non-opioid CNS receptors, including alpha-2 adrenergic receptor (α2R), adenosine A2a, dopamine D2, and the serotonin receptors 5-HT2C and 5-HT7, but the strength of these affinities has not been reported.33 Mitragynine analgesia has also been shown to be inhibited by α2R antagonist idazoxan and by the non-specific serotonin antagonist cyproheptadine.34
Considering the many possible confounding factors present at the tissue or system level, such in vivo and ex vivo tissue assays are not ideal for positive confirmation of a functional effect at a particular receptor. Although receptor-level functional activity studies ([35S]GTPγS binding) using cloned opioid receptors have recently been reported for several synthetic oxidized analogs35, no similar functional studies have been reported for mitragynine itself, or for other naturally occurring alkaloids in Mitragyna speciosa.36 Given the unique and still unclear molecular pharmacology surrounding Mitragyna alkaloids, we undertook a thorough examination of mitragynine and a number of natural and novel analogs at the opioid receptors, measuring receptor activation and intracellular signaling.
Results and Discussion
Mitragynine is a Partial Agonist of Human MOR
We isolated the four major alkaloids (Figure 1) from the Thai strain of Mitragyna speciosa and prepared 7-OH by photochemical oxidation of mitragynine (Scheme S1). Interestingly, only trace quantities of 7-OH were observed (by mass spectrometry) in our extractions of the raw plant material, and it was not possible to isolate any measurable quantity of this derivative. Therefore, it is doubtful that this alkaloid is a universal constituent of all Mitragyna speciosa preparations, and is unlikely to generally account for the psychoactive properties of this plant.
We then set out to evaluate the activity of these compounds in HEK cells expressing the MOR, delta-opioid receptor (DOR), or kappa-opioid receptor (KOR), using bioluminescence resonance energy transfer (BRET) functional assays. In these G protein-dependent assays, the Gα subunit is fused with a luciferase (RLuc8) donor, and the Gγ subunit is fused with a fluorescent protein (mVenus) acceptor. Thus, on receptor activation, the G protein subunits move apart and the observed BRET signal decreases (Figure 2A).
Figure 2.

Activity of mitragynine and 7-hydroxymitragyine (7-OH) at the human mu-opioid receptor (hMOR). (A) Conceptual representation of the G protein BRET assay employing atomistic cartoons derived from available x-ray crystal structures. To measure G protein activation, hMOR37 (brown) was co-expressed with GαoB38 (red) fused to RLuc839 (teal), β138 (blue), and mVenus40 (yellow-green) fused to γ238 (yellow). (B) Agonist activity at hMOR; positive control = [D-Ala2N-Me-Phe4, Gly-ol5]-enkephalin (DAMGO); curves represent the average of n≥3, with error bars representing ±SEM.
At the human opioid receptors (hMOR, hKOR, and hDOR), mitragynine acted as a partial agonist at hMOR (Table 1 and Figure 2B, EC50 = 339 ± 178 nM; maximal efficacy (Emax) = 34%). This result represents the first demonstration of functional opioid agonist activity at human receptors with a natural Mitragyna alkaloid, all prior functional studies having been conducted in vivo in rodents or ex vivo in rodent tissue. In contrast, at hKOR, mitragynine was a competitive antagonist (Table 1, IC50 = 8.5 ± 7.6 µM; pA2 = 1.4 ± 0.40 µM, and Figure S2 and S4), fully inhibiting the activity of the reference agonist U-50,488. Similarly, mitragynine acted as an antagonist at hDOR, but with very low potency (Figure S3). The other major natural alkaloids paynantheine, speciogynine, and speciociliatine, showed no measurable agonist activity at any of the human opioid receptors at concentrations up to 100 µM, and only weak antagonist effects were observed (Table 1 and Figures S1, S2, and S3). The oxidized analog, 7-OH, was also characterized and found to be a potent, partial agonist at hMOR (Table 1 and Figure 2B, EC50 = 34.5 ± 4.5 nM; Emax = 47%). Further, it acted as a competitive antagonist at both hKOR (Table 1, IC50 = 7.9 ± 3.7 µM; pA2 = 490 ± 131 nM, and Figures S2 and S5) and hDOR (Table 1, IC50 = 15.6 ± 9.1 µM, and Figure S3). The partial agonist activity of mitragynine and 7-OH at the human receptors was further confirmed in antagonist experiments, as both compounds were able to partially inhibit the response elicited by the full agonist [D-Ala2, N-Me-Phe4, Gly-ol5]-enkephalin (DAMGO) (Figure S1).
Table 1.
Functional Activity of Mitragyna Alkaloids at Human Opioid Receptors as Determined in G Protein BRET Assays.
| EC50 ± SEM (Emax)a or [IC50 ± SEM (pA2)]b (µM) |
|||
|---|---|---|---|
| Compound | hMOR | hKOR | hDOR |
| mitragynine |
0.339 ± 0.178 (34%) |
[8.5 ± 7.6 (1.4)] | [>10] |
| paynantheine | [2.2 ± 10] | [>10] | [>10] |
| speciogynine | [5.7 ± 2.8] | [>10] | [>10] |
| speciociliatine | [4.2 ± 1.6] | [>10] | [>10] |
| 7-OH |
0.0345 ± 0.0045 (47%) |
[7.9 ± 3.7 (0.49)] |
[>10] |
Agonist activity indicated by EC50 values, maximal efficacy (Emax) relative to DAMGO in parentheses;
Antagonist activity indicated by IC50 values for inhibition of a reference agonist, pA2 determined from Schild analysis in parentheses; All data points represent mean ± SEM (µM) of n ≥ 3.
In order to further confirm the opioid activity of the Mitragyna alkaloids, we performed radioligand binding studies to assess the affinity of the natural alkaloids and 7-OH for the opioid receptors (Table 2). As expected from their activity in our functional assays, both mitragynine and 7-OH exhibited significant binding affinities for hMOR. Similarly, the other natural alkaloids also bound to hMOR, consistent with their antagonist activity in the functional assays. Binding was also observed for mitragynine and 7-OH at hKOR and hDOR, again in agreement with the results of the functional assays. Further, the Ki values determined in the binding experiments were in accord with the EC50 and IC50 (after correction by Cheng-Prusoff equation) values determined in the functional experiments, or with the pA2 values (an estimate of Kd) determined by Schild analysis. Similar binding results were also obtained at the mouse opioid receptors (Table S2). Further, our binding data with 7-OH was consistent with previous literature reports (using guinea pig brain homogenates), but the affinities determined for mitragynine at MOR and DOR were much weaker than previously described.27,30 Given the better agreement between our in vitro data and the relative potencies of mitragynine and 7-OH in vivo, this new data may be more reliable. Taken together, the functional and binding results provide a rigorous and internally consistent assessment of the in vitro activity of the Mitragyna alkaloids at all three human opioid receptors.
Table 2.
Binding Affinities of Mitragyna Alkaloids at Human Opioid Receptors
| Ki ± SEM (µM)a |
|||
|---|---|---|---|
| Compound | hMOR | hKOR | hDOR |
| mitagynine | 0.233 ± 0.048 | 0.772 ± 0.207 | > 10 |
| paynantheine | 0.410 ± 0.152 | 2.56 ± 0.37 | > 10 |
| speciogynine | 0.728 ± 0.061 | 3.20 ± 0.36 | > 10 |
| speciociliatine | 0.560 ± 0.168 | 0.329 ± 0.112 | > 10 |
| 7-OH | 0.047 ± 0.018 | 0.188 ± 0.038 | 0.219 ± 0.041 |
All data points represent mean ± SEM (µM) of n ≥ 3.
Surprisingly, analogous assays using rodent receptors (mouse or rat) revealed that mitragynine exhibited no agonist activity, and it was instead found to act as a competitive antagonist at mouse MOR (mMOR) (Figure S6 and Table S1, IC50 = 1.1 µM; pA2 = 807 ± 573 nM), fully inhibiting the activation induced by the reference agonist DAMGO. The other isolated natural products, paynantheine, speciogynine, and speciociliatine, showed no notable agonist or antagonist activity at any of the rodent opioid receptors. In contrast, the activity of 7-OH was similar at the rodent receptors (Figure S6 and Table S1). These observations complicate the interpretation of prior reports of analgesic effects elicited by mitragynine in rodent models. Further, this interspecies variation may present significant challenges in the development of compounds in this class as novel therapeutics, as activity in rodent models may not be easily translatable to man. In this regard, it is fortunate that the more potent derivative, 7-OH, retains its agonist activity at mMOR, as the oxidized scaffold represents a more promising starting point for the development of analgesics in this class. With respect to mitragynine, it is also conceivable that the in vitro assays used in our study do not exactly replicate the receptor activation and signaling in native murine brain tissue. For example, a low efficacy partial agonist in the brain may appear as an antagonist (i.e. agonist with 0% efficacy) in the cell-based assays. Thus, consideration of both interspecies and interassay variability is advised for the future development of these compounds.
Mitragyna Alkaloids Bias Intracellular Signaling Toward G Proteins
Functionally selective (biased) agonists of the opioid receptors, which preferentially activate only certain downstream signaling pathways, hold promise as analgesics or antidepressants with reduced side effects. Given this potential to separate the well established therapeutic benefits of MOR activation from negative side effects, the study of functionally selective MOR ligands is a very active area of research at present.41–44 In particular, some evidence indicates that MOR agonists biased toward G protein signaling over β-arrestin signaling display less respiratory depression, tolerance development, and constipation, while remaining potent analgesics.45–48 Therefore, we were interested in exploring the potential for mitragynine’s unique molecular scaffold to serve as a starting point for the development of biased MOR agonists. To assess the level of β-arrestin recruitment induced by Mitragyna alkaloids, we employed a recently described BRET assay in transfected HEK cells, which was adapted for use with the opioid receptors.49 In this assay, the unlabeled receptor (in this case hMOR) is transfected alongside β-arrestin-2 (arrestin-3), fused with luciferase (RLuc8) and an SH3 binding domain (Sp1), and the membrane protein GAP43, fused with a fluorescent protein (citrine) and an SH3 domain. Accordingly, when an agonist induces β-arrestin recruitment to MOR, the Sp1 domain on β-arrestin is brought into proximity with the membrane-localized SH3 domain, and binding occurs. This results in concomitant association of the luciferase donor and fluorescent protein acceptor, and a BRET signal is observed (Figure 3A). An advantage of this format is that native receptor can be used, avoiding potential confounds related to C-terminal tagging on expression or function.
Figure 3.

Mitragynine and 7-OH do not recruit β-arrestin. (A) Conceptual representation of the β-arrestin BRET assay employing atomistic cartoons derived from available x-ray crystal structures. To measure β-arrestin recruitment, hMOR37 (brown) was co-expressed with β-arrestin-250 (purple) fused to RLuc839 (teal) and Sp1, GAP43 fused to citrine40 (yellow-green) and SH3, and G protein-coupled receptor kinase 2 (GRK2). On β-arrestin recruitment, association of the Sp1 and SH3 domains results in an increase in the BRET signal between RLuc8 and citrine. (B) β-arrestin recruitment at hMOR, positive control = DAMGO; curves represent the average of n≥3, with error bars representing ±SEM.
As measured in this cell signaling assay, DAMGO induced robust recruitment of β-arrestin. Interestingly, both mitragynine and 7-OH were found to elicit no measurable β-arrestin binding, even in the presence of G protein-coupled receptor kinase 2 (GRK2), which typically enhances coupling to β-arrestin (Figure 3B). Due to this extremely weak signal, we were not able to quantitate the magnitude of functional selectivity for mitragynine and 7-OH. However, since both compounds exhibit significant efficacy for activation in the G protein dissociation assay (Figure 2), there is a strong qualitative bias in favor of G protein signaling in these cells. Such selectivity for G protein-dependent signaling may explain the reduced respiratory depression previously reported for mitragynine compared to codeine32, although any such connection between upstream receptor signaling and physiological properties must be considered speculative. In any case, both mitragynine and 7-OH trigger downstream signaling significantly different from that induced by DAMGO. These results provide a good rationale for future experiments aimed at examining cellular signaling in relevant neuronal cells and mapping the effects of structural modifications on the level of functional selectivity.
SAR Exploration: Strategy
Having confirmed the MOR agonist activity of both mitragynine and 7-OH, we chose to explore several key structure-activity relationships (SAR) within this scaffold. In particular, we were interested in the effects of modifications at three positions: the methoxy group at position 9, the ethyl group on ring D, and the β-methoxyacrylate moiety (Figure 4). The aryl methoxy group at position 9 was of particular interest, as in the classical morphinan and benzomorphan opioid scaffolds, the change from aryl methoxy to phenol at key position 3 (e.g. codeine to morphine), results in a dramatic increase in potency. Likewise, unsubstituted phenyl analogs in these scaffolds also exhibit weak activity.51 The importance of a phenolic moiety in many known MOR ligands is proposed to be due to formation of a water-mediated hydrogen bonding network with H2976.52 (superscript numbers refer to Ballesteros and Weinstein’s generic numbering scheme52), as shown in two recent X-ray crystal structures of MOR.37,53 Accordingly, it was hypothesized that if mitragynine binds in a similar pose, then the corresponding desmethyl analog 1 (Figure 4) would be expected to exhibit significantly increased potency. We also envisioned simplification of the D ring substituents by selective deletion of the enol ether (2) or pendant ethyl group (3) (Figure 4), analogs which would probe previously unexplored SAR in this region.
Figure 4.

Key SAR locants of Mitragyna alkaloids (highlighted in green) and select analogs designed to interrogate each position.
Enantioselective Total Synthesis of Mitragynine and Analogs
Although 7-OH and the desmethyl derivative 1 were prepared from the isolated natural product (Scheme S1), we also desired a total synthesis of mitragynine that would allow for the construction of more extensively modified analogs. Accordingly, we envisioned a synthesis starting from 4-methoxyindole, where ring C could be installed via Bischler-Napieralski reaction to give a 3,4-dihydro-β-carboline. This intermediate would then be subjected to an enantioselective, proline-catalyzed Mannich-Michael-type cyclization with the appropriate enone to install ring D and set the stereocenter at position 3. This transformation was developed by Itoh and coworkers, and has been successfully employed in the total synthesis of several structurally similar indole alkaloids.54–56 The resulting tetracyclic ketone would serve as a versatile intermediate for the synthesis of a variety of analogs, including mitragynine itself (Figure 5).
Figure 5.

Retrosynthesis of mitragynine.
The required 3,4-dihydro-β-carboline (4) was successfully prepared in a six step sequence starting from commercially available 4-methoxyindole (21% overall yield, Scheme S2A). The required enone (5) was synthesized from methyl 2-ethyl-3-oxobutanoate according to previously described procedures (Scheme S2B).57
With the necessary building blocks in hand, we attempted the key proline-mediated step for formation of ring D. Initial experiments with crude β-carboline 4 treated with an excess of enone 5 in the presence of proline, afforded the desired ketones in moderate yield as a mixture of epimers 6 and 7. Importantly, we found that the major diastereomer 6 was obtained in high enantiomeric excess (ee), while the minor diastereomer 7 was obtained in much lower ee. After optimization of the reaction conditions (it proved crucial to purify the β-carboline by chromatography), the desired ketone isomer 6S could be obtained in good yield and excellent enantiomeric excess (Scheme 1A). To rationalize the stereoselectivity of this transformation, a two step mechanism (Scheme 1B, D-proline shown) can be imagined, in which a Mannich-like reaction between the proline enamine of enone 5 and the β-carboline 4, first sets the stereocenter at position 3. This is followed by intramolecular Michael addition to close the ring. In the last step, proline controls the face of protonation during hydrolysis, thereby also influencing the stereocenter at position 19. Proline is believed to be involved in control of the stereochemistry at the 19-position, as the diastereomeric ratio obtained in the reaction varies, and is different from that observed when either ketone product (6 or 7) is epimerized under basic conditions. Thus, the orientation of this stereocenter is not due simply to thermodynamic equilibration of the ketone product.
Scheme 1.

Stereoselective Formation of D-ring
With key intermediate ketone 6S in hand, the next challenge of the synthesis was to effect epimerization of the pendant ethyl group into the appropriate β stereochemical configuration (as in mitragynine). This could not be accomplished by thermodynamic equilibration of the ketone with base or acid, as the desired stereochemistry positions the ethyl group in the less favorable axial configuration. As an alternative strategy, we envisioned that pseudo-allylic strain in the ene-ester product of a Horner-Wadsworth-Emmons (HWE) reaction, or in the corresponding transition state, might be used to perturb the equilibrium away from the equatorial alignment.58 Therefore, ketone 6S was treated with the carbanion formed from methyl diethylphosphonoacetate, and partial epimerization was indeed observed (Scheme 2A). The ene-ester with the desired axial ethyl group was obtained as a mixture of E and Z isomers, 8E and 8Z (55 mol% of total product by NMR), while the ene-ester with retention of configuration at the ethyl group, 9, was obtained exclusively as the E isomer (45 mol% of total product by NMR). Unfortunately, the low reactivity of ketone 6S necessitated the use of large excesses of phosphonoaceate, a requirement that led to partial transesterification of the methyl ester (products 8 and 9 contained ~25 mol% of the corresponding ethyl ester).
Scheme 2.

Total Synthesis of Mitragynine in Both Enantiomeric Forms
With the ethyl group successfully epimerized to the appropriate position, the mixed ene-esters 8E/Z were converted to (-)-mitragynine by a sequence of several key steps, including the simultaneous reduction of the ene-ester and detosylation with magnesium, transesterification, Claisen condensation (formylation), and O-methylation59 of the enol-ester intermediate 10 (Scheme 2A). The final O-methylation step provided both (-)-mitragynine and the isomeric analog (Z)-mitragynine (11) in modest yields. Starting from ketone 6R and employing the same synthetic sequence, the unnatural enantiomer, (+)-mitragynine, was also obtained (Scheme 2B). Comparison of this material to the natural product by chiral HPLC confirmed their opposite stereochemistry, and thus, the absolute stereochemistry of the ketone products 6 and 7 obtained in the proline-catalyzed cyclization.
Synthesis of Desethylmitragynine
Considering that speciogynine, which is epimeric with mitragynine at the pendant ethyl group on ring D, was inactive as an opioid agonist in vitro, we reasoned that the nature and orientation of the substituent at this position was important for activity, and thus decided to further define the tolerance of the scaffold in this region via synthesis of the desethylmitragynine analog 3 (Figure 4). This dramatically modified analog was prepared by a synthetic sequence analogous to that applied for the synthesis of mitragynine, starting with the treatment of β-carboline 4 with methyl vinyl ketone in the presence of D-proline (Scheme S3).
Opioid Activity of Synthetic Analogs
With a variety of semi- and fully synthetic mitragynine derivatives in hand, key SAR of this molecular scaffold at the opioid receptors could be further explored. In particular, the synthesized analogs allowed investigation of the key moieties highlighted above, namely the aryl methoxy group at position 9, the β-methoxyacrylate moiety, and the ethyl group on ring D (see above, SAR Exploration: Strategy), as well as the importance of the absolute stereochemistry. As expected, the unnatural enantiomer (+)-mitragynine was found be a much weaker agonist at hMOR, in terms of both potency and maximal efficacy (Table 3). Surprisingly however, in contrast to naturally occurring (-)-mitragynine, it was found to be a low potency partial agonist at hKOR (EC50 = 9.1 ± 4.3 µM; Emax = 31%). Apparently, inversion of the stereochemistry in this scaffold is sufficient to switch from antagonist to agonist activity at hKOR.
Table 3.
Agonist Activity of Synthetic Analogs at hMOR as Determined in G Protein BRET Assays
| Compound | Structure |
EC50 ± SEM (% efficacy) (µM) a |
|---|---|---|
| (-)-mitragynine | 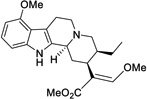 |
0.339 ± 0.178 (34%) |
| (+)-mitragynine | 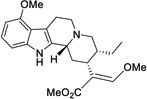 |
3.34 ± 1.1 (18%) |
| 1 | 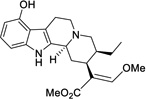 |
0.681 ± 0.379 (29%) |
| 2 | 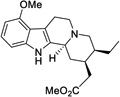 |
>50 |
| 11 | 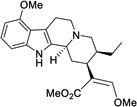 |
0.219 ± 0.071 (38%) |
| 3 | 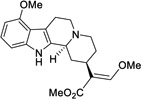 |
12 ± 7.6 (59%) |
Agonist activity at hMOR indicated by EC50 values, maximal efficacy (Emax) relative to DAMGO in parentheses; All data points represent mean ± SEM (µM) of n ≥ 3.
In examining the specific functional groups identified above (Figure 4), we observed several important trends. For one, the switch from a methoxy group to a hydroxy group at position 9 had very little effect on activity at hMOR, with the phenolic analog 1 being of similar potency and efficacy to mitragynine itself (Table 3). This result suggests that the mitragynine scaffold is likely to adopt a unique binding pose compared to classical morphinan- and benzomorphan-derived opioids, which display a dramatic relationship between their MOR activity and an exposed phenol group. Moving to the β-methoxyacrylate moiety, removal of the enol ether, as in the total synthesis intermediate 2, completely eliminated activity at hMOR (both agonist and antagonist). In contrast, the very close analog (Z)-mitragynine (11), was found to possess very similar activity to the natural product, indicating that stereochemical inversion of the β-methoxyacrylate moiety is tolerated (Table 3). Lastly, we turned our attention to the ethyl group on ring D. Results with speciogynine (Table 1) had already demonstrated that epimerization of this group was sufficient to switch from agonist to antagonist activity at hMOR, while also reducing binding affinity (see above, Tables 1 and 2, mitragynine vs. speciogynine). Examining the desethylmitragynine analog 3, we found that this derivative retained agonist activity at hMOR, but with much lower potency (Table 3). Thus, the substituent at position 19 is important for controlling both efficacy (agonist vs. antagonist) and potency. The synthetic derivatives described here ((+)-mitragynine, 1, 2, 3, and 11) were also found to be without activity at hKOR and hDOR (agonist and antagonist mode, EC50 and IC50 both >30 µM).
Molecular Docking Reveals a Unique Binding Pose
In order to rationalize our empirical SAR observations in the mitragynine scaffold and inform future rational design, we conducted preliminary molecular docking studies. Specifically, AutoDock60 was used to dock mitragynine and several of the analogs described above in the binding pocket of the agonist-bound X-ray crystal structure of mMOR (PDBID = 5C1M)37 (Figure 6).
Figure 6.

Docking of (-)-mitragynine and other analogs to the active µ-opioid receptor crystal structure. Top-scoring binding poses of (A) (-)-mitragynine, (B) (Z)-mitragynine, (C) 7-hydroxmitragynine, and (D) antagonists paynantheine (pink) and speciogynine (cyan). Only residue sidechains within 4 Å of the ligand are reported. Polar interactions are shown as dotted lines. TM helices are shown in cartoon representation (in gray). ECL2 and part of TM5 have been omitted for clarity. Residues are labeled using one-letter amino acid code and Ballesteros and Weinstein’s generic numbering scheme.
The top-scored binding pose of (-)-mitragynine in the orthosteric site of the receptor partially overlapped with the binding pose of the co-crystallized morphinan-derived agonist, BU72, but exhibited some dramatic differences (compare Figure 6A to Figure S7). Although (-)-mitragynine and BU72 share a polar interaction between their protonated amines and D1473.32, which is well known to be critical for the binding of classical opioid agonists and antagonists37,53, other important ligand-receptor contacts are significantly different. In BU72 (Figure S7), the phenol occupies a hydrophobic pocket formed by TM5 and TM6 and forms a water-mediated hydrogen bonding network with H2976.52, an interaction that is common to opioid ligands in the morphinan class (see above for discussion, SAR Exploration: Strategy).36,51,53 In contrast, our docking studies suggest that the methoxyindole domain of (-)-mitragynine is preferentially directed toward an alternative hydrophobic pocket formed by residues of TM2 and TM3 (Figure 6A), which appears to lack a means for forming an analogous hydrogen bonding network. This result is in agreement with the minimal activity difference observed in vitro between (-)-mitragynine and phenolic derivative 1 (Table 3). Instead, the enol ether domain (part of the β-methoxyacrylate) of mitragynine appears to be directed into the same region as the phenol of BU72 (toward TM5 and TM6). Thus, it can be speculated that this functional group participates in an analogous hydrogen bonding network with H2976.52, consistent with the complete loss of activity when this group was removed (see compound 2, Table 3).
The importance of the ethyl group on ring D is also explained by the predicted lowest-energy binding pose of (-)-mitragynine. In comparing the orientation of this ligand with that of BU72 (Figure 6A and Figure S7, respectively), it can be seen that the ethyl group occupies a nearly identical region as the N-methyl group of BU72, being in the vicinity of several hydrophobic residues (M1513.36, Y1483.33, and Y3267.43). Accordingly, loss of this group, as in compound 3, would be expected to diminish hydrophobic ligand-receptor interactions and reduce activity, as was observed (Table 3).
Docking of other mitragynine analogs described above also produced results that were in agreement with the in vitro activity data. For example, in accord with its similar in vitro activity to (-)-mitragynine, (Z)-mitragynine (11) adopted a nearly identical binding pose (Figure 6B). Likewise, the predicted low-energy binding pose of 7-OH was also similar, with all key functionalities (the acrylate, ethyl group, tertiary amine, and indole) occupying very similar positions (Figure 6C). Interestingly, the 7-position hydroxy group does not appear to make a close contact with the receptor surface, and thus, potential hydrogen bonding interactions do not seem to be involved in the significant increase in potency afforded by oxidation at position 7. Instead, the slight bend introduced to the core structure by this modification appears to be more important. Notably, while (-)-mitragynine interacts with W2936.48 through its acrylate group, 7-OH interacts with this residue through both ethyl and acrylate groups, according to the chosen cutoff for apolar interactions (4.5 Å between heavy atoms; see Table S3). In contrast, docking of the unnatural enantiomer, (+)-mitragynine, revealed a dramatically different binding pose, reversed in its overall orientation in the pocket (i.e. with the acrylate projecting toward TM2 and TM3, see Figure S8A and compare with Figure 6A), in agreement with its weak activity in the functional assays.
We also docked paynantheine, speciogynine, and speciociliatine, compounds which bind to hMOR, but exhibit no agonist activity, instead serving as competitive antagonists. Considering paynantheine and speciogynine, the inverted stereochemistry at position 19 (vinyl or ethyl group) induces a significant shift of the indole moiety toward TM1 and away from TM3 (Figure 6D), which results in the formation of additional contacts with TM1 (Y751.39) and TM6 (H3197.36) that are not seen in the docking of the active compounds. Moreover, the hydrophobic interactions between the indole methoxy group and V1433.28 and I1443.29 observed with (-)mitragynine are lost in these analogs, potentially resulting in reduced affinity for the active conformation of the receptor and corresponding antagonist activity. Consistent with the inverted stereochemistry of the core structure at position 3, docking of speciociliatine revealed a completely different binding pose with its indole group extending towards TM3 and forming a polar interaction with Y1483.33 (Figure S8B and Table S3). Notably, unlike all other compounds reported here, the acrylate moiety in speciociliatine is in axial rather than equatorial position, pointing towards TM5 and TM6. All in all, a number of different stereochemical configurations in the Mitragyna alkaloid family appear to be sufficient to retain binding at hMOR, while the absolute stereochemistry found in mitragynine is required for activation of the receptor.
Conclusion
In this work, we have presented the first thorough, in vitro characterization of the opioid receptor pharmacology and signaling of the natural Mitragyna alkaloids in receptor-level functional assays. The partial agonist activity of (-)-mitragynine at MOR is likely to account for many of the effects of Mitragyna speciosa extracts in humans. However, the other major alkaloids isolated from the plant, paynanthiene, speciogynine, and speciociliatine, all exhibited competitive antagonist activity at this receptor. Considering that cumulatively, these secondary alkaloids accounted for an approximately equal percentage of the total alkaloid content compared to mitragynine in our extracts, the gross psychoactive effects of crude plant material are likely to represent a complex interplay of competing agonist and antagonist effects at the opioid receptors. It was also found that the potent oxidized analog 7-OH is not necessarily present in all extracts of plant material (as previously suggested), so the potential contribution of this alkaloid to the effects of Mitragyna speciosa is not general, unless 7-OH is formed as a metabolite.
Exploration with several synthetic mitragynine analogs allowed us to define basic SAR at each of the existing substituents, including in regions (the D ring) that were only accessible to derivatization via total synthesis. This work revealed that opioid activity in the mitragynine scaffold is quite sensitive to structural modification and suggested that the Mitragya alkaloids adopt a unique binding pose at MOR, distinct from that of classical opioids. This possibility was further supported by molecular docking studies at MOR. The first total synthesis of the unnatural enantiomer, (+)-mitragynine, likewise allowed us to confirm that the naturally occurring isomer is the more active opioid agonist.
We also found that mitragynine and 7-OH display biased signaling at MOR, being selective for the G protein pathway. Thus, it is hoped that this scaffold may represent a starting point for the development of novel opioid analgesics with reduced side effects. Relatedly, the finding that, in addition to their activity at MOR, both mitragynine and 7-OH are KOR antagonists, suggests that Mitragyna alkaloids or their synthetic derivatives may also serve as leads for novel dual-action antidepressants. In this regard, mitragynine itself is of particular interest, as it acts as both a low efficacy MOR partial agonist, and a KOR antagonist. Both of these mechanisms have been shown to elicit antidepressant effects in animals, and in the case of MOR, humans as well (see above). However, modulators of the KOR have received considerably more attention in the psychiatry community in recent years, likely due to their apparent lack of abuse potential. Antagonists of this receptor are antidepressant in animal models and oppose a variety of stress-related responses that are believed to be mediated through the endogenous KOR agonists, dynorphins.61–63 Further, several agents in this class have entered clinical trials for MDD, but none have yet reached the market.64 Given the ability of mitragynine to perturb both of these opioid signaling systems in the direction consistent with mood enhancement, it is unsurprising that it has been shown to exhibit antidepressant activity in the forced swim test (a rodent model of depression).65 Therefore, Mitragyna alkaloids also hold promise in the treatment of mood disorders.
Supplementary Material
Acknowledgments
The authors would like to thank Kevin Wulf for illustration of Figure 1, and Professor Bryan L Roth and the National Institute of Mental Health’s Psychoactive Drug Screening Program for their assistance in determining binding affinities at the human opioid receptors.
Work in the Sames laboratory was supported by the Interdisciplinary Research Initiatives Seed (IRIS) Fund Program of Columbia University College of Physicians & Surgeons and G. L. Freeman. Work in the Javitch laboratory was supported by National Institutes of Health (NIH) grants MH054137 and DA022413 (to JJ). Work in the Filizola laboratory was supported by NIH grants DA026434 and DA034049 (to MF). Computations were run on resources available through the Scientific Computing Facility at Mount Sinai and the Extreme Science and Engineering Discovery Environment (XSEDE) under MCB080109N, which is supported by National Science Foundation grant number OCI-1053575. Work in the Majumdar laboratory was supported by NIH grant DA034106 (to SM) and in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Supporting Information
- Synthetic procedures and spectral data for all compounds, copies of NMR spectra, additional binding data at rodent receptors, additional functional data at rodent and human receptors
The authors declare no competing financial interest.
References
- 1.Pasternak GW, Pan Y-X. Pharmacol. Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inturrisi CE. Clin. J. Pain. 2002;18(4 Suppl):S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- 3.National Institute on Drug Abuse. [Accessed September 1, 2015];Overdose Death Rates. https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates.
- 4.Quinones S. Dreamland: The True Tale of America’s Opiate Epidemic. New York: Bloomsbury Press; 2015. [Google Scholar]
- 5.Pasternak GW. Clin. J. Pain. 2010;26(Supplement 10):S3–S9. doi: 10.1097/AJP.0b013e3181c49d2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grinnell SG, Majumdar S, Narayan A, Le Rouzic V, Ansonoff M, Pintar JE, Pasternak GW. J. Pharmacol. Exp. Ther. 2014;350:710–718. doi: 10.1124/jpet.114.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Largent-Milnes T, Yamamoto T, Nair P, Moulton J, Hruby V, Lai J, Porreca F, Vanderah T. Br. J. Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevenson GW, Luginbuhl A, Dunbar C, LaVigne J, Dutra J, Atherton P, Bell B, Cone K, Giuvelis D, Polt R, Streicher JM, Bilsky EJ. Pharmacol. Biochem. Behav. 2015;132:49–55. doi: 10.1016/j.pbb.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kraepelin E. Einführung in die psychiatrische Klinik: Zweiunddreissig Vorlesungen. Barth: Leipzig; 1905. [Google Scholar]
- 10.Gerner RH, Catlin DH, Gorelick DA, Hui KK, Li CH. Arch. Gen. Psychiatry. 1980;37:642–647. doi: 10.1001/archpsyc.1980.01780190040005. [DOI] [PubMed] [Google Scholar]
- 11.Stoll AL, Rueter S. Am. J. Psychiatry. 1999;156:2017. doi: 10.1176/ajp.156.12.2017. [DOI] [PubMed] [Google Scholar]
- 12.Dean AJ, Bell J, Christie MJ, Mattick RP. Eur. Psychiatry. 2004;19:510–513. doi: 10.1016/j.eurpsy.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Shapira NA, Verduin ML, DeGraw JD. J. Clin. Psychiatry. 2001;62:205–206. doi: 10.4088/jcp.v62n0312b. [DOI] [PubMed] [Google Scholar]
- 14.Shapira NA, Keck PE, Goldsmith TD, McConville BJ, Eis M, McElroy SL. Depress. Anxiety. 1997;6:170–173. [PubMed] [Google Scholar]
- 15.Emrich HM, Vogt P, Herz A. Ann. N. Y. Acad. Sci. 1982;398:108–112. doi: 10.1111/j.1749-6632.1982.tb39483.x. [DOI] [PubMed] [Google Scholar]
- 16.Karp JF, Butters MA, Begley AE, Miller MD, Lenze EJ, Blumberger DM, Mulsant BH, Reynolds CF. J. Clin. Psychiatry. 2014;75:e785–e793. doi: 10.4088/JCP.13m08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodkin JA, Zornberg GL, Lukas SE, Cole JO. J. Clin. Psychopharmacol. 1995;15:49–57. doi: 10.1097/00004714-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Besson A, Privat AM, Eschalier A, Fialip J. Psychopharmacology. 1996;123:71–78. doi: 10.1007/BF02246283. [DOI] [PubMed] [Google Scholar]
- 19.Rojas-Corrales MO, Berrocoso E, Gibert-Rahola J, Micó JA. Life Sci. 2002;72:143–152. doi: 10.1016/s0024-3205(02)02220-8. [DOI] [PubMed] [Google Scholar]
- 20.Fichna J, Janecka A, Piestrzeniewicz M, Costentin J, do Rego J-C. Neuropsychopharmacology. 2007;32:813–821. doi: 10.1038/sj.npp.1301149. [DOI] [PubMed] [Google Scholar]
- 21.Rojas-Corrales MO, Gibert-Rahola J, Micó JA. Life Sci. 1998;63:PL175–PL180. doi: 10.1016/s0024-3205(98)00369-5. [DOI] [PubMed] [Google Scholar]
- 22.Gassaway MM, Rives M-L, Kruegel AC, Javitch JA, Sames D. Transl. Psychiatry. 2014;4:e411. doi: 10.1038/tp.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takayama H. Chem. Pharm. Bull. 2004;52:916–928. doi: 10.1248/cpb.52.916. [DOI] [PubMed] [Google Scholar]
- 24.Adkins JE, Boyer EW, McCurdy CR. Curr. Top. Med. Chem. 2011;11:1165–1175. doi: 10.2174/156802611795371305. [DOI] [PubMed] [Google Scholar]
- 25.Raffa RB, Beckett JR, Brahmbhatt VN, Ebinger TM, Fabian CA, Nixon JR, Orlando ST, Rana CA, Tejani AH, Tomazic RJ. J. Med. Chem. 2013;56:4840–4848. doi: 10.1021/jm400143z. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto K. Ph.D. Dissertation. Chiba, Japan: Chiba University; 2006. Pharmacological Studies on 7-Hydroxymitragynine, Isolated from the Thai Herbal Medicine Mitragyna speciosa: Discovery of an Orally Active Opioid Analgesic. [Google Scholar]
- 27.Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, Watanabe K, Murayama T, Horie S. J. Med. Chem. 2002;45:1949–1956. doi: 10.1021/jm010576e. [DOI] [PubMed] [Google Scholar]
- 28.León F, Habib E, Adkins JE, Furr EB, McCurdy CR, Cutler SJ. Nat. Prod. Commun. 2009;4:907–910. [PMC free article] [PubMed] [Google Scholar]
- 29.Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai S. Planta Med. 1994;60:580–581. doi: 10.1055/s-2006-959578. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto K, Horie S, Ishikawa H, Takayama H, Aimi N, Ponglux D, Watanabe K. Life Sci. 2004;74:2143–2155. doi: 10.1016/j.lfs.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai S, Aimi N, Watanabe H. Life Sci. 1996;59:1149–1155. doi: 10.1016/0024-3205(96)00432-8. [DOI] [PubMed] [Google Scholar]
- 32.Macko E, Weisbach JA, Douglas B. Arch. Int. Pharmacodyn. Ther. 1972;198:145–161. [PubMed] [Google Scholar]
- 33.Boyer EW, Babu KM, Adkins JE, McCurdy CR, Halpern JH. Addiction. 2008;103:1048–1050. doi: 10.1111/j.1360-0443.2008.02209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai S, Aimi N, Watanabe H. Eur. J. Pharmacol. 1996;317:75–81. doi: 10.1016/s0014-2999(96)00714-5. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto K, Narita M, Muramatsu N, Nakayama T, Misawa K, Kitajima M, Tashima K, Devi LA, Suzuki T, Takayama H, Horie S. J. Pharmacol. Exp. Ther. 2014;348:383–392. doi: 10.1124/jpet.113.208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dale O, Ma G, Gemelli C, Husni A, McCurdy C, Avery B, Leon J, Furr E, Manly S, Cutler S. Planta Med. 2012;78 P_91. [Google Scholar]
- 37.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loening AM, Fenn TD, Gambhir SS. J. Mol. Biol. 2007;374:1017–1028. doi: 10.1016/j.jmb.2007.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rekas A, Alattia J-R, Nagai T, Miyawaki A, Ikura M. J. Biol. Chem. 2002;277:50573–50578. doi: 10.1074/jbc.M209524200. [DOI] [PubMed] [Google Scholar]
- 41.Luttrell LM, Maudsley S, Bohn LM. Mol. Pharmacol. 2015;88:579–588. doi: 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raehal KM, Schmid CL, Groer CE, Bohn LM. Pharmacol. Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donald J Kyle. In: Research and Development of Opioid-Related Ligands. Ko M-C, Husbands SM, editors. Vol. 1131. Washington, DC: ACS Symposium Series; American Chemical Society; 2013. pp. 177–197. [Google Scholar]
- 44.Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Br. J. Pharmacol. 2012;167:960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. Mol. Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamb K, Tidgewell K, Simpson DS, Bohn LM, Prisinzano TE. Drug Alcohol Depend. 2012;121:181–188. doi: 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen X-T, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. J. Pharmacol. Exp. Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 48.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 49.Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, Neve KA. J. Biol. Chem. 2014;289:33663–33675. doi: 10.1074/jbc.M114.605378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhan X, Gimenez LE, Gurevich W, Spiller BW. J. Mol. Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hedberg MH, Johansson AM, Fowler CJ, Terenius L, Hacksell U. Bioorg. Med. Chem. Lett. 1994;4:2527–2532. [Google Scholar]
- 52.Ballesteros JA, Weinstein H. Receptor Molecular Biology. Vol. 25. Elsevier: Methods in Neurosciences; 1995. pp. 366–428. [Google Scholar]
- 53.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itoh T, Yokoya M, Miyauchi K, Nagata K, Ohsawa A. Org. Lett. 2003;5:4301–4304. doi: 10.1021/ol030103x. [DOI] [PubMed] [Google Scholar]
- 55.Itoh T, Yokoya M, Miyauchi K, Nagata K, Ohsawa A. Org. Lett. 2006;8:1533–1535. doi: 10.1021/ol0530575. [DOI] [PubMed] [Google Scholar]
- 56.Nagata K, Ishikawa H, Tanaka A, Miyazaki M, Kanemitsu T, Itoh T. Heterocycles. 2010;81:1791–1798. [Google Scholar]
- 57.Surber W, Schinz H. Helv. Chim. Acta. 1954;37:1239–1251. [Google Scholar]
- 58.Openshaw HT, Whittaker N277. J. Chem. Soc. 1963:1461–1471. [Google Scholar]
- 59.Bárczai-Beke M, Dörnyei G, Tóth G, Tamás J, Szántay C. Tetrahedron. 1976;32:1153–1159. [Google Scholar]
- 60.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jones RM, Portoghese PS, Carlezon WA. J. Pharmacol. Exp. Ther. 2003;305(1):323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 62.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. J. Neurosci. 2008;28(2):407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bruijnzeel AW. Brain Res. Rev. 2009;62(1):127–146. doi: 10.1016/j.brainresrev.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Urbano M, Guerrero M, Rosen H, Roberts E. Bioorg. Med. Chem. Lett. 2014;24(9):2021–2032. doi: 10.1016/j.bmcl.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 65.Idayu NF, Hidayat MT, Moklas MAM, Sharida F, Raudzah ARN, Shamima AR, Apryani E. Phytomedicine. 2011;18:402–407. doi: 10.1016/j.phymed.2010.08.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.