

Abstract
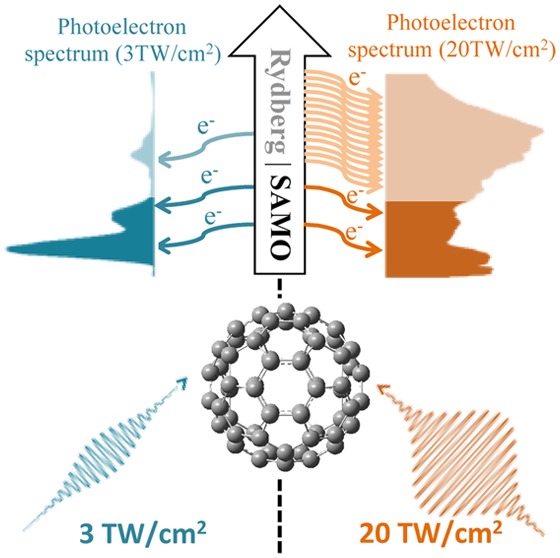
The transition between two distinct ionization mechanisms in femtosecond laser fields at 785 nm is observed for C60 molecules. The transition occurs in the investigated intensity range from 3 to 20 TW/cm2 and is visualized in electron kinetic energy spectra below the one-photon energy (1.5 eV) obtained via velocity map imaging. Assignment of several observed broad spectral peaks to ionization from superatom molecular orbitals (SAMOs) and Rydberg states is based on time-dependent density functional theory simulations. We find that ionization from SAMOs dominates the spectra for intensities below 5 TW/cm2. As the intensity increases, Rydberg state ionization exceeds the prominence of SAMOs. Using short laser pulses (20 fs) allowed uncovering of distinct six-lobe photoelectron angular distributions with kinetic energies just above the threshold (below 0.2 eV), which we interpret as over-the-barrier ionization of shallow f-Rydberg states in C60.
Studying ultrafast electronic and nuclear dynamics in complex systems has become a key research field in recent years.1−9 Among the large variety of target systems, the C60 fullerene has been an archetypical nanoscale system for the investigation of many-body problems. C60 exhibits hybrid properties that bridge the gap between molecular and bulk systems and has a complex response to laser excitation. The behavior of C60 in femtosecond (fs) laser fields usually exhibits an interplay of single-active-electron dynamics in atoms and a multiactive-electron response of polyatomic molecules.10−13 Furthermore, external fields can lead to strong coupling of electronic and nuclear degrees of freedom.14 The ionization and fragmentation of C60 have been investigated with tailored laser pulses.15−17 It was found that the response of C60 to intense laser pulses heavily depends on pulse duration. When excited by ns laser pulses, delayed ionization on microsecond time scales can be observed, which arises from efficient energy redistribution between the electronic and vibrational degrees of freedom.16,18 In fs laser fields (down to tens of fs), prominent peak structures can be observed superimposed on a “thermal” background.19,20 Within time scales of about 100 fs, the excitation energy mostly remains in electronic degrees of freedom, yielding emitted photoelectrons with a much higher effective temperature compared to those obtained in ps laser fields, where significant energy can be transferred to vibrations.14,19,21 The nature of electron emission has been shown to depend on laser fluence for femtosecond pulses at moderate intensities.14,19 Earlier work assigned peaks to ionization from a series of Rydberg states (with n = 3, 5, 7).11 Recently, electronic states arising from excitation of atomic-like molecular orbitals, in which the electronic density is localized both inside and outside of the fullerene cage and that exhibit the nodal patterns of nanometric size hydrogenic orbitals, have been discovered in C60 molecules adsorbed on copper surfaces with scanning tunneling microscopy (STM).22,23 These states were named SAMOs for superatom molecular orbitals. They also have been characterized for isolated gas-phase C60 molecules using photoelectron imaging techniques.24
The properties of SAMO states in fullerenes are of interest for designing nanodevices because they can form nearly free electron bands in solids.23 Exploring SAMO states in the gas phase provides an approach that is free of molecule–substrate interactions and therefore is more easily compared with theoretical results. Systematic theoretical studies of the excited electronic states of C60 based on time-dependent density functional theory (TDDFT) have revealed that the SAMO excited states differ from the Rydberg states in many aspects.25 The SAMO excited states have a significant electronic density inside of or in close vicinity to the fullerene cage, whereas the charge density for Rydberg orbitals localizes tens of Å outside of the fullerene cage. The density inside of the cage stabilizes the excited states, generating low-lying SAMO excited states. SAMO and Rydberg states are also characterized by markedly different photoionization rates.25 Therefore, their photoionization lifetimes, which represent the time that it takes an electronic state population to ionize with a yield of 63% (1 – 1/e) through the interaction with the pulse electric field, are also markedly different.
While both SAMO and Rydberg states have been observed for photoexcited gas-phase C60 molecules,10,24 studies of their relative contribution to the photoelectron spectra (PES) as a function of laser intensity have been hampered by a background of thermal electron emission for longer pulses (of about 100 fs and above). Here, we used 20 fs near-infrared (NIR) laser pulses centered at 785 nm to circumvent this problem and selectively photoionize SAMO or Rydberg states in C60 by tuning the laser intensity. The intensity dependence of the resulting PES clearly shows the transition between ionization of SAMO and Rydberg states for the studied intensity range of 3–20 TW/cm2. Furthermore, the low background in thermal electron emission allowed uncovering of a six-lobe angular distribution at low kinetic energies, the origin of which is illuminated with our calculations.
The experimental setup is introduced in detail in the Supporting Information (SI). Briefly, a beam of 20 fs pulses at a central wavelength of 785 nm is focused into a velocity map imaging (VMI) spectrometer, where it intersects a beam of neutral C60 molecules generated from a homemade oven. The laser intensity is adjusted by a rotatable neutral density filter. The 3-D momentum distributions of the resulting photoelectrons after laser–fullerene interaction are measured with the VMI and postprocessed by Abel inversion26 to extract the 2-D momentum distribution in the plane around pz = 0, where the laser is polarized along the y-axis and propagating along the x-axis (see the SI). The peak intensities in the focal volume are estimated by measuring the above-threshold ionization (ATI) electron emission from Xe under identical experimental conditions.27
The theoretical interpretation of the PES is based on the computed values of binding energies and photoionization lifetimes of 17 bands of SAMO and Rydberg states. The SAMO states are not optically active; therefore, they cannot be accessed directly from the ground state. Several mechanisms including vibronic coupling to optically active valence states, which is estimated to take 50–100 fs,16,18 and dipole coupling between optically active and dark excited states can lead to the population of the SAMO and Rydberg states during the pulse (see the SI for more details). The SAMO and Rydberg states also have large dipole coupling with the p-SAMO states that are in turn vibronically coupled to quasi-isoenergetic bright valence states. Modeling the electron–nuclear dynamics, required to describe the photoexcitation or vibronic coupling during the pulse, is currently out of reach for a large molecule such as C60. Therefore, we do not attempt to explicitly describe the dynamics of the photoexcitation process here. Once the SAMO and Rydberg states are transiently populated during the pulse, they can be ionized by one-photon ionization, leading to sharp peaks in the PES spectrum or by over-the-barrier ionization (OTB) if the lowering of the ionization potential (IP) due to the Stark shift is larger than the binding energy of the states so that they become metastable.
The electronic structure required for the computation of the binding energies and photoionization lifetimes of the SAMO and Rydberg states is carried out at the TDDFT level using the 6-31G(d) basis set augmented by a set of 12 diffuse atomic orbitals of s, p, d, and f symmetry to account for the very diffuse s, p, d, and f-Rydberg states close to the IP. The band of excited electronic states was limited to 500 states by restricting the number of allowed transitions to the excited states.28 The LC-BLYP functional was used with a tuned parameter of 0.21 (ref (30)) so that the Rydberg series converged to the IP. The computed binding energies of the Rydberg states are in good agreement with the Rydberg series of C60 (ref (10)) with a quantum defect of 0.84 (ref (31)). From the electronic states, we computed the photoionization lifetimes and angular distributions.25,32,33 The lifetimes depend on the orbital from which the electron is ionized (i.e., the Dyson orbital), on the kinetic energy of the ionized electron (given by the difference between the IP of the ionized state and the energy of the photon), and on the field strength of the laser. The lifetimes strongly depend on the properties of the Dyson orbitals, in particular, their nodal patterns and diffuse character. The SAMO and Rydberg states have Dyson orbitals25,32,34 with a simple hydrogenic electron distribution and a few nodes, which results in large photoionization coupling elements and small lifetimes. In comparison, for the same kinetic energy and laser intensity, the lifetimes of SAMO and Rydberg states are more than 3 orders of magnitude lower than the ones of the valence excited states. Photoionization lifetimes sharply increase with photoelectron kinetic energy for the SAMO and Rydberg states.25,35 Because the SAMOs have larger binding energies than Rydberg states, the kinetic energy of the ionized electron corresponding to the one-photon photoionization of a SAMO electronic state is lower, and therefore, its lifetime is shorter. The lifetimes are computed for a laser intensity corresponding to the maximum of the laser pulse. However, the laser only reaches its maximum intensity during a limited time, so that only the electronic states with a lifetime much smaller than the pulse duration will ionize during the pulse. Once the pulse is over, the states are stable because they are below the IP and therefore they cannot ionize, except by tunnelling ionization, which is a much slower process.
The momentum distributions of the photoelectron emission
from C60 are measured at several laser intensities in the
range of
3–20 TW/cm2. Four corresponding VMI images are shown
in Figure 1a–d.
The raw VMI images are shown on the left and the Abel-inverted images
on the right half. Rich information can be extracted from these VMI
images. First, characteristic peaks from ATI electron emission with
spacing equal to the photon energy can be observed for radial momenta pr > 0.3 a.u., 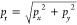 . ATI electron emission
in C60 has been investigated for various pulse durations
down to sub-10
fs.16 The reducing contrast and smoothing
of the angular distribution of the ATI peaks with increasing laser
intensity can be attributed to many-electron effects in ultrafast
ionization of C60 (ref (13)).
. ATI electron emission
in C60 has been investigated for various pulse durations
down to sub-10
fs.16 The reducing contrast and smoothing
of the angular distribution of the ATI peaks with increasing laser
intensity can be attributed to many-electron effects in ultrafast
ionization of C60 (ref (13)).
Figure 1.

(a–d) Momentum distributions of the photoelectron emission from C60 in a 20 fs laser field at intensities of 3.0, 7.5, 10, and 20 TW/cm2, respectively. The uncertainty for the intensity is about 10%. Raw VMI images are shown in the left half. The images in the right half are Abel-inverted and correspond to the 2-D momentum distributions around the pz = 0 plane. The laser is polarized along the y-axis, as indicated by the arrow in (a).
We focus here on the momentum range pr < 0.3 au (which corresponds to the energy range in the PES shown in Figure 2 below 1.5 eV). The PES along the laser polarization axis are obtained by integrating the signal along the py axis within an angular range of ±10° and are shown in Figure 2 for the indicated laser intensities (in units of TW/cm2). For the lowest intensity, that is, 3.0 TW/cm2, three distinct peaks can be distinguished below the one-photon energy. They are around 0.2, 0.5, and 1.0 eV (marked with Peak I, II, and III), with the peak height decreasing for higher kinetic energies and the peak at 0.2 eV dominating.
Figure 2.
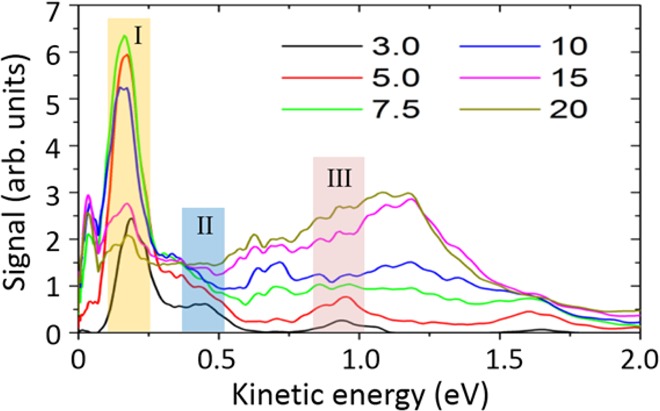
PES for different laser intensities (indicated numbers are in units of TW/cm2) obtained from integration over Abel-inverted experimental VMI images.
On the basis of the binding energies and photoionization lifetimes, we assign the first peak to the p-SAMO, the second one to the d-SAMO, and the third one to the 2p-Rydberg states. The kinetic energy of the peaks corresponds qualitatively to the one-photon ionization of the p- and d-SAMOs and 2p-Rydberg states computed at the TDDFT level (see Figure 3a). The excitation energy and, therefore, the kinetic energy of the ionized electron for the SAMO and Rydberg states are systematically underestimated by 0.1–0.2 eV compared to the experiment, as expected in TDDFT.36 Furthermore, the assignment of the p- and d-SAMO agrees with former work10,11,24 (within 0.1 eV). Among the dense manifold of SAMO and Rydberg states, only the three states listed in Table 1 have a photoionization lifetime shorter than 10 fs for an intensity of 3 TW/cm2, such that they dominate the PES. The other excited states can be populated during the pulse, but they will not photoionize significantly during its ∼20 fs duration because of their longer lifetimes. The computed average lifetime of the Rydberg states is around 40 fs (Table 1). The relative height of the peaks is governed by the excitation process that is not modeled here. The computed value of the lifetimes only allows determining if the electronic state can ionize or not during the pulse.
Figure 3.
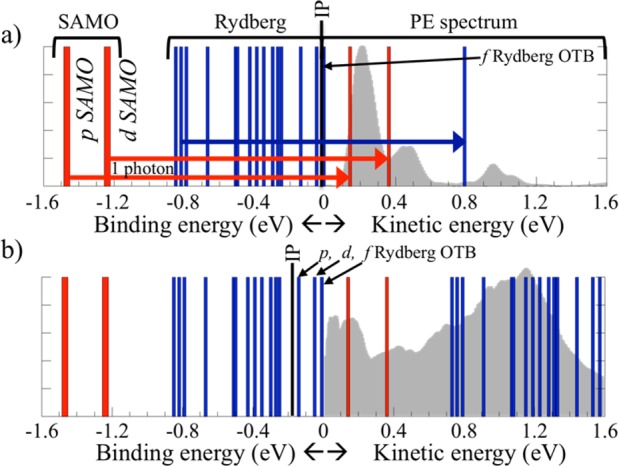
Binding energy of the SAMO and Rydberg states (left) and the experimental photoelectron spectrum (right) for laser intensities of 3 (a) and 20 TW/cm2 (b). In both panels, a bar corresponding to the kinetic energy of the ionized electron is shown when the one-photon photoionization lifetime of an electronic state is shorter than 10 fs. The IP lowered by the Stark shift (0.02 and 0.21 eV for intensities of 3 and 20 TW/cm2, respectively) is shown by a black bar. All of the states above the lowered IP that can photoionize by the OTB mechanism are indicated. The values of the OTB lifetimes have not been computed.
Table 1. Lifetimes (in fs) of C60’s p-SAMO, d-SAMO, and 2p-Rydberg States for the Lowest and Highest Applied Intensitiesa.
| lifetime
(in fs) |
||
|---|---|---|
| 3 TW/cm2 | 20 TW/cm2 | |
| p-SAMO | 3.48 | 0.49 |
| d-SAMO | 4.58 | 0.64 |
| 2p-Rydberg | 9.25 | 1.31 |
| average on the rydberg series | 37.56 | 5.31 |
The mean lifetime of the Rydberg states, computed as the average over the 17 bands of Rydberg states present in the TDDFT computation, is also shown for comparison.
As the laser intensity increases, a contribution in the spectrum at around 0.7 eV appears. For higher laser intensities, a broad peak between 1.0 and 1.5 eV becomes more intense and dominates the spectrum. This peak reflects the ionization of a large band of Rydberg states. For a laser intensity of 20 TW/cm2, both SAMO and Rydberg states have lifetimes shorter than 6 fs (Figure 3b). The density of Rydberg states is significantly larger than that of the SAMO states, which is reflected by the broad and intense peak observed experimentally. The lowest energy peak at around 0.2 eV first increases and then decreases with increasing laser intensity and is strongest at around 5.0–10 TW/cm2 with about 3 times higher peak signal. This trend may be qualitatively understood by the excitation dynamics. When the field is very low, the p-SAMO states, which are lower in energy, should have a larger population than the higher d-SAMO or Rydberg states. As the field strength increases but still is not too strong, the peak grows with population of the p-SAMO state. At higher intensities, all states are populated and the relative height of the p-SAMO peak decreases.
The experimental conditions in our work are similar to those in refs (13), (24), and (37). However, the six-lobe angular distributions for electrons below 0.2 eV have not been reported. The angular distribution can only be clearly observed for low laser intensities and with sufficient signal-to-background ratio. Here, by using a very short pulse duration and low laser intensity, thermal electron emission can be significantly suppressed,19 as can be seen by the signal in the direction perpendicular to the laser polarization (cf. Figure 1). In contrast, in earlier work,24 NIR pulses with about 100 fs duration were used to investigate the ionization from SAMO states of C60. On the time scale of 100 fs, significant nuclear motion, for example via the ag(1)-breathing mode with a period of about 100 fs,15 may play a role and influence the observations by redistribution of the population from optically active states to the SAMO states.
We now turn to the potential origin of the observed six-lobe photoelectron angular distribution (PAD) at low kinetic energy (see Figure 1a). If this contribution comes from photoionization of a p-SAMO (for a representation of the Dyson orbitals of s-, p-, d-, and f-SAMOs, see Figure 4a), the final angular distribution should be a superposition of s and d distributions,33 which cannot explain the six-lobe pattern. The six-lobe structure can also not originate from single-photon ionization from an f-SAMO state as proposed in ref (38) because the corresponding PAD should in this case be a superposition of d and g orbitals.29−41 For the intensities used here, the Stark shift, computed to the second order using C60’s quadrupole moments (the dipole moment is zero in the neutral and cationic ground states), varies between 0.02 and 0.21 eV for a laser intensity varying between 3 and 20 TW/cm2, respectively, which means that several bands of shallow Rydberg states can ionize over the barrier; see Figure 3. Among the 500 excited states computed in TDDFT, there is a band of f-, d-, and p-Rydberg states with binding energies of 0.01, 0.08, and 0.16 eV, respectively. In this strong-field ionization process, the PAD corresponds to the angular distribution of the orbitals from which the electron is ionized.29 Because the barrier is lowered more in the field direction, the ionization probability will be larger along the polarization direction. The unique six-lobe PAD can therefore be attributed to OTB ionization from an f-Rydberg state. The energy-integrated PAD for the range of 0.1–0.2 eV at intensities varying from 3 to 20 TW/cm2 are plotted in Figure 4c. The intensity of the peaks at 60 and 120° increases with the laser intensity because of the isotropic thermal electron emission that also increases with the laser intensity. The agreement of the energy-integrated PAD (for a laser intensity of 3 TW/cm2, where thermal contributions are smallest) with the computed angular distribution of an f orbital (Figure 4b) is excellent in terms of both amplitudes and peak locations, and only small angle offsets can be seen. The separation between the two humps of the f orbital is about 60°, while the measured one is about 50°. The small shift could arise from the Coulomb interaction between the ejected electron and the fullerene cage and/or be due to the extra contribution of a d-type angular distribution from the photoionization of a partly overlapping p-SAMO.
Figure 4.
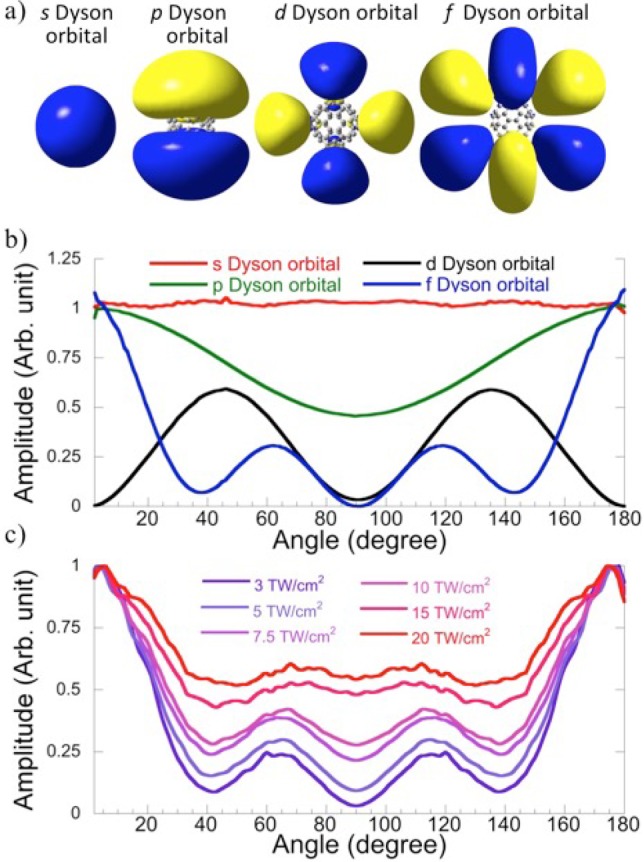
(a) Isocontour amplitude of the Dyson orbitals of a s-, p-, d-, and f-SAMO or Rydberg states. (b) The angular distributions for the Dyson orbitals from (a). (c) Measured PAD from C60 (0.1–0.2 eV) for laser intensities ranging from 3 to 20 TW/cm2 as indicated.
To summarize, we have studied the transition from SAMO to Rydberg state photoionization from C60 fullerenes in intense 20 fs laser fields. By comparison with TDDFT calculations, the peaks between 0.2 and 1.5 eV in the PES can be assigned to ionization from p- and d-SAMOs and Rydberg states. The intensity dependence is interpreted with the photoionization lifetimes of SAMO and Rydberg states, which are selectively ionized when changing the laser intensity. The spectra at low laser intensity are dominated by a peak at kinetic energies below 0.2 eV, which exhibits a six-lobe pattern in the momentum distribution. We interpret the origin of the peak as OTB from f-Rydberg states. Our general findings about the transition from SAMO to Rydberg ionization should also apply to other fullerenes and can help to improve understanding of the photoionization of complex multielectron systems.
Acknowledgments
We acknowledge support by C. Fehrenbach in operating the PULSAR laser system. We are grateful for support by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, Grants DE-FG02-86ER13491 (JRML umbrella grant), DE-SC0012376 and DE-SC0012628 (support of B.M. and F.R.), and DE-SC0008146 (early career award, H.L. and M.F.K.). M.F.K. is grateful for support by the European Union via the ERC grant ATTOCO (Grant No. 307203) and by the DFG via the Cluster of Excellence: Munich Center for Advanced Photonics (MAP). B.M. and F.R. acknowledge support from the Fonds National de la Recherche Scientifique, Belgium. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under Grant No. 2.5020.11.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b02139.
The Supporting Information contains details of the experimental setup and excitation mechanisms of the SAMO and Rydberg states (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fennel T.; Döppner T.; Passig J.; Schaal C.; Tiggesbäumker J.; Meiwes-Broer K. H. Plasmon-Enhanced Electron Acceleration in Intense Laser Metal-Cluster Interactions. Phys. Rev. Lett. 2007, 98, 143401. 10.1103/PhysRevLett.98.143401. [DOI] [PubMed] [Google Scholar]
- Xu H.; Okino T.; Kudou T.; Yamanouchi K.; Roither S.; Kitzler M.; Baltuska A.; Chin S.-L. Effect of Laser Parameters on Ultrafast Hydrogen Migration in Methanol Studied by Coincidence Momentum Imaging. J. Phys. Chem. A 2012, 116, 2686–2690. 10.1021/jp207483y. [DOI] [PubMed] [Google Scholar]
- Xie X.; Doblhoff-Dier K.; Roither S.; Schöffler M. S.; Kartashov D.; Xu H.; Rathje T.; Paulus G. G.; Baltuška A.; Gräfe S.; Kitzler M. Attosecond-Recollision-Controlled Selective Fragmentation of Polyatomic Molecules. Phys. Rev. Lett. 2012, 109, 243001. 10.1103/PhysRevLett.109.243001. [DOI] [PubMed] [Google Scholar]
- Wells E.; Rallis C. E.; Zohrabi M.; Siemering R.; Jochim B.; Andrews P. R.; Ablikim U.; Gaire B.; De S.; Carnes K. D.; Bergues B.; de Vivie-Riedle R.; Kling M. F.; Ben-Itzhak I. Adaptive strong-field control of chemical dynamics guided by three-dimensional momentum imaging. Nat. Commun. 2013, 10.1038/ncomms3895. [DOI] [PubMed] [Google Scholar]
- Kraus P. M.; Mignolet B.; Baykusheva D.; Rupenyan A.; Horný L.; Penka E. F.; Grassi G.; Tolstikhin O. I.; Schneider J.; Jensen F.; Madsen L. B.; Bandrauk A. D.; Remacle F.; Wörner H. J. Measurement and laser control of attosecond charge migration in ionized iodoacetylene. Science 2015, 350, 790–795. 10.1126/science.aab2160. [DOI] [PubMed] [Google Scholar]
- Li H.; Mignolet B.; Wachter G.; Skruszewicz S.; Zherebtsov S.; Süßmann F.; Kessel A.; Trushin S. A.; Kling N. G.; Kübel M.; Ahn B.; Kim D.; Ben-Itzhak I.; Cocke C. L.; Fennel T.; Tiggesbäumker J.; Meiwes-Broer K. H.; Lemell C.; Burgdörfer J.; Levine R. D.; Remacle F.; Kling M. F. Coherent Electronic Wave Packet Motion in C60 Controlled by the Waveform and Polarization of Few-Cycle Laser Fields. Phys. Rev. Lett. 2015, 114, 123004. 10.1103/PhysRevLett.114.123004. [DOI] [PubMed] [Google Scholar]
- Li H.; Kling N. G.; Förg B.; Stierle J.; Kessel A.; Trushin S. A.; Kling M. F.; Kaziannis S. Carrier-envelope phase dependence of the directional fragmentation and hydrogen migration in toluene in few-cycle laser fields. Struct. Dyn. 2016, 3, 043206. 10.1063/1.4941601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kübel M.; Siemering R.; Burger C.; Kling N. G.; Li H.; Alnaser A. S.; Bergues B.; Zherebtsov S.; Azzeer A. M.; Ben-Itzhak I.; Moshammer R.; de Vivie-Riedle R.; Kling M. F. Steering Proton Migration in Hydrocarbons Using Intense Few-Cycle Laser Fields. Phys. Rev. Lett. 2016, 116, 193001. 10.1103/PhysRevLett.116.193001. [DOI] [PubMed] [Google Scholar]
- Schütte B.; Arbeiter M.; Mermillod-Blondin A.; Vrakking M. J. J.; Rouzée A.; Fennel T. Ionization Avalanching in Clusters Ignited by Extreme-Ultraviolet Driven Seed Electrons. Phys. Rev. Lett. 2016, 116, 033001. 10.1103/PhysRevLett.116.033001. [DOI] [PubMed] [Google Scholar]
- Boyle M.; Hoffmann K.; Schulz C. P.; Hertel I. V.; Levine R. D.; Campbell E. E. B. Excitation of Rydberg Series in C60. Phys. Rev. Lett. 2001, 87, 273401. 10.1103/PhysRevLett.87.273401. [DOI] [PubMed] [Google Scholar]
- Boyle M.; Laarmann T.; Hoffmann K.; Hedén M.; Campbell E. E. B.; Schulz C. P.; Hertel I. V. Excitation dynamics of Rydberg states in C60. Eur. Phys. J. D 2005, 36, 339–351. 10.1140/epjd/e2005-00281-7. [DOI] [Google Scholar]
- Scully S. W. J.; Emmons E. D.; Gharaibeh M. F.; Phaneuf R. A.; Kilcoyne A. L. D.; Schlachter A. S.; Schippers S.; Müller A.; Chakraborty H. S.; Madjet M. E.; Rost J. M. Photoexcitation of a Volume Plasmon in C60 Ions. Phys. Rev. Lett. 2005, 94, 065503. 10.1103/PhysRevLett.94.065503. [DOI] [PubMed] [Google Scholar]
- Huismans Y.; Cormier E.; Cauchy C.; Hervieux P. A.; Gademann G.; Gijsbertsen A.; Ghafur O.; Johnsson P.; Logman P.; Barillot T.; Bordas C.; Lépine F.; Vrakking M. J. J. Macro-atom versus many-electron effects in ultrafast ionization of C60. Phys. Rev. A 2013, 88, 013201. 10.1103/PhysRevA.88.013201. [DOI] [Google Scholar]
- Johansson J. O.; Campbell E. E. B. Probing excited electronic states and ionisation mechanisms of fullerenes. Chem. Soc. Rev. 2013, 42, 5661–5671. 10.1039/c3cs60047e. [DOI] [PubMed] [Google Scholar]
- Laarmann T.; Shchatsinin I.; Stalmashonak A.; Boyle M.; Zhavoronkov N.; Handt J.; Schmidt R.; Schulz C. P.; Hertel I. V. Control of Giant Breathing Motion in C60 with Temporally Shaped Laser Pulses. Phys. Rev. Lett. 2007, 98, 058302. 10.1103/PhysRevLett.98.058302. [DOI] [PubMed] [Google Scholar]
- Campbell E. E. B.; Hansen K.; Hoffmann K.; Korn G.; Tchaplyguine M.; Wittmann M.; Hertel I. V. From Above Threshold Ionization to Statistical Electron Emission: The Laser Pulse-Duration Dependence of C60 Photoelectron Spectra. Phys. Rev. Lett. 2000, 84, 2128–2131. 10.1103/PhysRevLett.84.2128. [DOI] [PubMed] [Google Scholar]
- Hertel I. V.; Shchatsinin I.; Laarmann T.; Zhavoronkov N.; Ritze H. H.; Schulz C. P. Fragmentation and Ionization Dynamics of C60 in Elliptically Polarized Femtosecond Laser Fields. Phys. Rev. Lett. 2009, 102, 023003. 10.1103/PhysRevLett.102.023003. [DOI] [PubMed] [Google Scholar]
- Campbell E. E. B.; Levine R. D. Delayed Ionization and Fragmentation en Route to Thermionic Emission: Statistics and Dynamics. Annu. Rev. Phys. Chem. 2000, 51, 65–98. 10.1146/annurev.physchem.51.1.65. [DOI] [PubMed] [Google Scholar]
- Kjellberg M.; Johansson O.; Jonsson F.; Bulgakov A. V.; Bordas C.; Campbell E. E. B.; Hansen K. Momentum-map-imaging photoelectron spectroscopy of fullerenes with femtosecond laser pulses. Phys. Rev. A 2010, 81, 023202. 10.1103/PhysRevA.81.023202. [DOI] [Google Scholar]
- Johansson J. O.; Fedor J.; Goto M.; Kjellberg M.; Stenfalk J.; Henderson G. G.; Campbell E. E. B.; Hansen K. Anisotropic hot electron emission from fullerenes. J. Chem. Phys. 2012, 136, 164301. 10.1063/1.4704828. [DOI] [PubMed] [Google Scholar]
- Hansen K.; Hoffmann K.; Campbell E. E. B. Thermal electron emission from the hot electronic subsystem of vibrationally cold C60. J. Chem. Phys. 2003, 119, 2513–2522. 10.1063/1.1584671. [DOI] [Google Scholar]
- Feng M.; Zhao J.; Petek H. Atomlike, Hollow-Core–Bound Molecular Orbitals of C60. Science 2008, 320, 359–362. 10.1126/science.1155866. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Feng M.; Yang J.; Petek H. The Superatom States of Fullerenes and Their Hybridization into the Nearly Free Electron Bands of Fullerites. ACS Nano 2009, 3, 853–864. 10.1021/nn800834k. [DOI] [PubMed] [Google Scholar]
- Johansson J. O.; Henderson G. G.; Remacle F.; Campbell E. E. B. Angular-resolved Photoelectron Spectroscopy of Superatom Orbitals of Fullerenes. Phys. Rev. Lett. 2012, 108, 173401. 10.1103/PhysRevLett.108.173401. [DOI] [PubMed] [Google Scholar]
- Mignolet B.; Johansson J. O.; Campbell E. E. B.; Remacle F. Probing Rapidly-Ionizing Super-Atom Molecular Orbitals in C60: A Computational and Femtosecond Photoelectron Spectroscopy Study. ChemPhysChem 2013, 14, 3332–3340. 10.1002/cphc.201300585. [DOI] [PubMed] [Google Scholar]
- Vrakking M. J. J. An iterative procedure for the inversion of two-dimensional ion/photoelectron imaging experiments. Rev. Sci. Instrum. 2001, 72, 4084–4089. 10.1063/1.1406923. [DOI] [Google Scholar]
- Kling M. F.; Rauschenberger J.; Verhoef A. J.; Hasović E.; Uphues T.; Milošević D. B.; Muller H. G.; Vrakking M. J. J. Imaging of carrier-envelope phase effects in above-threshold ionization with intense few-cycle laser fields. New J. Phys. 2008, 10, 025024. 10.1088/1367-2630/10/2/025024. [DOI] [Google Scholar]
- Mignolet B.; Remacle F.. Time-efficient computation of the electronic structure of the C60 super-atom molecular orbital (SAMO) states in TDDFT. AIP Conference Proceedings; AIP, 2016; in press. Preprint: http://hdl.handle.net/2268/201371. [Google Scholar]
- Refaely-Abramson S.; Baer R.; Kronik L. Fundamental and excitation gaps in molecules of relevance for organic photovoltaics from an optimally tuned range-separated hybrid functional. Phys. Rev. B 2011, 84, 075144. 10.1103/PhysRevB.84.075144. [DOI] [Google Scholar]
- Boyle M.Energy Absorption and Redistribution Dynamics in Isolated C60 Molecules. Dissertation, FU Berlin, 2010. [Google Scholar]
- Seabra G. M.; Kaplan I. G.; Zakrzewski V. G.; Ortiz J. V. Electron propagator theory calculations of molecular photoionization cross sections: The first-row hydrides. J. Chem. Phys. 2004, 121, 4143–4155. 10.1063/1.1773135. [DOI] [PubMed] [Google Scholar]
- Mignolet B.; Levine R. D.; Remacle F. Localized electron dynamics in attosecond-pulse-excited molecular systems: Probing the time-dependent electron density by sudden photoionization. Phys. Rev. A 2012, 86, 053429. 10.1103/PhysRevA.86.053429. [DOI] [Google Scholar]
- Reisler H.; Krylov A. I. Interacting Rydberg and valence states in radicals and molecules: experimental and theoretical studies. Int. Rev. Phys. Chem. 2009, 28, 267–308. 10.1080/01442350902989170. [DOI] [Google Scholar]
- Bohl E.; Sokół K. P.; Mignolet B.; Thompson J. O. F.; Johansson J. O.; Remacle F.; Campbell E. E. B. Relative Photoionization Cross Sections of Super-Atom Molecular Orbitals (SAMOs) in C60. J. Phys. Chem. A 2015, 119, 11504–11508. 10.1021/acs.jpca.5b10339. [DOI] [PubMed] [Google Scholar]
- Peach M. J. G.; Benfield P.; Helgaker T.; Tozer D. J. Excitation energies in density functional theory: An evaluation and a diagnostic test. J. Chem. Phys. 2008, 128, 044118. 10.1063/1.2831900. [DOI] [PubMed] [Google Scholar]
- Olof Johansson J.; Bohl E.; Henderson G. G.; Mignolet B.; Dennis T. J. S.; Remacle F.; Campbell E. E. B. Hot electron production and diffuse excited states in C70, C82, and Sc3N@C80 characterized by angular-resolved photoelectron spectroscopy. J. Chem. Phys. 2013, 139, 084309. 10.1063/1.4818987. [DOI] [PubMed] [Google Scholar]
- Zhang G. P.; Zhu H. P.; Bai Y. H.; Bonacum J.; Wu X. S.; George T. F. Imaging superatomic molecular orbitals in a C60 molecule through four 800-nm photons. Int. J. Mod. Phys. B 2015, 29, 1550115. 10.1142/S0217979215501155. [DOI] [Google Scholar]
- Reid K. L. Photoelectron Angular Distributions. Annu. Rev. Phys. Chem. 2003, 54, 397–424. 10.1146/annurev.physchem.54.011002.103814. [DOI] [PubMed] [Google Scholar]
- Zare R. N.Angular Momentum: Understanding Spatial Aspects in Chemistry and Physics; Wiley: New York, 1988. [Google Scholar]
- Cooper J.; Zare R. N. Angular Distribution of Photoelectrons. J. Chem. Phys. 1968, 48, 942–943. 10.1063/1.1668742. [DOI] [Google Scholar]
- Oana C. M.; Krylov A. I. Cross sections and photoelectron angular distributions in photodetachment from negative ions using equation-of-motion coupled-cluster Dyson orbitals. J. Chem. Phys. 2009, 131, 124114. 10.1063/1.3231143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.