


Abstract
Differences between species have been suggested to largely reside in the network of connections among the genes. Nevertheless, the rate at which these connections evolve has not been properly quantified. Here, we measure the extent to which co-regulation between pairs of genes is conserved over large phylogenetic distances; between two eukaryotes Caenorhabditis elegans and Saccharomyces cerevisiae, and between two prokaryotes Escherichia coli and Bacillus subtilis. We first construct a reliable set of co-regulated genes by combining various functional genomics data from yeast, and subsequently determine conservation of co-regulation in worm from the distribution of co-expression values. For B.subtilis and E.coli, we use known operons and regulons. We find that between 76 and 80% of the co-regulatory connections are conserved between orthologous pairs of genes, which is very high compared with previous estimates and expectations regarding network evolution. We show that in the case of gene duplication after speciation, one of the two inparalogous genes tends to retain its original co-regulatory relationship, while the other loses this link and is presumably free for differentiation or sub-functionalization. The high level of co-regulation conservation implies that reliably predicted functional relationships from functional genomics data in one species can be transferred with high accuracy to another species when that species also harbours the associated genes.
INTRODUCTION
With the availability of complete genome sequences, it has become clear that the perceived large differences in the phenotype of organisms do not correspond to equally large differences in their gene repertoire (1–3). Instead, it has been proposed that these differences largely reside in the network of connections among genes (2–7). This implies that the wiring must evolve fast: e.g. transcription regulation is supposed to evolve relatively fast (3,6,8,9) and the gene regulatory network across organisms reveals extensive variations (4).
We are flooded with high-throughput experiments to determine the functional links between genes and proteins: functional genomics data such as those derived from proteomics or transcriptomics (10–12). As these data come from multiple species, they open the possibility of studying the evolution of functional links. At the same time, these data raise the question as to what extent findings from functional genomics experiments are transferable from, e.g. yeast, where most of these techniques are piloted, to other organisms such as human. Co-regulation is one such type of connection and it is an important facet of the cellular network for which abundant data is available (10,11). Here, we study the evolution of co-regulation, with the aim of determining the degree of conservation of the co-regulatory link between two genes across species.
Previous studies that compared the co-regulation between species did not focus on the degree of conservation of co-regulatory links, but rather on the large-scale properties of the regulatory network or on gene function prediction. The studies that focussed on the network did not address the level of conservation of co-regulation between genes, but did report similarities in terms of global network features (5,13–15). The function-prediction studies have shown that the conservation of co-expression, or arguably an approximation of that, drastically increase the accuracy of microarray data for gene function prediction (4,5,16,17). To the extent that these function-prediction studies did address conservation, they implied that co-regulation is poorly conserved except for functionally tightly associated genes, e.g. genes that code for physically interacting proteins.
At any rate, it is not trivial to measure the degree of co-regulation conservation from functional genomics data or genome sequence data. There are two important issues when measuring the degree of evolutionary conservation of co-regulation between two species A and B. First, the set of genes that are selected as co-regulated in species A might actually contain too many genes that are not co-regulated (false positives). For example, even when using a high threshold of correlation in co-expression (r), genes could still be spuriously expressed as suggested by the poor performance of using co-expression alone for the prediction of functional interaction (12,16), and by its limited correlation with shared transcription factor binding sites (TFBSs) (18). Second, assessing the absence of co-regulation in species B is not trivial. When, e.g. the same strict co-expression threshold to find co-regulated genes in species A is subsequently used to define the absence in species B, many truly co-regulated pairs below this threshold are missed (i.e. many false negatives). A similar example is to equate the lack of gene order conservation in prokaryotes to the lack of co-regulation conservation because this indicates operon disruption. Yet, this also can lead to many false negatives because genes that are not in the same operon can still belong to the same regulon and thus can still be co-regulated (Figure 1). The low level of co-regulation conservation that has been previously hinted at might thus very well be the result of methodological pitfalls.
Figure 1.
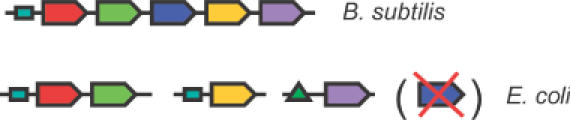
Measuring conservation of co-regulation between E.coli and B.subtilis. This figure illustrates the various contexts in E.coli that the genes from an operon in B.subtilis can occur in. The various possibilities should be taken into account when measuring conservation of co-regulation between the two species: genes can be absent from E.coli (the blue gene), genes can still be together in an operon (the red and green gene); genes can be in different operons but still share the same transcription factor (the red/green genes versus the yellow gene) or a gene can be in another operon and be regulated by a different transcription factor (the purple gene versus the others).
Here, we directly measure the extent to which co-regulation is conserved between two eukaryotes, the nematode Caenorhabditis elegans and the budding yeast Saccharomyces cerevisiae, and between two well-studied prokaryotes, the proverbial workhorse of bacterial molecular genetics, Escherichia coli, and its Gram-positive counterpart Bacillus subtilis. We seek to answer a simple question: if two genes are co-regulated in one species, how often are they also co-regulated in the other, distantly related species? To overcome the potential pitfalls for defining co-regulation mentioned above, we obtain a reliable set of co-regulated genes for the eukaryotes by comparison of two functional genomics data sets, namely the TFBSs as determined by chromatin immunoprecipitation (ChIP-on-chip) (10) and co-expression from microarray experiments (11). To prevent spurious detection of the absence of co-regulation, we analyse regulons rather than only operons when comparing the two prokaryotes, and in the comparison of the two eukaryotes, we analyse the distribution of co-expression values rather than take an a priori threshold of co-expression. In order to choose a criterion of what conservation entails in the case of inparalogs (19), we test for the preferential retention of a co-regulation link by one of the inparalogs.
METHODS
Genomes, KEGG and orthology: eukaryotes
The S.cerevisiae genome was obtained from the EMBL database (20). The C.elegans genome was obtained from wormbase (21). Genes from these genomes are linked to KEGG (22) via the kegg gene name files. Orthologies between C.elegans and S.cerevisiae were assigned through the construction of gene trees of homologues across multiple eukaryotic genomes and subsequent analysis of each tree for orthology between our two species based on the phylogenetic tree [see (16,23) for a detailed description]. Note that this procedure can result in multi-to-one and multi-to-multi co-orthologous relationships.
Expression data, TFBS data and correlations
For the co-expression in S.cerevisiae, we use the microarray expression data from Hughes et al. (11). For the co-expression in C.elegans, we use the Kim et al. (24) data set. Correlation coefficients between genes are computed using uncentred correlation (25). The TFBS data for S.cerevisiae were obtained from Lee et al. (10) on ChIP using a cut-off of P < 0.001, which the authors propose as a reliable indicator that the transcription factor binds that upstream region.
Genomes, operons and regulons in E.coli and B.subtilis
For operons in B.subtilis, we use the data set created by Itoh et al. (26). For regulons and operons in E.coli, we use the data set of known regulons from RegulonDB (27). Genes from these files were linked to orthologous groups as defined by the latest release of the COG database (28) through gene names therein, and through the SwissProt proteome files (29).
Testing for differentiation of inparalogs
We test for the preferential retention of co-regulation of one of the paralogs by analysing the 130 yeast gene pairs that have exactly two ortholgous gene pairs in C.elegans. We then introduce a threshold of r (0.45) such that half of the 260 ortholgous pairs in C.elegans fall below and the other half above this threshold. This allows us to count the occurrences of the scenarios depicted in Figure 3 (complete conservation, partial conservation, complete loss). These counts allow us to test whether the losses tend to be independent by observing the deviation from the expected number of occurrences. Given that half of the gene pairs is conserved (i.e. in this case, their r is >0.45) the expected number of occurrences is as follows: 32.5 (0.25 × 130) for complete conservation, 65 (0.5 × 130) for partial conservation, and 32.5 (0.25 × 130) for complete loss. The deviations are summed according to the χ2 formula and tested for significance.
Figure 3.
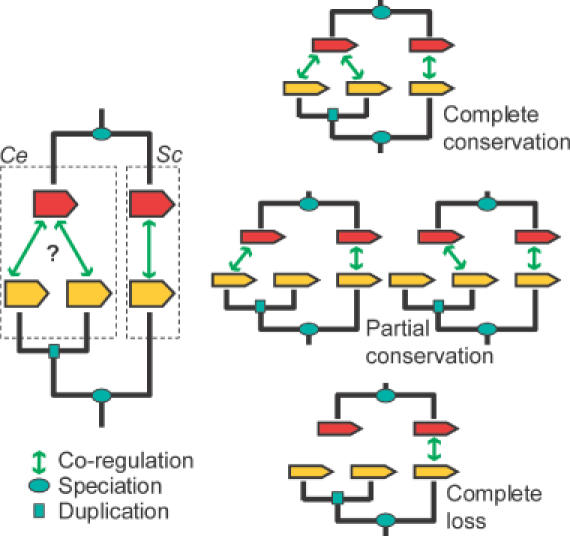
Possible outcomes of co-regulation evolution after gene duplication. This figure illustrates the dilemma with respect to inparalogs, which occurs when studying co-regulation. We delineate three different situations: complete conservation, partial conservation and complete loss. We argue (see text) that gene co-regulation is conserved, not only in the case of complete conservation but also in the case of partial conservation. Ce denotes C.elegans and Sc denotes S.cerevisiae.
RESULTS
Determining co-regulation in eukaryotes
Co-regulation between genes has successfully been defined as a threshold in the correlation coefficient, r, between microarray-based expression profiles in a single species for the purpose of function prediction (11,25). This definition has also been used for detecting conservation of co-regulation between different species of eukaryotes to improve function prediction (4,16,17). However, here we argue that such a definition of co-regulation is not well suited for measuring the amount of co-regulation conservation, because even above a high correlation of co-expression threshold (such as 0.6 or 0.7), there still might be genes that are not truly co-regulated but only spuriously co-expressed.
There are two empirical lines of evidence that suggest that gene pairs whose co-expression is above 0.6 are not necessarily co-regulated, despite statistical considerations to the opposite (25). First, the recent advance of the large-scale elucidation of TFBSs by ChIP-on-chip experiments (10) reveal very limited correlation between the co-expression and the number of shared TFBS (18). Here, we find a correlation coefficient of only 0.034 between the degree of co-expression of pairs of yeast genes versus the similarity in the transcription factor binding profile (Figure 2). The second reason why a set of gene pairs whose co-expression is above 0.6 are not necessarily co-regulated comes from the poor performance of co-expression by itself as a predictor of functional relationships and functional co-regulation between genes. The probability that two highly co-expressed genes have a functional relationship is only ∼50% when defined according to a database of known cellular processes, such as KEGG and when compared to a database of genes sharing functional transcription factor binding such as SCPD (16,22,30) (Table 1). This leaves the question why the other 50% has a high co-expression, unless one assumes that either our ‘database’ knowledge of processes does not agree with the cellular point of view or that some of these seemingly highly co-expressed genes are in fact not co-regulated. We observe the same poor correlation between functional relations and sharing of one more TFBS in the ChIP-on-chip-based functional genomics data set of Lee et al. (10) (Table 1), leading to the same question. In contrast, >90% of the genes that have a high co-expression and share a TFBS, function in the same cellular pathway, while 80% are regulated by the same transcription factor (Table 1). This suggests that the more pedestrian explanation, i.e. that there is plenty of noise (false positives) both in the TFBS data and in the highly co-expressed genes, plays a much larger role in explaining the relatively poor performance of co-expression for the prediction of functional relations, than the explanation that the human point of view of what constitutes a functional relation, as e.g. implemented in KEGG, differs widely from the cellular point of view.
Figure 2.
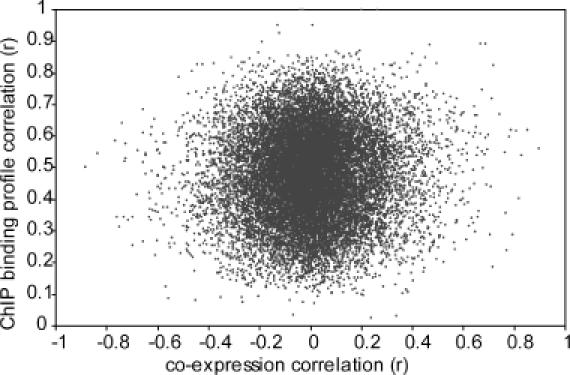
A scatter plot of co-expression correlations from microarray experiments and correlation in transcription factor binding profiles from ChIP experiments. Correlations between expression profiles are computed in the normal fashion (see Methods). Correlations between the ChIP binding profiles are used here to allow a quantitative evaluation of the relation between both data sets, which would be conceptually complicated if the ChIP-on-chip data were treated by the presence/absence of transcription factor binding. The correlation coefficient is 0.034. This is a very weak correlation, but it nevertheless is very significant with P < 10−16 when tested against the normal distribution.
Table 1. Correlation of various measures of co-regulation with functional relations.
| Data set of gene pairs | Fraction of gene pairs on the same KEGG mapa | Fraction of gene pairs sharing a TF according to SCPDa | Number of gene pairs in this set |
|---|---|---|---|
| r > 0.5 | 0.43 | 0.35 | 169 768 |
| r > 0.6 | 0.52 | 0.49 | 65 430 |
| r > 0.7 | 0.51 | 0.53 | 22 459 |
| Sharing ≥ 1 TFBS | 0.50 | 0.45 | 356 947 |
| Sharing ≥ 2 TFBS | 0.77 | 0.59 | 39 818 |
| Sharing ≥ 1 TFBS and r > 0.3 | 0.86 | 0.57 | 19 386 |
| Sharing ≥ 1 TFBS and r > 0.4 | 0.88 | 0.64 | 11 434 |
| Sharing ≥ 1 TFBS and r > 0.5 | 0.90 | 0.71 | 6 687 |
| Sharing ≥ 1 TFBS and r > 0.6 | 0.90 | 0.80 | 3 382 |
| Sharing ≥ 1 TFBS and r > 0.7 | 0.86 | 0.87 | 1 156 |
aThe fraction is computed by taking only those pairs where both genes are present in the database they are compared to.
The increased likelihood of genes to be involved in the same cellular pathway when they are co-expressed and share upstream transcription factors according to high-throughput experiments (Table 1), in addition, suggests that this definition of co-regulation is a promising method for function prediction. Its accuracy is higher than that of the composing data sets (Table 1) while still predicting a substantial number of links. In fact, when we lower the co-expression correlation threshold to 0.3, there are over 19 000 links that still have a probability of 85% to be involved in the same cellular pathway (Table 1). Combining the co-expression data with the TFBS data thus promises to be a very successful venue for vertical data integration of functional genomics data for function prediction.
Possible outcomes of evolution of co-regulation in the case of gene duplication
To accurately measure the conservation of co-regulation, we need to take into account recent gene duplications, i.e. inparalogs (19) that result in multi-to-multi and one-to-multi co-orthology relations (Figure 3). For example, when do we define a co-regulatory link to be conserved in the case that a co-regulated pair of genes from S.cerevisae has multiple, equivalent ‘orthologous pairs’ in C.elegans (because of inparalogs)? Do we consider the link conserved when all pairs in C.elegans are still co-regulated, or do we consider it conserved when at least one of the pairs is still co-regulated? In line with the current work on function and gene duplication (31), here, we use the latter: if one of the multiple orthologous pairs still displays co-regulation, then the co-regulation is conserved. The underlying model for this statement is that in the case of gene duplication, one gene would maintain the relations of the ancestral gene, while the other gene would be ‘free’ of selection constraints and could differentiate and/or undergo sub-functionalization.
We can validate this proposition by testing for the independence of the loss of co-expression of inparalogous gene pairs (Figure 3). Assuming that the loss of co-expression of inparalogous gene pairs is independent, we can compute the expected number of occurrences of all possible scenarios: complete conservation, partial conservation and complete loss of co-regulation (Figure 3). Subsequently, we compare the observed number of each scenario against this expected value (see Methods). We find that the observed number of cases in which one of the inparalogs retained co-regulation is significantly higher than expected (P < 0.005; χ2 test). Thus, the loss of co-expression of paralogs is not independent: selection tends to prefer that one of the paralogs loses a connection, while the other retains its connection. This retention of one of the connections while losing the other probably reflects differentiation. When measuring the conservation of co-regulation (see below), we will thus consider this differentiation scenario to still indicate conservation of the ancestral co-regulatory link.
High level of conservation of co-regulation between two prokaryotes
We study the co-regulation evolution in prokaryotes by taking an operon map from one species and comparing it to the regulon map in the other species. We do not use conservation of gene order for the study of co-regulation evolution as has been performed before, because genes that are neighbours on the genome are not always co-regulated as they are not necessarily part of the same operon, and, more importantly even when an operon from one species is disrupted in another species, the genes from that operon can still both belong to the same regulon in that species (32). Previous studies that did not take this approach possibly interpreted the repeatedly noted rapid pace of gene order shuffling (33–36) in prokaryotes as the lack of co-regulation conservation (4).
Here, we compare, B.subtilis and E.coli, two bacteria in which at least a reasonable operon map is available for one (36) and a decent regulon map is available for the other (27). We would, of course, prefer to use regulon data from both species, but unfortunately such a database exists only for E.coli. Of the 1023 gene pairs in operons in the B.subtilis data set, 755 are amenable for analysis because they both have orthologs in E.coli. The regulation of 276 of those 755 E.coli gene pairs has been elucidated and has been documented in regulonDB (27). We can thus assess the conservation of the co-regulation link for 276 gene pairs, assuming that this is an unbiased sample for the entire pool of 755 gene pairs. Analysis of these 276 gene pairs in the regulon data set reveals that 222 of them are in the same regulon in E.coli. Thus, ∼80% of the gene pairs that are in an operon in B.subtilis are also co-regulated in E.coli, and the conservation of co-regulation between these two species is 80%. When we perform the same analysis using only gene order information and not regulon information, only 139 gene pairs of the 276 gene pairs can be linked in so-called runs (37,38), which would have entailed an estimate of 50% conservation. Taking regulon data into account, thus, leads to a ∼2-fold increase in conservation and it yields a level of conservation that is substantially higher than previous estimates based on gene-order conservation (4).
High level of conservation of co-regulation between two eukaryotes
To measure the extent of conservation of co-regulation between two eukaryotes, we analyse a set of yeast gene pairs that can be reliably said to be co-regulated. We obtain such a set by taking only those gene pairs that have both a co-expression r > 0.6 and share the upstream binding of at least one transcription factor. These criteria result in 3382 gene pairs in S.cerevisiae. Not all of the 3382 yeast gene pairs that have a reliable co-regulation link can be analysed in C.elegans: for 618 gene pairs, neither of the genes has an ortholog in C.elegans; for 703 gene pairs, only one of the genes has an ortholog; for 2061 gene pairs, both have an ortholog in C.elegans. Of these 2061 gene pairs, 1267 gene pairs are suitable for analysis in C.elegans. Among the rest, either the C.elegans orthologs are not in the microarray data or the S.cerevisae gene pairs are inparalogous and thus share the same orthologous C.elegans gene pair. The 1267 gene pairs still contain inparalogous worm pairs. To decide for each yeast pair, the worm pair that should count for the determination of conservation, we select the pair with the highest correlation. The choice of the highest correlation is based on the rationale outlined above that one inparalogous gene typically has a higher level of co-regulation than the others. We plot the frequency distribution of correlation coefficients of the remaining 975 C.elegans gene pairs (Figure 4). This reveals an asymmetric distribution with a median r of 0.65.
Figure 4.
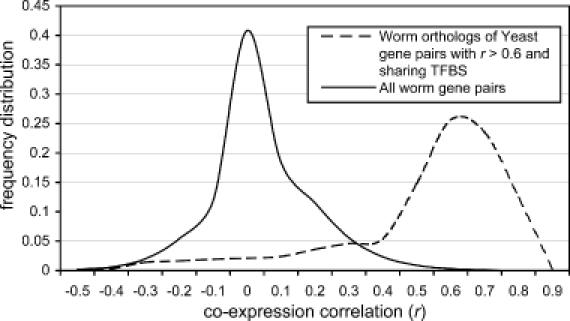
Distribution of rs of C.elegans orthologous pairs. We plot the r of the C.elegans orthologous pairs of reliably co-regulated yeast pairs to measure the degree of conservation. The distribution shows a very clear tendency to high co-expression correlation coefficients. The amount of conservation can be measured by comparing this distribution to the distribution of rs among all C.elegans genes.
Rather than defining an a priori threshold for what constitutes a not co-regulated gene pair in C.elegans, we compare the distribution of co-expression values of the 975 gene pairs to the distribution between all C.elegans gene pairs (i.e. the total genome), which represent the expected distribution for any random set of C.elegans gene pairs. This comparison provides a natural definition of conserved co-regulation: the fraction of the distribution of the 975 gene pairs that is above this expected distribution, i.e. the part of the distribution that does not intersect with the distribution of the total genome. We thereby find that 76% of the S.cerevisiae gene pairs retain co-regulation when both genes are also present in C.elegans. In order to investigate whether our choice of the inparalogous pair with the highest correlation unduly biases this estimate, we repeated the above procedure for gene pairs where both genes have a one-to-one orthology. As there are only 79 such pairs, the distribution is rugged (Supplementary Material Figure 1), and the resulting estimate might be unreliable because it is based on too few cases. Nevertheless, we obtain a similar value (70%) for the degree of co-regulation conservation. We, thus, reliably find a high level of conservation relative to previous estimates (4,16) and to existing expectations regarding the regulatory network.
DISCUSSION
Co-regulation evolves quite slowly
Here, we study the evolution of co-regulation between genes, to reveal more about the rate of evolution of connections between genes. Although these links are thought to evolve relatively fast (3,6), we find that co-regulation is relatively well conserved among eukaryotes and prokaryotes. Between S.cerevisiae and C.elegans, which have been estimated to have diverged 1.5 billion years ago (39), 76% of the gene pairs that can be reliably said to be co-regulated in S.cerevisiae and that are present in C.elegans are co-regulated in C.elegans. Similarly, between B.subitlis and E.coli, 80% of the gene pairs that are co-regulated in an operon in B.subtilis and are also present in E.coli are co-regulated in E.coli. Note that both estimates of the level of conservation are very similar, despite the fact that they were obtained using radically different data sets: regulons and operons for the prokaryotes versus microarray co-expression and ChIP-on-chip data for eukaryotes. This independence solidifies the reliability of our estimates. We, thus, find a much higher level of co-regulation conservation than previous estimates, because co-regulation evolution apparently was not properly measured and the resulting estimates fitted the existing expectations regarding regulatory network evolution.
Our estimate of the level of conservation is uncertain with respect to gene pairs, where one or both of the genes are not present in the other species. We did not include these pairs when estimating the level of conservation, because we can only analyse the co-regulation of gene pairs that are present as pairs in two species. On the one hand, it can be argued that such pairs constitute cases of disruption of co-regulation. We, on the other hand, argue that the fate of a co-regulatory link is unknown for gene pairs when one or both of the genes have been deleted. The co-regulation link could have existed right up until the deletion, or the loss of the link could predate, or even have caused, the subsequent deletion of one or both genes. The main justification for our approach, however, is that connections, in general, are evolutionary observables that are secondary to the evolutionary observable of nodes (i.e. gene presence and absence), hence our approach to measuring conservation relative to gene content.
Implications for co-expression correlations extracted from microarray data
We confirm here that there is only a small correlation between co-expression and sharing more than one TFBS as already noted (18). This could indicate either a fundamental intrinsic non-linearity in transcription regulation (e.g. a transcription factor binds upstream of a gene but this does not always result in upregulation of that gene) or simply a substantial level of noise in both data sets. Here, we observe that for both data sets, individually, only 50% of the gene pairs is involved in the same cellular pathway, while this fraction is 90% for the combined data set. This observation suggests that it is largely the latter, more pedestrian, explanation: the poor correlation between the two data sets is largely the result of noise in both data sets.
Interestingly, the combined data set performs so well in predicting whether two genes are involved in the same cellular pathway that this integrated definition of co-regulation is a very promising method for function prediction with a high coverage and a high reliability.
The poor correlation between the TFBS data and the co-expression data, as well as the performance for function prediction of the overlap versus the individual data sets on KEGG maps, also helps to explain why the use of conservation of co-expression for function prediction is so successful: it suggests that the co-expression data from multiple species filters out (experimental) noise. This explanation thereby complements the analysis of Stuart et al. (17), who found that conservation of co-expression works not only because it uses more data, but also because it uses data from different species.
Implications of a high level of conservation of co-regulation
The high level of conservation of co-regulation at first glance seems to leave little room for evolutionary flexibility. It, moreover, seems to contradict our finding that functional modules display a lot of plasticity in their evolution (40) and the finding of Wagner (9) that the expression of genes after duplication diverges rapidly. The former apparent contradiction is explained by the fact that the plasticity of functional modules was measured by the presence of their components, and, in fact, the observed high level of co-regulation conservation solidifies our hypothesis that when two genes are conserved, their functional association is also conserved. The latter contradiction can be explained by the fact that we measure the conservation of co-regulation after speciation (i.e. between orthologs), while Wagner (9) measured co-expression after gene duplication (between paralogs). This explanation is fortified by the tendency of recent gene duplicates to have one inparalog retain the original co-regulatory connection, while the other differentiates, as we observe here. One potentially more important implication of this tendency is that it suggests that we can use functional genomics data to pinpoint the ortholog that retained the ancestral function in the case of multiple inparalogs.
Analogous to the transfer of protein structures and molecular function between homologous proteins, the high level of conservation of co-regulation suggests that we can transfer knowledge on co-regulation in one species to another species: if we have determined that two genes in yeast are co-regulated, and they are also present in worm, then they are also likely to be co-regulated in worm. Here, we describe this conservation for co-regulation relationships, but expect it to hold for protein–protein interactions and other functional interactions. As worms and humans are evolutionarily equidistant to yeast, we have now provided a basis to reliably transfer findings on functional genomics from yeast to humans.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by a grant from the Netherlands organization for scientific research (NWO).
REFERENCES
- 1.Copley R.R., Schultz,J., Ponting,C.P. and Bork,P. (1999) Protein families in multicellular organisms. Curr. Opin. Struct. Biol., 9, 408–415. [DOI] [PubMed] [Google Scholar]
- 2.Brett D., Pospisil,H., Valcarcel,J., Reich,J. and Bork,P. (2002) Alternative splicing and genome complexity. Nature Genet., 30, 29–30. [DOI] [PubMed] [Google Scholar]
- 3.Levine M. and Tjian,R. (2003) Transcription regulation and animal diversity. Nature, 424, 147–151. [DOI] [PubMed] [Google Scholar]
- 4.Teichmann S.A. and Babu,M.M. (2002) Conservation of gene co-regulation in prokaryotes and eukaryotes. Trends Biotechnol., 20, 407–410; discussion 410. [DOI] [PubMed] [Google Scholar]
- 5.Bergmann S., Ihmels,J. and Barkai,N. (2004) Similarities and differences in genome-wide expression data of six organisms. PLoS Biol., 2, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll S.B. (2003) Genetics and the making of Homo sapiens. Nature, 422, 849–857. [DOI] [PubMed] [Google Scholar]
- 7.Wagner A. (2001) The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol. Biol. Evol., 18, 1283–1292. [DOI] [PubMed] [Google Scholar]
- 8.Wray G.A., Hahn,M.W., Abouheif,E., Balhoff,J.P., Pizer,M., Rockman,M.V. and Romano,L.A. (2003) The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol., 20, 1377–1419. [DOI] [PubMed] [Google Scholar]
- 9.Wagner A. (2000) Decoupled evolution of coding region and mRNA expression patterns after gene duplication: implications for the neutralist-selectionist debate. Proc. Natl Acad. Sci. USA, 97, 6579–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee T.I., Rinaldi,N.J., Robert,F., Odom,D.T., Bar-Joseph,Z., Gerber,G.K., Hannett,N.M., Harbison,C.T., Thompson,C.M., Simon,I. et al. (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science, 298, 799–804. [DOI] [PubMed] [Google Scholar]
- 11.Hughes T.R., Marton,M.J., Jones,A.R., Roberts,C.J., Stoughton,R., Armour,C.D., Bennett,H.A., Coffey,E., Dai,H., He,Y.D. et al. (2000) Functional discovery via a compendium of expression profiles. Cell, 102, 109–126. [DOI] [PubMed] [Google Scholar]
- 12.von Mering C., Krause,R., Snel,B., Cornell,M., Oliver,S.G., Fields,S. and Bork,P. (2002) Comparative assessment of large-scale data sets of protein–protein interactions. Nature, 417, 399–403. [DOI] [PubMed] [Google Scholar]
- 13.Alter O., Brown,P.O. and Botstein,D. (2003) Generalized singular value decomposition for comparative analysis of genome-scale expression data sets of two different organisms. Proc. Natl Acad. Sci. USA, 100, 3351–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ueda H.R., Hayashi,S., Matsuyama,S., Yomo,T., Hashimoto,S., Kay,S.A., Hogenesch,J.B. and Iino,M. (2004) Universality and flexibility in gene expression from bacteria to human. Proc. Natl Acad. Sci. USA, 101, 3765–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarroll S.A., Murphy,C.T., Zou,S., Pletcher,S.D., Chin,C.S., Jan,Y.N., Kenyon,C., Bargmann,C.I. and Li,H. (2004) Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nature Genet., 36, 197–204. [DOI] [PubMed] [Google Scholar]
- 16.van Noort V., Snel,B. and Huynen,M.A. (2003) Predicting gene function by conserved co-expression. Trends Genet., 19, 238–242. [DOI] [PubMed] [Google Scholar]
- 17.Stuart J.M., Segal,E., Koller,D. and Kim,S.K. (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science, 302, 249–255. [DOI] [PubMed] [Google Scholar]
- 18.Yu H., Luscombe,N.M., Qian,J. and Gerstein,M. (2003) Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet., 19, 422–427. [DOI] [PubMed] [Google Scholar]
- 19.Sonnhammer E.L. and Koonin,E.V. (2002) Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet., 18, 619–620. [DOI] [PubMed] [Google Scholar]
- 20.Kulikova T., Aldebert,P., Althorpe,N., Baker,W., Bates,K., Browne,P., van den Broek,A., Cochrane,G., Duggan,K., Eberhardt,R. et al. (2004) The EMBL Nucleotide Sequence Database. Nucleic Acids Res., 32 (Database issue), D27–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris T.W., Chen,N., Cunningham,F., Tello-Ruiz,M., Antoshechkin,I., Bastiani,C., Bieri,T., Blasiar,D., Bradnam,K., Chan,J. et al. (2004) WormBase: a multi-species resource for nematode biology and genomics. Nucleic Acids Res., 32 (Database issue), D411–D417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata H., Goto,S., Sato,K., Fujibuchi,W., Bono,H. and Kanehisa,M. (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res., 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabaldon T. and Huynen,M.A. (2003) Reconstruction of the proto-mitochondrial metabolism. Science, 301, 609. [DOI] [PubMed] [Google Scholar]
- 24.Kim S.K., Lund,J., Kiraly,M., Duke,K., Jiang,M., Stuart,J.M., Eizinger,A., Wylie,B.N. and Davidson,G.S. (2001) A gene expression map for Caenorhabditis elegans. Science, 293, 2087–2092. [DOI] [PubMed] [Google Scholar]
- 25.Eisen M.B., Spellman,P.T., Brown,P.O. and Botstein,D. (1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA, 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itoh T., Takemoto,K., Mori,H. and Gojobori,T. (1999) Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol. Biol. Evol., 16, 332–346. [DOI] [PubMed] [Google Scholar]
- 27.Salgado H., Santos-Zavaleta,A., Gama-Castro,S., Millan-Zarate,D., Diaz-Peredo,E., Sanchez-Solano,F., Perez-Rueda,E., Bonavides-Martinez,C. and Collado-Vides,J. (2001) RegulonDB (version 3.2): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res., 29, 72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatusov R.L., Fedorova,N.D., Jackson,J.D., Jacobs,A.R., Kiryutin,B., Koonin,E.V., Krylov,D.M., Mazumder,R., Mekhedov,S.L., Nikolskaya,A.N. et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruess M., Fleischmann,W., Kanapin,A., Karavidopoulou,Y., Kersey,P., Kriventseva,E., Mittard,V., Mulder,N., Phan,I., Servant,F. et al. (2003) The Proteome Analysis database: a tool for the in silico analysis of whole proteomes. Nucleic Acids Res., 31, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J. and Zhang,M.Q. (1999) SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics, 15, 607–611. [DOI] [PubMed] [Google Scholar]
- 31.Hughes A.L. (2002) Adaptive evolution after gene duplication. Trends Genet., 18, 433–434. [DOI] [PubMed] [Google Scholar]
- 32.Manson McGuire A. and Church,G.M. (2000) Predicting regulons and their cis-regulatory motifs by comparative genomics. Nucleic Acids Res., 28, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mushegian A.R. and Koonin,E.V. (1996) Gene order is not conserved in bacterial evolution. Trends Genet., 12, 289–290. [DOI] [PubMed] [Google Scholar]
- 34.Tamames J., Casari,G., Ouzounis,C. and Valencia,A. (1997) Conserved clusters of functionally related genes in two bacterial genomes. J. Mol. Evol., 44, 66–73. [DOI] [PubMed] [Google Scholar]
- 35.Huynen M.A. and Bork,P. (1998) Measuring genome evolution. Proc. Natl Acad. Sci. USA, 95, 5849–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe H., Mori,H., Itoh,T. and Gojobori,T. (1997) Genome plasticity as a paradigm of eubacteria evolution. J. Mol. Evol., 44 (Suppl. 1), S57–S64. [DOI] [PubMed] [Google Scholar]
- 37.Overbeek R., Fonstein,M., D'Souza,M., Pusch,G.D. and Maltsev,N. (1999) The use of gene clusters to infer functional coupling. Proc. Natl Acad. Sci. USA, 96, 2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overbeek R., Fonstein,M., D'Souza,M., Pusch,G.D. and Maltsev,N. (1998) Use of contiguity on the chromosome to predict functional coupling. In Silico Biol., 1, 0009. [PubMed] [Google Scholar]
- 39.Wang D.Y., Kumar,S. and Hedges,S.B. (1999) Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc. R. Soc. Lond., B. Biol. Sci., 266, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snel B. and Huynen,M.A. (2004) Quantifying modularity in the evolution of biomolecular systems. Genome Res., 14, 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.