


Abstract
Muscle-type carnitine palmitoyltransferase 1 (CPT1β) is considered to be the gene that controls fatty acid mitochondrial β-oxidation. A functional peroxisome proliferator-activated receptor (PPAR) responsive element (PPRE) and a myocite-specific (MEF2) site that binds MEF2A and MEF2C in the promoter of this gene had been previously identified. We investigated the roles of the PPRE and the MEF2 binding sites and the potential interaction between PPARα and MEF2C regulating the CPT1β gene promoter. Mutation analysis indicated that the MEF2 site contributed to the activation of the CPT1β promoter by PPAR in C2C12 cells. The reporter construct containing the PPRE and the MEF2C site was synergistically activated by co-expression of PPAR, retinoid X receptor (RXR) and MEF2C in non-muscle cells. Moreover, protein-binding assays demonstrated that MEF2C and PPAR specifically bound to one another in vitro. Also for the synergistic activation of the CPT1β gene promoter by MEF2C and PPARα-RXRα, a precise arrangement of its binding sites was essential.
INTRODUCTION
The incorporation of activated long-chain fatty acids into the mitochondria, which are then catabolized through β-oxidation, is carried out by the mitochondrial carnitine palmitoyltransferase (CPT) system. CPT1 [reviewed in (1)], the outer membrane component of this system, is the main regulatory step in the β-oxidation pathway. CPT1 is thus a suitable site for the pharmacological control of fatty acid oxidation, potentially useful in situations such as diabetes (2) or heart disease (3). CPT1 is encoded by at least two genes known as L-CPT1 (or -α) and M-CPT1 (or -β) on the basis of the tissues, liver (L-) or muscle (M-), where the expression of each one was first described. However, CPT1β is expressed, in addition to skeletal muscle, in heart, testis and brown and white adipose tissue, whereas CPT1α has a more widespread distribution. CPT1β expression increases in the heart after birth [in terms of Vmax, (4)] or after fasting [in terms of mRNA (5)] concomitant with an increase in circulating levels of fatty acids. This expression pattern may be of great significance since fatty acids are the major source of energy for heart, skeletal muscle and brown adipose tissue.
We (6), and others (5,7), had shown that CPT1β is a target gene for the action of peroxisome proliferator-activated receptors (PPARs), nuclear receptor transcription factors that are regulated by fatty acids and derivative metabolites (8–13), and had localized a PPAR responsive element (PPRE) upstream of the first exon of this gene. There are three related PPAR family members, PPARα, PPARγ and PPARδ. According to several lines of evidence, PPARα, through the regulation of CD36/FAT (14), acyl-CoA synthetase (15), CPT1β (5–7), CPTII (16) and medium-chain acyl-CoA dehydrogenase (17) stimulates, respectively, fatty acid transport into the cell, its activation, import into the mitochondria and β-oxidation.
In the human CPT1β gene promoter, the PPRE is flanked by one E-box motif and a myocite-specific (MEF2) binding site; this organization is highly conserved in the mouse, sheep and rat CPT1β gene (18). The MEF2 site of the CPT1β gene binds MEF2A and MEF2C (19), which are members of the MADS family of transcription factors that have been implicated in the regulation of muscle-selective gene expression (20–25). The PPARα-mediated regulation of the CPT1β gene is enhanced by the PPARγ coactivator-1 (PGC-1) (26) that also interacts with MEF2C to up-regulate GLUT4 expression (27) or MEF2A to stimulate CPT1β expression (28). To gain more insight into the tissue-specific control of CPT1β gene expression, we examined the basis of the specific expression of this gene in muscle cells. We conclude that the activity of the CPT1β promoter is controlled by a synergistic mechanism involving MEF2C and the heterodimer PPARα–RXRα, through a physical interaction between PPAR and MEF2C in a distance-dependent manner with sequences closely localized upstream of exon 1A.
MATERIALS AND METHODS
Plasmids
pCPTluc containing a 380 bp fragment of the human CPT1β gene was constructed by PCR using a pair of oligonucleotide primers, CPTF2 and CPTR2 (for sequences of all oligonucleotides used in this work, see Table 1), corresponding to coordinates −905 to −883 and −525 to −541 from the translation origin, respectively, of the human CPT1β gene, and pCPT-CAT (6) as template. After 10 cycles (94°C for 1 min; 58°C for 1 min and 72°C for 1.5 min), the amplified product was purified in a 1% agarose gel, and the KpnI–SmaI digest was cloned into pGL3Basic (Promega).
Table 1. Oligonucleotides used for EMSA and constructions (see Materials and Methods for details).
| Oligonucleotide | Sequence |
|---|---|
| CPTF2 | GGGGTACCTGCAGCTTAGAATAATAAATAC |
| CPTR2 | TCCCCCGGGCCACGTCCTTCAGGCCTA |
| Mef-F | AGCTTTTGGCTATTTTTAGCTCTAAAG |
| Mef-R | TCGACTTTAGAGCTAAAAATAGCCAAA |
| mutMef-F | AGCTTTTGGATCATTGTTGCTCTAAAG |
| mutMef-R | TCGACTTTAGAGCATCAATGAACCAAA |
| CAS-F1 | CTAGCAGCAGCTGACACATCGGTGACCTTTTCCCTACATTTGGCTATTTTTAGC |
| CAS-R1 | GCTAAAAATAGCCAAATGTAGGGAAAAGGTCACCGATGTGTCAGCTGCTG |
| CAS-F3 | GTGACCTTTTCCCTACATTTGGATCATTGTTGC |
| CAS-R3 | TCGAGCAACAATGATCCAAATGTAGGGAAAAG |
| CPT-M5-F | ACATTTGACGTCGCTATTTTTAGCTCTAATGC |
| CPT-M5-R | AATAGCGACGTCAAATGTAGGGAAAAGGTC |
| CPT-M10-F | TTTGACGTCTCTACGCTATTTTTAGCTCTAATGC |
| CPT-M10-R | AGCCTAGAGACGTCAAATGTAGGGAAAAGGTC |
| PPAR-MEF-F | ATCGGTGACCTTTTCCCTACATTTGGCTATTTTTAGCTCTAA |
| PPAR-MEF-R | TTAGAGCTAAAAATAGCCAAATGTAGGGAAAAGGTCACCGAT |
| PPAR-mutMEF-F | CGGTGACCTTTTCCCTACATTTGGATCATTGTTGCTC |
| PPAR-mutMEF-R | GAGCAACAATGATCCAAATGTAGGGAAAAGGTCACCG |
pCPTmutMEF, containing point mutations at the MEF-2 sequence, was obtained by site-directed mutagenesis overlapping extension PCR, as described previously (29), using the oligonucleotides CPT-F2, CPT-R2, and mutMef-F and mutMef-R (which introduce a mutation in MEF-2 site).
pCPT-B211, containing 46 bp flanking de PPRE upstream of exon 1A of the CPT1β gene, was constructed by cloning in NheI–SmaI-digested pGL3Basic (Promega), two complementary oligonucleotides, CAS-F1 and CAS-R1. pCPT-B211mutMEF2 was generated by digesting pCPT-B211 with BstEII and XhoI and cloning the oligonucleotides CAS-F3 and CAS-R3.
pCPT-M5 and pCPT-M10, containing either 5 or 10 bp between the MEF2 and the PPRE sites, respectively, were obtained by site-directed mutagenesis overlapping extension PCR, as described in (29), using the oligonucleotides CPT-F2, CPT-R2, CPT-M5-F, CPT-M5-R, CPT-M10-F and CPT-M10-R
pMEF2, containing the putative MEF-2 site, was obtained by cloning a pair of oligonucleotides (Mef-F and Mef-R) into the NheI–XhoI-digested pGL3Promoter. To confirm the sequence, all constructs were automatically sequenced using the fluorescent terminator kit (Perkin–Elmer).
pSG5-PPARα, pJCXRα and pCDNA-MEF2C contained the cDNAs for mouse PPARα, human RXRα and human MEF2C, respectively. pRL-CMV (Promega) contained the Renilla luciferase gene under the control of the cytomegalovirus (CMV) intermediate-early enhancer/promoter.
PGEXhPPARα, producing a glutathione S-transferase (GST–full-length human PPARα fusion protein), has been described previously (16).
Cell growth and differentiation
The CV-1 cells were cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS). Chinese hamster ovary (CHO) cells were cultured in Ham's F12 medium supplemented with 10% FBS. C2C12 myoblasts were grown in DMEM supplemented with 10% FBS (growth medium), and shifted to DMEM supplemented with 10% horse serum (HS) (differentiation medium) to allow acquisition of the myotube phenotype. All cells and subsequent experiments were maintained under 5% CO2 at 37°C.
Transient transfection assays
Typically, 0.5–1 × 105 cells (CV-1 and CHO) or 2.5–3 × 105 cells (C2C12) were cotransfected in 6-well plates with 1.5 μg of the reporter gene construct and 0.2 μg (except when indicated) of effector plasmids expressing full-length cDNAs for PPARα, RXRα, MEF2C or PGC-1; 40 ng of plasmid pRL-CMV was included as internal control. Cotransfections were carried out by the calcium phosphate method for CV-1 and CHO cells as described previously (30), and cells were harvested 48 h after transfection. For C2C12 cells, transfection FuGENE-6 reagent (Roche) was used, as indicated by the manufacturer; after 24 h in DMEM + 10% FBS, the medium was changed to DMEM + 10% HS to allow differentiation from myoblasts to myotubes, and 48 h later, cell extracts were obtained.
Firefly luciferase and Renilla luciferase assays
The cell extracts were prepared using the Passive Lysis Buffer method (Promega), and firefly and Renilla luciferase activities were determined in a TD-20/20 Luminometer (Turner Designs) using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer's recommendations.
In vitro transcription and translation
MEF2C, PPARα and RXRα were transcribed and translated by using commercially available kits according to the manufacturer's instructions (Promega).
Electrophoretic mobility shift assay
An aliquot of 2.5 μl of each factor synthesized in vitro was preincubated on ice for 10 min in 10 mM Tris–HCl (pH 8.0), 40 or 100 mM KCl for PPAR or MEF2C, respectively, 0.05% (v/v) Nonidet P-40, 6% glycerol, 1 mM dithiotheitol and 2 μg of poly(dI–dC). The total amount of reticulocyte lysate was kept constant in each reaction through the addition of unprogrammed lysate. When indicated, 1.5 μl of specific antibody (anti-MEF2 from SantaCruz Biotechnology) was added to the reaction mixture. Next, 2 ng of probe, 32P-labeled by 5′ end-labeling with T4 polynucleotide kinase, was added and the incubation was continued for 15 min at room temperature. The final volume for all the reactions was 20 μl. Samples were electrophoresed at 4°C on a 4.5% polyacrylamide gel in 0.5% TBE buffer [45 mM Tris, 45 mM boric acid and 1 mM EDTA (pH 8.0)], and the gel was dried and exposed to an autoradiographic film.
GST-pull down assay
GST and GST–PPARα fusion proteins were expressed in Escherichia coli and purified on glutathione–sepharose beads (Amersham Pharmacia Biotech) as described previously (31). Amounts and integrity of GST were checked by SDS–PAGE and Comassie Blue staining. 35S-methionine labeled MEF2C (4 μl) was incubated in the presence of equivalent quantities of immobilized GST or GST-PPARα in 1 ml of binding buffer [NETN + 0.5% milk + protease inhibition cocktail (Sigma)] for 4 h at 4°C with agitation. Then the samples were centrifuged for 1 min at 2000 r.p.m. and the resin was washed twice with NETN at room temperature. After that the samples were boiled, mixed with 2× Laemli Buffer and resolved by SDS–PAGE. Labeled proteins were visualized by fluorograpy.
RESULTS
The response of CPT1β promoter to PPARα is cell line specific
In order to examine the responsiveness of the CPT1β gene to PPARα on different tissular contexts, we performed a series of transient transfections in several cell lines derived from different tissues: CV-1 (kidney), CHO (ovary) and C2C12 (muscle). pCPTluc, a plasmid containing a 380 bp fragment of the human CPT1β promoter (Figure 1A), was used as reporter. In all cells studied, the basal expression of the construct was similar (data not shown) but, unexpectedly, induction mediated by PPARα/RXRα was higher in C2C12 myotubes, in which a 38-fold induction from basal activity was observed (Figure 1B). Analysis of the 5′ flanking region of the human CPT1β gene by the TFSEARCH routine showed the presence of a putative myogenic binding sequence (MEF-2 site) located at coordinates −759 to −744 from initial ATG, and neighboring the PPRE (Figure 1A). The higher PPARα responsiveness in C2C12 cells was correlated with the presence of proteins able to bind to MEF2 sites in the C2C12 nuclear extracts (data not shown). The positive correlation between the presence of such myogenic proteins and the increase in the response to PPARα observed in C2C12 suggested a mechanism of synergy between myogenic proteins and the heterodimer PPARα/RXRα.
Figure 1.
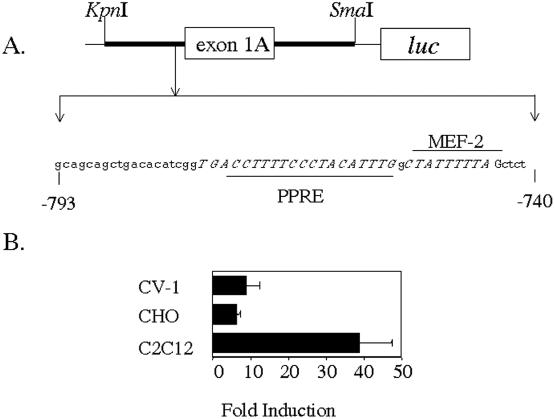
Increased response of the CPT1β promoter to PPARα in C2C12 cells. (A) Schematic diagram of pCPTluc reporter, which contains the gene luc under control of the 5′ flanking region of the human CPT1β gene. Sequences of interest are depicted in italics. (B) CV-1, CHO and C2C12 cells were transiently transfected with pCPTluc (1.5 μg) in the absence or in the presence of PPARα/RXRα, as indicated (0.2 μg each). At 48 h (CV-1 and CHO) or 72 h (C2C12) after transfection, reporter activity was assayed in cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments.
The MEF-2 sequence binds MEF2C: mutation of these sequences affects PPARα responsiveness
We performed gel mobility shift assays to analyze whether MEF2C binds to the putative MEF2 site of the CPT1β gene. Figure 2A shows how, when the wild-type sequence (lanes 1–3) was used as a probe in electrophoretic mobility shift assays (EMSAs), in vitro-transcribed and in vitro-translated MEF2C was able to promote shifted bands, whereas the mutation of the MEF2 site (lanes 4 and 5) abolished such binding. Moreover, specific MEF2 antibodies promote a supershifted band (lane 3). To confirm the functional importance of this cis element on the transcriptional activity of the CPT1β promoter, we generated a construct in which one copy of the MEF-2 sequence was cloned upstream of an SV40-driven luc reporter and then transfected this plasmid in C2C12 cells. This plasmid showed an increased basal expression when compared to the empty vector (Figure 2B), thus identifying these sequence as a functional MEF2 binding site. To confirm the function of this site on the CPT1β gene promoter activity, we analyzed the effect of the overexpression of MEF2C on the activity of a luciferase reporter construct driven either by the wild type or a MEF2 site mutant version of the CPT1β promoter on CV-1 cells. Figure 2C shows how the response to MEF2C is eliminated by the mutation of the MEF2 site. To further investigate the synergy between MEF2C and PPARα, a luc reporter construct was generated in which a 46 bp fragment of the CPT1β gene promoter flanking the PPRE was cloned directly upstream of the luc gene (pCPT-B211). Furthermore, mutation of the MEF-2 site (pCPT-B211mutMEF2) was performed as well. When assaying relative basal activities for each plasmid in C2C12 cells (Figure 2D), we found that the mutation of the MEF-2 element, which did not affect the PPAR-RXR binding (Figure 2A; lanes 6–9), surprisingly, did not affect basal activity in C2C12 cells but, in contrast, dramatically reduced the activity observed in the presence of PPARα/RXRα when compared to the wild-type promoter construct.
Figure 2.
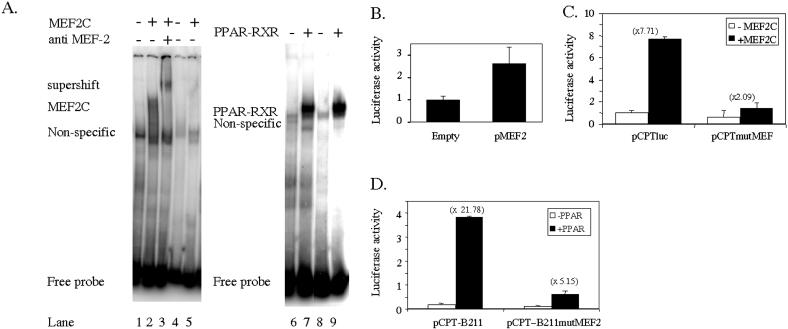
MEF-2 site bind MEF2C, and abolition of this sequence affects MEF2C responsiveness and PPARα transactivation. (A) In vitro-translated MEF2C, PPARα and RXRα were incubated with labeled probes containing the wild-type sequence (lanes 1–3, 6 and 7) (PPAR-MEF-F/R) or the wild-type PPRE sequence and a mutated version of the MEF2 site (lanes 4, 5, 8 and 9) (PPAR-mutMEF-F/R) (Table 1). When indicated, specific antibodies were added to the reaction mix. (B) The C2C12 cells were transiently transfected with plasmids containing one copy of the MEF-2 site upstream of an SV40-driven luc reporter, and basal activity of the constructs was assayed against the empty vector. (C) The CV-1 cells were transiently transfected with pCPTluc or pCPTmutMEF, a construct containing a mutated MEF2 site in the absence or in the presence of a MEF2C expression vector, as indicated. At 48 h after transfection, the reporter activity was assayed in the cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments. (D) A 46 bp fragment of the 5′ flanking region of the human CPT1β gene was cloned in pGL3Basic and subsequent MEF-2 site scrambling was performed (see Materials and Methods for details), generating the indicated reporter constructs. The C2C12 cells were transfected with these plasmids in the absence or in the presence of 0.2 μg of PPARα, and 72 h later, the reporter activities were assayed in the cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments.
PPARα/RXRα and MEF2C cooperate at the transcriptional level in the CPT1β gene promoter
In order to check the synergy between MEF2C and the PPARα/RXRα, we carried out transient transfections in CV-1 cells using pCPTluc as reporter and assaying the effect of cotransfection with expression vectors for PPARα, RXRα and MEF2C. To analyze the effect of the presence of ligand, we performed these experiments using charcoal-stripped serum in combination with the presence or absence of a synthetic ligand for PPARα. As shown in Figure 3, MEF2C induces the expression of CPT1β promoter (lane 5), confirming the role of these proteins in muscle-specific transcription of this gene. It also promotes a synergistic activation when coexpressed with PPARα/RXRα in the presence of ligand (compare lanes 4, 5 and 7). Taken together, results from Figures 1–3 confirm the existence of a synergy between MEF2C and the heterodimer PPARα/RXRα. Because PGC-1 is known to interact with both PPAR and MEF2C, we examined whether PGC-1 might further augment the response of the CPT1β promoter to PPAR in C2C12 cells. As shown in Figure 4, the presence of PGC-1 actually increases the response to PPAR.
Figure 3.
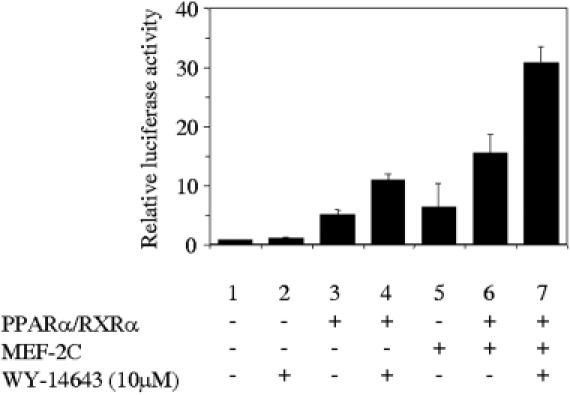
PPARα/RXRα and MEF2C synergistically activate the human CPT1β gene promoter. The CV-1 cells were transiently transfected with pCTPluc in the absence or in the presence of 0.2 μg PPARα, RXRα and 1 μg of MEF2C expression vectors, as indicated. At 16 h after transfection, cells were changed to charcoal-stripped serum and 8 h later Wy-14643, a specific ligand for PPARα, was added. At 48 h after transfection, the reporter activity was assayed in the cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments.
Figure 4.
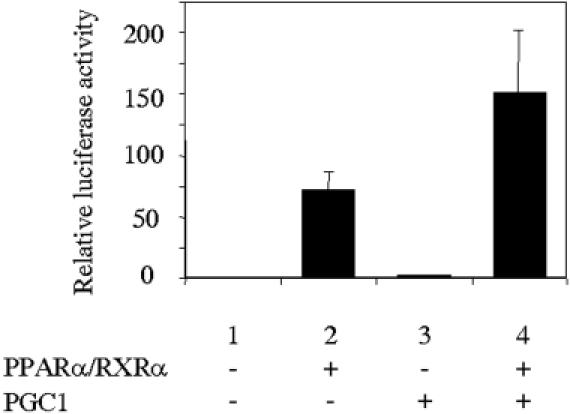
PGC-1 increases the response of the human CPT1β gene promoter to PPAR in C2C12 cells. The C2C12 cells were transiently transfected with pCTPluc in the absence or in the presence of 0.2 μg of PPARα, RXRα and 1 μg of PGC-1 expression vectors, as indicated. At 72 h after transfection, the reporter activity was assayed in the cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments.
MEF2C and PPARα physically associate in vitro
The cooperative effects of PPAR and MEF2C on transcriptional activation raise the possibility that these two proteins physically interact with each other. The direct and specific physical interaction of PPARα and MEF2C was examined by using in vitro protein-binding assays. Radioactive in vitro-translated MEF2C was incubated with GST-PPARα. After incubation, proteins bound to the GST fusion protein were subjected to SDS–PAGE and fluorography. As shown in Figure 5A, the MEF2C band was observed when the products of the specific in vitro translation were incubated with GST–PPARα but not with GST beads. In order to check whether this interaction was ligand dependent, we performed the same experiment with limiting amounts of the GST–PPARα protein in the presence or in the absence of a PPARα specific ligand. Figure 5B shows how this interaction is independent of the presence of the ligand.
Figure 5.
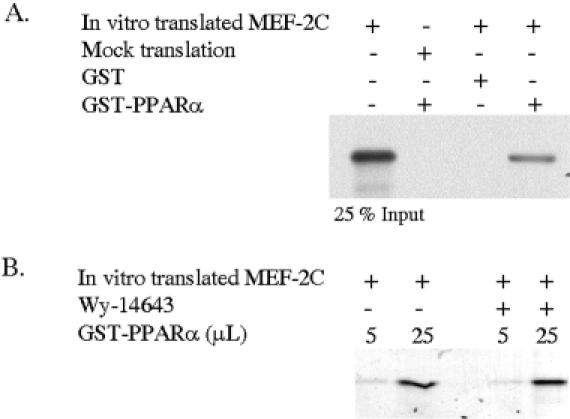
MEF2C and PPAR physically associate in vitro. (A) In vitro-transcribed and in vitro-translated 35S-methionine-labeled MEF2C or the result of a mock translation with the empty vector were incubated with GST or GST–PPARα fusion protein in the presence or in the absence of Wy-14643 as indicated. The bound proteins were subjected to SDS–PAGE and fluorography. (B) The in vitro-translated MEF2C (25% input) represents the fourth part of the total radiolabeled translated protein used in the pull down assay.
Synergy between MEF2C and PPARα is influenced by the separation between them
To further investigate the synergy between MEF2C and PPARα, a collection of luc reporter constructs were generated in which the mutation of the MEF-2 site that abolished the binding of MEF2C was performed in the context of the human CPT1β gene promoter (pCPTmutMEF). We also made different versions in which either a sequence of 5 nt (half turn) (pCPT-M5) or 10 nt (a whole turn) (pCPT-M10) of DNA helix were introduced to separate the MEF2 site from the PPRE sequences in pCPTluc. While assaying relative basal activities for each plasmid in C2C12 cells (Figure 6A), we found that the mutation of the MEF2C responsive sequence produces a little decrease in basal activity and a clear reduction of the activity in the presence of PPAR.
Figure 6.
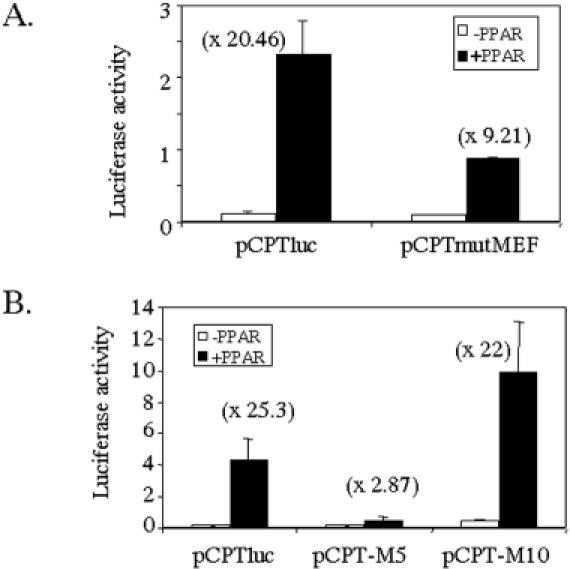
MEF-2 site contribution to muscle-specific expression of the human CPT1β gene. The C2C12 cells were transiently transfected with (A) either pCPTluc or pCPTmutMEF, a construct containing a mutated MEF2 site, (B) mutated versions of pCPTluc, pCPT-M5 or pCPT-M10 containing, respectively, a half (5 nt) or a whole turn (10 nt) of DNA helix between the MEF2 site and the PPRE sequences. When indicated, 0.2 μg of PPARα and RXRα expression vectors were added. At 72 h after transfection, the reporter activities were assayed in the cell lysates after normalization with Renilla luciferase (internal control). Bars represent normalized means ± SD of three independent experiments.
Showing the functionality of the interaction between PPAR and MEF2C, only those constructs that maintained the natural spatial disposition of the binding sites for these proteins respect de DNA helix (pCPT and pCPT-M10) were able to show high PPARα responsiveness, whereas those in which the spatial arrangement was disrupted (pCPT-M5) failed (Figure 6B). Taken together, these results demonstrate that the interaction and synergy between MEF2C and PPARα/RXRα show a strong dependence on a precise arrangement of activator recognition sites.
DISCUSSION
Interaction between transcription factors that bind to different sequences within a promoter can lead to synergistic effects on transcriptional activation. In this report, we show a synergistic activation of the CPT1β gene promoter by the heterodimer of nuclear receptors PPARα-RXRα and the myogenic factor MEF2C. We and others had previously demonstrated that the human CPT1β gene is a target for PPARs and had localized the PPAR responsive element (PPRE) upstream of the first exon (6–8). It has been previously shown that the CPT1β gene promoter contains a MEF2 binding site upstream of exon 1A, flanking the PPRE (19). The vicinity of these DNA elements and the enhanced PPARα responsiveness in muscle cells led us to assess the effect of this nuclear receptor in the presence of myogenic factors. Here, we show that MEF2C and PPAR interact to synergistically activate the CPT1β gene expression. Mutation of the MEF2 binding site dramatically affects PPARα responsiveness in C2C12 cells, or after the coexpression of MEF2C in non-muscle (CV-1) cells; the alteration of the natural arrangement of the PPAR and MEF2 binding sites also influences the response to PPAR.
It has been suggested recently that PPARδ plays a pivotal role in the control of both the program for fatty acid oxidation and the CPT1β promoter in the skeletal muscle (32,33). Although not shown in this paper, PPARδ and PPARγ are also able to physically interact with MEF2C in vitro, and therefore, this interaction could be physiologically relevant in different tissues with different levels of the PPAR isotypes.
The identity of the protein that binds to the MEF2 sequence of the CPT1β gene in muscle cannot be entirely established. Till date, four different MEF2 proteins have been identified MEF2A, MEF2B, MEF2C and MEF2D. In skeletal muscle cells in culture, MEF2D has been reported to be expressed in proliferating myoblasts prior to the onset of differentiation, MEF2A protein appears as cells enter the differentiation pathway and MEF2C is expressed late in the differentiation program (34). In agreement with the expression patterns of MEF2 proteins in myotube culture and with previously published observations that both MEF2A and MEF2C bind to the CPT1β MEF2 binding site (19), we believe that MEF2C is responsible for the activation of the CPT1β gene.
PGC-1 is a coactivator of MEF2C and can control the level of endogenous GLUT4 gene expression in muscle (27). It has also been shown that PGC-1 and MEF2A synergistically activate the CPT1β gene promoter (28). Our results point to the possibility of a simultaneous interaction between PPAR, MEF2C and PGC1 forming a ternary complex and creating a unique surface that interacts with the transcription machinery more efficiently that any of the individual factors alone. The localization of the regions involved in the interaction between PPAR and MEF2C will help us to solve this issue. Alternatively, the interaction of each of these factors could modify the chromatin structure, facilitating the action of the next. In this sense, Huang et al. (35) demonstrated that promoter context affects the transcriptional activation by MyoD through E-boxes, so that neighboring proteins can recruit different associated factors to the promoter, or influence the conformation of the promoter DNA and/or of the protein-E-box complex.
A number of studies have examined the transactivating properties of the MEF2 proteins, which were originally studied in their role as transcriptional regulators of myogenic cells, including interactions with other transcription factors such as myogenin and MyoD (36,37). In particular, several studies have reported the physical or functional interaction between myogenic proteins and members of the nuclear receptor superfamily. Thus, it has been demonstrated that MEF-2A and the TR receptor interact synergistically to activate the α-cardiac MHC gene expression (38). A functional cooperation between MyoD, MEF2 and TRα1 is sufficient for the induction of GLUT4 gene transcription (39). The interaction between MyoD and RARα has also been demonstrated (40) and a functional interaction between MyoD and PPAR proposed (41). The transcriptional coactivator PGC-1 mediates an increase in GLUT4 expression, by binding to and coactivating MEF2C (27). Other coactivators such as CBP/p300 (25) or the Glucocorticoid Receptor Interacting Protein 1 (GRIP-1) (42), which functions as a cofactor for several nuclear receptors, has been shown to physically interact with both MyoD and MEF-2. The particular spatial arrangement of PPAR-, MEF2- and MyoD-binding sequences along the human CPT1β promoter is highly conserved in mouse, sheep and rat CPT1β genes (18). Besides the interaction between PPARα and MEF2C shown in this paper, interactions between RXR or PPAR and MyoD are probable, but they are yet to be demonstrated.
In summary, the combination of cis elements in the promoter of the CPT1β gene maximally induces the expression of this gene in response to a combination of different signals. Thus, it seems likely that the concurrence of myogenic and metabolic signals generates a transcriptionaly permissive conformation of the CPT1β gene promoter that gives rise to a synergistic maximal transcription of the gene in those tissues containing the corresponding transcription factors (MEF2C, PPARα/RXRα) and in the presence of the metabolic substrate of the enzyme, the fatty acids that activate PPARα.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to Drs S. Green, R. M. Evans, E. N. Olson, and to B. M. Spiegelman for supplying the expression vectors for PPARα, RXRα, MEF2C and PGC-1. We are also grateful to Robin Rycroft of the Language Service for valuable assistance in the preparation of the English manuscript. This research was supported by Grants PB97-0958 and SAF2001-2923-C02-01 from Dirección General de Investigación Científica y Técnica, a Grant from the FIS of the Instituto de Salud Carlos III, Red de Centros RCMN (C03/08) and the Fundació la Marató de TV3. A.B. and J.R. were Predoctoral fellows from Generalitat de Catalunya.
REFERENCES
- 1.McGarry J.D. and Brown,N.F. (1997) The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem., 244, 1–14. [DOI] [PubMed] [Google Scholar]
- 2.Prentki M. and Corkey,B.E. (1996) Are the beta-cell signaling molecules malonyl-CoA and cystolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes, 45, 273–283. [DOI] [PubMed] [Google Scholar]
- 3.Corr P.B. and Yamada,K.A. (1995) Selected metabolic alterations in the ischemic heart and their contributions to arrhythmogenesis. Herz, 20, 156–168. [PubMed] [Google Scholar]
- 4.Brown N.F., Weis,B.C., Husti,J.E., Foster,D.W. and McGarry,J.D. (1995) Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J. Biol. Chem., 270, 8952–8957. [DOI] [PubMed] [Google Scholar]
- 5.Yu G., Lu,Y. and Gulick,T. (1998) Co-regulation of tissue-specific alternative human carnitine palmitoyltransferase Ibeta gene promoters by fatty acid enzyme substrate. J. Biol. Chem., 273, 32901–32909. [DOI] [PubMed] [Google Scholar]
- 6.Mascaró C., Acosta,E., Ortiz,J.A., Marrero,P.F., Hegardt,F.G. and Haro,D. (1998) Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. J. Biol. Chem., 273, 8560–8563. [DOI] [PubMed] [Google Scholar]
- 7.Brandt J.M., Djouadi,F. and Kelly,D.P. (1998) Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J. Biol. Chem., 273, 23786–237892. [DOI] [PubMed] [Google Scholar]
- 8.Kliewer S.A., Sundseth,S.S., Jones,S.A., Brown,P.J., Wisely,G.B., Koble,C.S., Devchand,P., Wahli,W., Willson,T.M., Lenhard,J.M. and Lehmann,J.M. (1997) Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl Acad. Sci. USA, 94, 4318–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forman B.M., Ruan,B. Chen,J., Schroepfer,G.J.,Jr and Evans,R.M. (1997) The orphan nuclear receptor LXRalpha is positively and negatively regulated by distinct products of mevalonate metabolism. Proc. Natl Acad. Sci. USA, 94, 10588–10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kliewer S.A., Forman,B.M., Blumberg,B., Ong,E.S., Borgmeyer,U., Mangelsdorf,D.J., Umesono,K. and Evans,R.M. (1994) Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl Acad. Sci. USA, 91, 7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoonjans K., Staels,B. and Auwerx,J. (1996) Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J. Lipid Res., 37, 907–925. [PubMed] [Google Scholar]
- 12.Gottlicher M., Widmark,E., Li,Q. and Gustafsson,J.A. (1992) Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl Acad. Sci. USA, 89, 4653–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu K., Bayona,W., Kallen,C.B., Harding,H.P., Ravera,C.P., McMahon,G., Brown,M. and Lazar,M.A. (1995) Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem., 270, 23975–23983. [DOI] [PubMed] [Google Scholar]
- 14.Sato O., Kuriki,C., Fukui,Y. and Motojima,K. (2002) Dual promoter structure of mouse and human FAT/CD36 genes and unique transcriptional activation by PPAR alpha and gamma ligands. J. Biol. Chem., 277, 15703–15711. [DOI] [PubMed] [Google Scholar]
- 15.Schoonjans K., Watanabe,M., Suzuki,H., Mahfoudi,A., Krey,G., Wahli,W., Grimaldi,P., Staels,B., Yamamoto,T. and Auwerx,J. (1995) Induction of the acyl-coenzyme A synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J. Biol. Chem., 270, 19269–19276. [DOI] [PubMed] [Google Scholar]
- 16.Barrero M.J., Camarero,N., Marrero,P.F. and Haro,D. (2003) Control of human carnitine palmitoyltransferase II gene transcription by peroxisome proliferator-activated receptor through a partially conserved peroxisome proliferator-responsive element. Biochem. J., 369, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulick T., Cresci,S., Caira,T., Moore,D.D. and Kelly,D.P. (1994) The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl Acad. Sci. USA, 91, 11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Der Leij F.R., Cox,K.B., Jackson,V.N., Huijkman,N.C., Bartelds,B., Kuipers,J.R., Dijkhuizen,T., Terpstra,P., Wood,P.A., Zammit,V.A. and Price,N.T. (2002) Structural and functional genomics of the CPT1B gene for muscle-type carnitine palmitoyltransferase I in mammals. J. Biol. Chem., 277, 26994–27005. [DOI] [PubMed] [Google Scholar]
- 19.Moore M.L., Wang,G.-L., Belaguli,N.S., Schwartz,R.J. and McMillin,J.B. (2001) GATA-4 and serum response factor regulate transcription of the muscle-specific carnitine palmitoyltransferase I beta in rat heart. J. Biol. Chem., 276, 1026–1033. [DOI] [PubMed] [Google Scholar]
- 20.Edmonson D.G. and Olson,E.N. (1993) Helix–loop–helix proteins as regulators of muscle-specific transcription. J. Biol. Chem., 268, 755–758. [PubMed] [Google Scholar]
- 21.Murre C., McCaw,P.S., Vaessin,H., Caudy,M., Jan,L.Y., Jan,Y.N., Cabrera,C.V., Buskin,J.N., Hauschka,S.D. and Lassar,A.B. (1989) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell, 58, 537–44. [DOI] [PubMed] [Google Scholar]
- 22.Lassar A.B., Davis,R.L., Wright,W.E., Kadesch,T., Murre,C., Voronova,A., Baltimore,D. and Weintraub,H. (1991) Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell, 66, 305–315. [DOI] [PubMed] [Google Scholar]
- 23.Olson E.N., Perry,M. and Schulz,R.A. (1995) Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev. Biol., 172, 2–14. [DOI] [PubMed] [Google Scholar]
- 24.Martin J.F., Schwarz,J.J. and Olson,E.N. (1993) Myocyte enhancer factor (MEF) 2C: a tissue-restricted member of the MEF-2 family of transcription factors. Proc. Natl Acad. Sci. USA, 90, 5282–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sartorelli V., Huang,J., Hamamori,Y. and Kedes,L. (1997) Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell. Biol., 17, 1010–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vega R.B., Huss,J.M. and Kelly,D.P. (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol., 20, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michael L.F., Wu,Z., Cheatham,R.B., Puigserver,P., Adelmant,G., Lehman,J.J., Kelly,D.P. and Spiegelman,B.M. (2001) Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl Acad. Sci. USA, 98, 3820–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore L.M., Park,E.A. and McMillin,J.B. (2003) Upstream stimulatory factor represses the induction of carnitine palmitoyltransferase-Iβ expression by PGC-1. J. Biol. Chem., 278, 17263–17268. [DOI] [PubMed] [Google Scholar]
- 29.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 30.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1987) Current Protocols in Molecular Biology. Green Publishing Associates/Wiley-Interscience, NY. [Google Scholar]
- 31.Elbrecht A., Chen,Y., Adams,A., Berger,J., Griffin,P., Klatt,T., Zhang,B., Menke,J., Zhou,G., smith,R.G. and Moller,D. (1999) L-764406 is a partial agonist of human peroxisome proliferator-activated receptor γ. The role of Cys313 in ligand binding. J. Biol. Chem., 274, 7913–7922. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T., Yamamoto,J., Iwasaki,S., Asaba,H., Hamura,H., Ikeda,Y., Watanabe,M., Magoori.K., Ioka,R.X., Tachibana,K., Watanabe,Y., Uchiyama,Y., Sumi,K., Iguchi,H., Ito,S., Doi,T., Hamakubo,T., Naito,M., Auwerx,J., Yanagisawa,M., Kodama,T. and Sakai,J. (2003) Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl Acad. Sci. USA, 100, 15924–15929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dressel U., Allen,T.L., Pippal,J.B., Rohde,P.R., Lau,P. and Muscat,G.E. (2003) The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol., 17, 2477–2493. [DOI] [PubMed] [Google Scholar]
- 34.Black B.L. and Olson,E.N. (1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol., 14, 167–196. [DOI] [PubMed] [Google Scholar]
- 35.Huang J., Blackwell,T.K., Kedes,L. and Weintraub,H. (1996) Differences between MyoD DNA binding and activation site requirements revealed by functional random sequence selection. Mol. Cell. Biol., 16, 3893–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gossett L.A., Kelvin,D.J., Sternberg,E.A. and Olson,E.N. (1989) A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol., 9, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaushal S., Schneider,J.W., Nadal-Ginard,B. and Mahdavi,V. (1994) Activation of the myogenic lineage by MEF2A, a factor that induces and cooperates with MyoD. Science, 266, 1236–1240. [DOI] [PubMed] [Google Scholar]
- 38.Lee Y., Nadal-Ginard,B., Mahdavi,V. and Izumo,S. (1997) Myocyte-specific enhancer factor 2 and thyroid hormone receptor associate and synergistically activate the alpha-cardiac myosin heavy-chain gene. Mol. Cell. Biol., 17, 2745–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santalucia T., Moreno,H., Palacin,M., Yacoub,M.H., Brand,N.J. and Zorzano,A. (2001) A novel functional co-operation between MyoD, MEF2 and TRalpha1 is sufficient for the induction of GLUT4 gene transcription. J. Mol. Biol., 314, 195–204. [DOI] [PubMed] [Google Scholar]
- 40.Froesché A., Alric,S., Kitzmann,M., Carnac,G., Auradé,F., Rochette-Egly,C. and Bonnieu,A. (1998) Retinoic acid receptors and muscle b-HLH proteins: partners in retinoid-induced myogenesis. Oncogene, 16, 3369–3378. [DOI] [PubMed] [Google Scholar]
- 41.Solanes G., Pedraza,N., Iglesias,R., Giralt,M. and Villarroya,F. (2003) Functional relationship between MyoD and peroxisome proliferator-activated receptor-dependent regulatory pathways in the control of the human uncoupling protein-3 gene transcription. Mol Endocrinol., 17, 1944–1958. [DOI] [PubMed] [Google Scholar]
- 42.Chen S.L., Dowhan,D.H., Hosking,B.M. and Muscat,G.E. (2000) The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev., 10, 1209–1214. [PMC free article] [PubMed] [Google Scholar]