


Abstract
The major challenge to identifying natural sense– antisense (SA) transcripts from public databases is how to determine the correct orientation for an expressed sequence, especially an expressed sequence tag sequence. In this study, we established a set of very stringent criteria to identify the correct orientation of each human transcript. We used these orientation-reliable transcripts to create 26 741 transcription clusters in the human genome. Our analysis shows that 22% (5880) of the human transcription clusters form SA pairs, higher than any previous estimates. Our orientation-specific RT–PCR results along with the comparison of experimental data from previous studies confirm that our SA data set is reliable. This study not only demonstrates that our criteria for the prediction of SA transcripts are efficient, but also provides additional convincing data to support the view that antisense transcription is quite pervasive in the human genome. In-depth analyses show that SA transcripts have some significant differences compared with other types of transcripts, with regard to chromosomal distribution and Gene Ontology-annotated categories of physiological roles, functions and spatial localizations of gene products.
INTRODUCTION
Gene regulation by natural antisense RNA in prokaryotes has been well known for many years (1,2). The first example was reported in the plasmid ColE1, in which DNA replication was regulated by an antisense RNA (3,4). Since then, antisense RNAs have been observed in various organisms, from prokaryotes (1,2) to eukaryotes (5,6) including plants (7) and animals (8). Mounting evidence suggests that it is a conserved feature within the genomes of all species from archaebacterials to humans (4,9,10). There are two different kinds of endogenous antisense RNAs in eukaryotes: one is called cis-encoded antisense, which is transcribed from the opposite strand of the same genomic locus as the sense RNA and has a long (or perfect) overlap with the sense transcript; the other is called trans-encoded antisense, which is transcribed from the genomic locus different from the sense RNA and has a short (or imperfect) overlap with the sense transcript (5,6). Most of the natural antisense transcripts are cis-encoded antisenses (5,6) and they are the focus of this study.
In the human genome, it was predicted that from 1 to 2% (11–13) to 8% (14) of the human genes were influenced by antisense transcripts. Kiyosawa et al. (15) predicted that ∼15% of the mouse genes formed sense–antisense (SA) transcript pairs. These data support the view that antisense regulation may be more widespread in the mammalian genome than appreciated previously (16,17). Various criteria were used in the prediction of SA transcripts in previous studies. For example, Yelin et al. (14) mainly collected the expressed sequences that span intron(s) and predicted 8.4% of human transcription clusters formed SA pairs, the largest SA data set reported to date. Do more SA transcripts exist in the human genome that are presently unidentified by previous criteria? If this is the case, an alternative prediction method with high sensitivity and specificity is needed.
The major challenge to identifying natural SA transcripts from public databases is how to determine the correct orientation of candidate sequences, especially expressed sequence tag (EST) sequences. EST sequences are single-pass and partial sequencing reads generated from either the 5′ or 3′ end of cDNA clones (18). As the cDNA libraries were generated from either normal or pathological samples from different tissues and different developmental stages, ESTs provide a tremendous resource for transcript analysis at the genome level. Currently, there are over 5 million human ESTs in dbEST [(19), dbEST release 090503] and over 4 million human ESTs clustered in UniGene [(20), Build #167]. However, uncertainty regarding the correct orientation of ESTs (11,12) has been the major obstacle in using the EST database for antisense transcript identification (14). Most of ESTs and many mRNAs do not span introns, but they may contain reliable 3′ ends. Therefore, we established a set of very stringent criteria based on the presence of a polyadenylation site [i.e. poly(A) signal] and the poly(A) tail at 3′ end of the sequence and the sequence annotation to determine the correct orientation of each transcript. Consequently, 386 415 orientation-reliable transcripts were collected. A combined use of CAP3 (21) and Blat (22) restricted with the specific criteria, identified 26 741 transcription clusters derived from 346 168 transcript sequences in the whole human genome, of which 22% (5880) form putative SA pairs, higher than previous prediction. Note that neither non-polyadenylated transcripts nor trans-encoded antisenses were included in our antisense set, thus, the percentage of SA pairs is likely to be even higher. Our orientation-specific RT–PCR test together with the comparison with the relative published data confirms that this SA data set is reliable, indicating that our method is very efficient for antisense transcript prediction.
MATERIALS AND METHODS
Data source
The human transcript sequences (4 409 214 in total) were downloaded from the human UniGene database (http://www.ncbi.nlm.nih.gov/UniGene; Build #167). UniGene (20) is an experimental system for automatically partitioning GenBank sequences into a non-redundant set of gene-oriented clusters. In UniGene, vector contaminants are removed and repetitive elements or low-complexity sequences are masked and EST sequences with very poor sequence quality are discarded. Besides mRNAs and ESTs, many gene Models are also collected into human UniGene. There are different levels of evidence for Models, ranging from fully transcript-supported to just ab initio prediction, and only those that match other transcripts in the same species are included in UniGene. Thus, compared with the raw expressed sequence data deposited in GenBank, human UniGene provides a more reliable base for our study. Since Models are helpful for EST sequence assembling and genome mapping, we kept them through our genome-mapping step, but excluded them from our transcription clusters. The human genome sequences downloaded from UCSC goldenPath (http://genome.cse.ucsc.edu/goldenPath; NCBI build #34) were used for genome mapping.
Selection of orientation-reliable human transcripts
Poly(A) signals and poly(A) tails are two indicators in determining the reliable 3′ end for a given expressed sequence. We adapted the criteria of Caron et al. (23) to determine whether a sequence contains a poly(A) signal and/or the poly(A) tail (see Table 1). Once the 3′ end is determined, the correct orientation of the sequence is determined. In addition, CDS (protein coding sequence) is an important sign appearing in the sequence annotation for a well-annotated sequence. Basically, a sequence with the annotated CDS indicates it is well annotated and in the ‘sense’ form (i.e. the sequence's orientation is from the 5′ end to the 3′ end). Table 1 shows the criteria used for selecting different types of sequences. It is notable that, for a 3′ labeled EST sequence, we accepted the sequence only if it contains both a poly(A) signal and the poly(A) tail. These criteria are more stringent than those used by previous studies in selecting reliable 3′ end clones (23–25). After passing our filters, 386 415 sequences were collected. All the collected sequences are in the correct sense form (i.e. the sequence has been determined to be in the correct orientation and formed from the 5′ end to the 3′ end) with reliable 3′ ends (we called them ‘sense-orientation-reliable’ transcripts).
Table 1. Criteria for selection of sense-orientation-reliable transcripts.
| Clone labela | Has CDS? | Sequence form(s) checkedb | Requirement(s) of poly(A) signalc and poly(A) taild for a sequence to be accepted as sense-orientation-reliable form |
|---|---|---|---|
| mRNA | Yes | Original | No requirement |
| No | Original | Either poly(A) signal or poly(A) tail | |
| 3′ EST | No | Original and RC | Both poly(A) signal and poly(A) tail |
| Others | Yes | Original | Either poly(A) signal or poly(A) tail |
| No | Original and RC | Both poly(A) signal and poly(A) tail |
aNone of the 3′ ESTs contain CDS; ‘Others’ refers to a sequence except for mRNAs and 3′ ESTs in the UniGene database.
bExcept for mRNAs, for a sequence without CDS, we checked both its original and reverse complementary (RC) forms.
cPoly(A) signal: containing AATAAA, ATTAAA, AATTAA, AATAAT, CATAAA or AGTAAA within the last 50 bp of 3′ end of a sequence [adapted from Caron et al.'s (23) criteria].
dPoly(A) tail: containing a stretch of at least 10 As at 3′ end of a sequence [adapted from Caron et al.'s (23) criteria].
Genome mapping and clustering of orientation-reliable transcripts
To reduce the workload and improve the mapping quality, we first applied the selected sense-orientation-reliable transcripts for assembling by CAP3 (21). The generated contigs and singlets were then mapped to the human genome sequences using Blat (22) restricted with the conditions of Identity ≥94, Coverage ≥0.80 and Alignment ≥0.97. All imperfect alignments and uncertain multiple alignments were removed. The mapped genomic fragments were combined if they were overlapping and then were used as the target to map the selected sense-orientation-reliable sequences. The Blat conditions for this step are: Identity ≥96, Coverage ≥0.70 and Alignment ≥0.97. The transcript sequences that were aligned to more than one genomic fragment were discarded as suspected chimeras. All mapped genomic loci with overlaps were combined. An exact genomic locus was determined for each transcript. Splice junctions are very strong evidence regarding the strand of origin of expressed sequences. Splice donor and acceptor sites are GT-AG for 98.12% of human introns, or GC-AG and AT-AC for most of the remaining human introns (26,27). Because all the selected sequences were in the ‘sense form’, the start and end sites of the introns they span should be GT-AG, GC-AG or AT-AC. If an intron starts with CT (the reverse complement of AG) and ends with AC or GC (the reverse complement of GT or GC), or starts with GT (the reverse complement of AC) and ends with AT (the reverse complement of AT), the sequence might be oriented in the wrong direction. Only 558 sequences (∼0.3% of the selected intron-spanning sequences) had suspicious splice sites and were discarded. Since both intron-spanning sequences and unspliced sequences were selected based on the same rules and their sequence numbers are comparable, we could expect that all the sequences, regardless of intron-spanning or not, might have a similar false-positive rate, i.e. ∼0.3%. In addition, all sequences with an average intron length over 300 kb, all Models and highly abundant and tandem duplicate genes such as immunoglobulins and T-cell receptors were excluded from further study.
All transcript sequences aligned to the same genomic locus were assembled into one transcription cluster. After assembly, all (4071) clusters that contained only one sequence that did not span an intron were excluded (intron length and the flanking exon length must be ≥50 and ≥10 nt, respectively). Finally, 22 341 transcription clusters were identified, which contained a total of 346 168 transcript sequences.
Classification of bi-directional transcription cluster pairs
A bi-directional cluster, whether a SA cluster (Figure 1) or a non-exon-overlapping bi-directional cluster (NOB; Figure 1), contains the sequences transcribed from both strands of the same genomic locus. For the sequences in such a cluster, we further separated them into two new clusters (a cluster pair) based on their alignments to the genome. If the two new clusters originated from a SA cluster, they were called either ‘sense cluster’ (S) or ‘antisense cluster’ (A); if they originated from a NOB cluster, they were called either ‘sense-like cluster’ (SL) or ‘antisense-like cluster’ (AL). The S and A or SL and AL clusters in a cluster pair were determined using the following rules: (i) define the one containing more sequences as the S or SL cluster, the other as the A or AL cluster; (ii) if the sequence numbers were the same, define the one with more mRNA sequences as the S or SL cluster, the other as the A or AL cluster; (iii) if their mRNA sequence numbers were still the same, define the one with intron-spanning sequence(s) as the S or SL cluster while the other one without such intron-spanning sequence(s) would be the A or AL cluster. If none of the above conditions was satisfied, define the one mapped to the sense strand of chromosome as the S or SL cluster and the other as the A or AL cluster.
Figure 1.

Classification of the transcription clusters in the human genome. (A) Eight categories and sub-categories of the transcription cluster are shown. The categories are classified according to the transcribed patterns of how the transcripts are mapped on the genome sequences. (B) The descriptions of each category are shown briefly. Total cluster counts as well as sequence number for each category are presented. ‘mRNA’, ‘Intron’ or ‘CDS’ refers to the number of the clusters which contain known mRNA(s), intron-spanning sequnce(s) that span at least one ‘consensus’ intron (flanked by consensus donor and acceptor splice sites) or protein-coding sequence(s), respectively.
Assessment of transcriptional directionality via orientation-specific RT–PCR assay
Twenty-five SA pairs, longer than 250 bp of exon overlaps, were randomly selected from our data set. Ten antisense-negative controls were chosen from highly abundant genes. Candidate primers were designed to amplify a 180–400 bp sequence that was internal to a predicted region of bi-directional transcription (i.e. overlapped exon region). Control primers were designed to amplify a 180–400 bp sequence as well, either from randomly selected regions of non-SA (i.e. NBD or NOB; Figure 1) transcripts or from non-overlapping regions of SA transcripts. Total RNA from human brain tissue was used as template (Clontech), which was treated with DNase I to remove all potential genomic DNA contamination before RT–PCR. We used a Qiagen One Step RT–PCR kit according to a procedure modified from Shendure and Church (13), with 25 μl per reaction. Orientation of a transcript was assessed by restricting which primer was present during RT single-strand synthesis. For each candidate or control, four RT–PCR reactions were carried out. In the first reaction, only the primer complementary to the antisense-orientation of the PCR product was present during RT-single-strand synthesis (i.e. the sense primers listed in Supplementary Table 2 to assay for antisense transcription). In the second reaction, only the primer complementary to the sense-orientation of the PCR product was present during RT-single-strand synthesis (i.e. the antisense primers listed in Supplementary Table 2 to assay for sense transcription). In the third reaction, regular oligo(dT)20 (Invitrogen) was present during RT-single-strand synthesis (positive control). In the fourth reaction, no primer was present during RT-single-strand synthesis (control for genomic contamination). In all four reactions, both primers (sense and antisense) were present during the subsequent PCRs. The cycling parameters were as follows: (i) 50°C × 30 min, reverse transcription single-strand synthesis; (ii) 95°C × 15 min, activate AmpliTaq polymerase, inactivate RT enzymes; (iii) 4°C, add missing primers; (iv) 94°C × 30 s, commence PCR cycling; (v) 60°C × 30 s; (vi) 72°C × 35 s; (vii) go to step iv (35 cycles in total); and (viii) 72°C × 10 min.
Characterization of Gene Ontology (GO) annotations for SA and non-SA genes
The GO project is a collaborative effort to address the need for consistent descriptions of gene products in different databases (28). The GO consists of three separate ontologies (vocabularies) that describe molecular function, biological process and cellular component. We obtained the Locus IDs and GO IDs of known human genes from the NCBI LocusLink website (http://www.ncbi.nlm.nih.gov/LocusLink; April 1, 2004), and all GO terms and annotations from Gene Ontology Consortium (http://www.geneontology.org; April 1, 2004). By comparing the Locus IDs, we identified 10 498 (2174 SA; 8324 non-SA) transcription clusters that were annotated by human GO terms, of which 9248 (1931 SA; 7317 non-SA) participated in ‘molecular function’, 8539 (1778 SA; 6761 non-SA) in ‘biological process’ and 7248 (1471 SA; 5777 non-SA) in ‘cellular component‘. For each ontology category, we set up our own GO slims (i.e. sub-trees) for GO terms that matched to our data set. We defined a sub-tree covering a given set of GO terms as the one rooted by the deepest common ancestor of all the given GO terms; we also tried to avoid having a given GO sub-tree cover too great a percentage of genes in our data set. Each GO sub-tree was represented by the GO term of the root. 13, 9 and 7 sub-trees were built up for three ontologies of biological process, molecular function and cellular component, respectively (Figure 4). For each sub-tree, the number of genes (i.e. transcription clusters) participating was recorded. The number of genes represented by the sub-tree was divided into the total number of annotated clusters in the ontology to arrive at the percentage coverage. ‘Chi-square test’ was used to determine the significance of difference between the percentages of SA and non-SA genes in each sub-tree.
Figure 4.
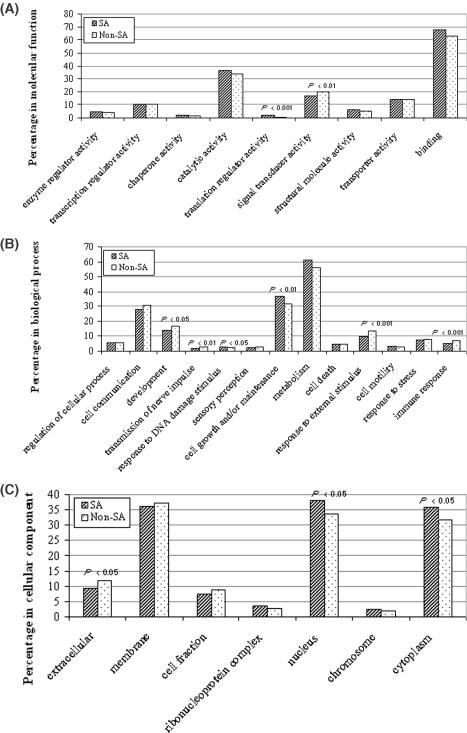
GO analysis. Because one gene (i.e. transcription cluster) might match several different GO terms, the sum of gene percentages of all sub-trees in a given ontology would be higher than 100%. (A) In molecular functions, SA genes have a significantly higher percentage participating in ‘translation regulator activity’ (1.6 versus 0.7%), but a significantly lower percentage in ‘signal transducer activity’ (16.7 versus 20.0%) than other genes. (B) In biological process, SA genes have a significantly higher percentage in ‘response to DNA damage stimulus’ (2.8 versus 1.9%) and ‘cell growth and/or maintenance’ (36.8 versus 32.0%), but a significantly lower percentage in ‘development’ (13.7 versus 16.4%), ‘transmission of nerve impulse’ (1.7 versus 2.9%), ‘response to external stimulus’ (9.6 versus 13.1%) and ‘immune response’ (4.6 versus 7.2%) than other genes. (C) In cellular component, SA genes have a significantly higher percentage in ‘nucleus’ (38.1 versus 33.9%) and ‘cytoplasm’ (36.0 versus 31.9%), but a significantly lower percentage in ‘extracellular’ (9.2 versus 11.8%) than other genes.
RESULTS
Genome-wide identification of transcription clusters
A total of 22 341 transcription clusters were created based on the mRNA and EST alignments to the human genome. The sequences and alignments were filtered stringently to ensure the correct orientation. The clusters were classified according to the categories depending on the transcribed pattern in the genome. A total of 4400 clusters containing at least one pair of sequences transcribed from opposite strands of the same genomic locus were called ‘bi-directional clusters‘ (BD; Figure 1); the remaining 17 941 clusters only containing one-directional transcripts were called ‘non-bi-directional clusters’ (NBD; Figure 1). Of the 4400 BD clusters, 2940 containing at least one opposite-strand transcribed sequence pair that had exon overlaps (identity ≥94%) were further classified as ‘SA clusters’ (Figure 1); the remaining 1460 BD clusters without such exon-overlapping pair(s) were then referred to NOB (Figure 1). Each SA or NOB cluster was further separated into two new clusters (a ‘cluster pair’); thus SA became sense (S) and antisense (A), and NOB included sense-like (SL) and antisense-like (AL). Such separation finally resulted in a total of 26 741 clusters (S, A, SL, AL and NBD; Figure 1), each of which contained single-direction transcribed sequences and represented a single or partial gene. The categories and the number of clusters and sequences in each category are summarized in Figure 1. One transcript pair was selected as the representative for each SA cluster pair (Supplementary Table 1). The length of exon overlaps between the paired SA representative transcripts varied from 30 (the minimum set by our program) to 4223 nt, with an average of 388 and a median of 282. We found that most of the exon overlaps occur in the 5′-untranslated regions (5′-UTRs) and especially in the 3′-UTRs of the sense and antisense genes, consistent with the previous observations (12,14,29).
Ninety-eight percentage of the SA and of the NOB cluster pairs had at least one cluster containing known mRNA(s), and 96% of the SA and more than 99% of the NOB pairs had at least one cluster containing intron-spanning sequence(s). Of the 2940 SA cluster pairs, 91% of the S and 38% of the A clusters contained CDS (protein-coding sequences), while in the 1460 NOB cluster pairs, 91% of the SL and 27% of the AL clusters contained CDS (Figure 1B). The result indicates that most of the S and SL clusters represent protein-coding genes, whereas the majority of the A and AL transcripts represent non-coding RNAs (8,30,31).
Experimental validation of SA pairs by orientation-specific RT–PCR
To validate our methodology, we sought to confirm a subset of our SA predictions experimentally. We randomly selected 25 SA candidate pairs from our data set and performed orientation-specific RT–PCR (see Materials and Methods). As shown in Figure 2 (summarized in Supplementary Table 2), 24 (96%) out of the 25 antisense transcriptions were successfully detected in normal brain tissue compared with none out of 10 antisense-negative controls. For 23 (92%) of the 25 SA pairs, both sense and antisense transcripts were detected.
Figure 2.
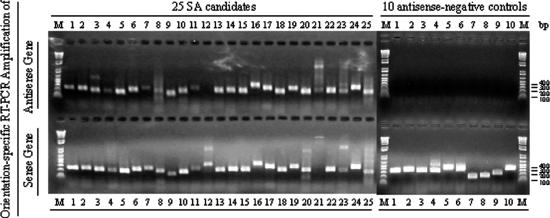
Assessment of transcriptional directionality by orientation-specific RT–PCR. SA-1 to SA-25 are 25 randomly selected human SA candidates, and NC-1 to NC-10 are 10 negative controls (see Materials and Methods). A 1.5% agarose gel was used for RT–PCR product checking, and ‘1 kb Plus DNA Ladder’ (Invitrogen) was used as DNA molecular weight marker (M). For each candidate or negative control, four RT–PCR reactions were carried out (see Materials and Methods). All the positive controls were detected as positive and all the negative controls were detected as negative (data not shown). Except for SA-12, 24 out of the 25 SA candidate primer sets and no negative control primer sets were positive for antisense transcription over the regions queried. Of these 24 sets, 23 were also positive for sense transcription, except for SA-21. All the 10 negative controls were positive only for their sense transcription, not for antisense transcription. In addition, only one of the SA candidate genes (i.e. the antisense gene of the SA-6 pair; NM_002643) was included in Yelin et al.'s (14) micro-array data set; it was not detected by their micro-array, but was detected by our RT–PCR. In addition, sense gene (NM_003275) of the SA-15 and antisense gene (NM_032622) of the SA-17 were also included in Shendure and Church's (13) RT–PCR analysis, and both of them were detected as positive in both RT–PCR analyses.
Chromosomal distribution of bi-directional transcription clusters
All bi-directional cluster loci mapping to the whole genome are summarized in Figure 3. The distribution and coverage of all cluster loci are listed in Table 2. As presented in Figure 3, except for some regions like centromeres that are essentially not sequenced and annotated, SA and NOB clusters are widespread on every chromosome except for chromosomes X and Y. Our result that the total cluster loci (22 341) covered 38.7% of the whole genome (Table 2) is in accord with Lander et al.'s (27) prediction that over one-third of the genome might be transcribed in genes, although only ∼1.5% of the human genome would consist of coding sequences.
Figure 3.
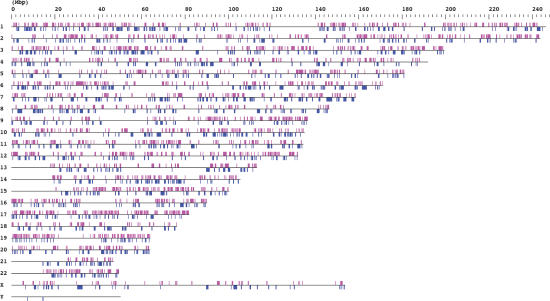
Chromosome map of SA clusters and NOB clusters. All of the mapped positions of the bi-directional transcription clusters are represented schematically. The SA and NOB clusters (above and below) are in magenta and in blue, respectively. Note that SA and NOB clusters are widespread on every chromosome except for chromosomes X and Y; there are no SA clusters on the Y chromosome. Centromeres, the short arms of chromosomes 13, 14, 15, 21 and 22, the variable heterochromatic regions on chromosomes 1, 9 and 16, and the variable region at the q-terminus end of chromosome Y are essentially not sequenced and annotated (42). Thus, no SA or NOB loci are observed in these regions.
Table 2. Distribution of bi-directional clusters on each chromosomea.
| Chromosome | Total number of loci | SA loci/total loci (%) | NOB loci/total loci (%) | Chromosome length (kb) | Coverage of SA loci/chromosome length (%) | Coverage of NOB loci/chromosome length (%) | Coverage of total loci/chromosome length (%) |
|---|---|---|---|---|---|---|---|
| 1 | 2214 | 12.9 | 6.1 | 246 128 | 8.4 | 10.1 | 42.9 |
| 2 | 1604 | 14 | 6.5 | 243 616 | 9.6 | 7.3 | 40.7 |
| 3 | 1263 | 15.3 | 8.1 | 199 344 | 12.0 | 10.1 | 43.9 |
| 4 | 980 | 12.6 | 6.9 | 191 732 | 8.5 | 7.3 | 36 |
| 5 | 1083 | 13.1 | 6.9 | 181 035 | 7.6 | 7.4 | 35.8 |
| 6 | 1151 | 12.7 | 8.2 | 170 915 | 9.5 | 10.1 | 41 |
| 7 | 1053 | 12.8 | 7.2 | 158 546 | 8.3 | 14.5 | 44 |
| 8 | 865 | 10.1 | 7.3 | 146 309 | 6.1 | 11.2 | 38.3 |
| 9 | 877 | 15.1 | 6.4 | 136 372 | 8.3 | 6.1 | 35.2 |
| 10 | 891 | 13.6 | 7.6 | 135 037 | 10.6 | 10.6 | 44.5 |
| 11 | 1250 | 13.3 | 6 | 134 483 | 8.9 | 7.8 | 40.7 |
| 12 | 1190 | 13.9 | 6.3 | 132 078 | 12.3 | 5.3 | 44.5 |
| 13 | 438 | 13 | 7.5 | 113 043 | 8.4 | 7.9 | 30.3 |
| 14 | 678 | 15.8 | 7.2 | 105 311 | 6.8 | 9.3 | 34.7 |
| 15 | 723 | 14.8 | 7.3 | 100 257 | 9.3 | 8.1 | 38.9 |
| 16 | 900 | 13.7 | 6.2 | 90 042 | 9.4 | 6.3 | 38.7 |
| 17 | 1232 | 16.4 | 4.1 | 81 860 | 14.9 | 7.3 | 50.7 |
| 18 | 388 | 10.6 | 8.2 | 76 115 | 7.6 | 10 | 38.8 |
| 19 | 1336 | 10.9 | 3.2 | 63 812 | 7.7 | 2.9 | 43.4 |
| 20 | 629 | 11.8 | 8.4 | 63 742 | 7.2 | 11 | 40.3 |
| 21 | 286 | 10.8 | 9.8 | 46 976 | 5.7 | 9.8 | 28.2 |
| 22 | 494 | 15 | 6.5 | 49 397 | 8.8 | 6.9 | 36.4 |
| X | 782 | 7.5 | 4.7 | 153 692 | 3.2 | 5.2 | 28 |
| Y | 34 | 0 | 5.9 | 50 287 | 0.0 | 0.6 | 4.5 |
| Overall | 22 341 | 13.2 | 6.5 | 3 070 128 | 8.6 | 8.4 | 38.7 |
aFor statistical significance, we used ‘Chi-square test’ to determine P-values. SA loci had a significantly (P < 0.05) higher percentage on chromosomes 3 (15.3%), 14 (15.8%) and 17 (16.4%), but a significantly lower percentage on chromosomes 8 (10.1%), 19 (10.9%), X (7.5%) and Y (0%), compared to its overall percentage (13.2%) on the whole genome; whereas, NOB loci had a significantly higher percentage on chromosomes 3 (8.1%), 6 (8.2%) and 21 (9.8%), but a significantly lower percentage on chromosomes 17 (4.1%), 19 (3.2%) and X (4.7%), compared to its overall percentage (6.5%) on the whole genome. On chromosome Y, all bi-directional cluster loci were NOB loci. Although SA loci had a significantly higher percentage (P < 10−4) than NOB loci (13.2 versus 6.5%) in overall loci, their overall coverage of chromosome length are very close (8.6 versus 8.4%), no significant difference.
There are more than twice as many SA loci (2940) compared with NOB loci (1460), whereas their total coverage (8.6 versus 8.4%) of the genome is not significantly different (Chi-square P > 0.7; Table 2), which suggests that the NOB transcripts may span longer introns than the SA transcripts. The distribution of SA and NOB loci on the whole genome is not random. The percentage of SA or NOB loci on some chromosomes is significantly (P < 0.05) higher or lower than the overall rate (13.2% for SA loci and 6.5% for NOB loci), such that (i) SA loci had a significantly higher percentage on chromosomes 3 (15.3%), 14 (15.8%) and 17 (16.4%), but a significantly lower percentage on chromosomes 8 (10.1%), 19 (10.9%), X (7.5%) and Y (0%); and (ii) NOB loci had a significantly higher percentage on chromosomes 3 (8.1%), 6 (8.2%) and 21 (9.8%), but a significantly lower percentage on chromosomes 17 (4.1%), 19 (3.2%) and X (4.7%). However, the percentage of SA and NOB loci on most chromosomes was comparable to the relative overall rate. These facts indicate that the SA and NOB clusters share similarities as well as differences in their distribution patterns on the genome.
Comparison of GO annotations for SA and non-SA genes
Compared with other genes, SA genes have significantly different distribution in some sub-trees of the three ontologies. As shown in Figure 4, in molecular functions, SA genes have a significantly higher percentage participating in ‘translation regulator activity’ (P < 0.001), but a significantly lower percentage in ‘signal transducer activity’ (P < 0.01) (Figure 4A); in biological process, SA genes have a significantly higher percentage in ‘response to DNA damage stimulus’ (P < 0.05) and ‘cell growth and/or maintenance’ (P < 0.01), but a significantly lower percentage in ‘development’ (P < 0.05), ‘transmission of nerve impulse’ (P < 0.01), ‘response to external stimulus’ (P < 0.001) and ‘immune response’ (P < 0.001) (Figure 4B); in cellular component, SA genes have a significantly higher percentage in ‘nucleus’ (P < 0.05) and ‘cytoplasm’ (P < 0.05), but a significantly lower percentage in ‘extracellular’ (P < 0.05) (Figure 4C).
DISCUSSION
Previous large-scale predictions (11–14) have significantly expanded our knowledge about the prevalence of natural human antisense transcription. Particularly, Yelin et al. (14) predicted 2667 SA pairs in the human genome, far more than the other studies (11–13). In this study, we tried to identify additional human antisense transcripts that have not been identified before. With direct sequence accession number matching, we observed that ∼62% of our SA pairs did not appear in Yelin et al.'s (14) SA set although the two data sets have comparable SA pairs. In comparison with two other SA sets, we found that 98% (85 out of the total 87) of Lehner et al. (12) and 76% (110 out of 144) of Shendure and Church's (13) candidates were included in our SA set. Because these two SA sets were predicted from a limited number of reliable sequences, their numbers are relative small. We could not compare our data with Fahey et al.'s (11) because their data lack candidate sequence IDs. These results confirmed our hypothesis that more SA transcripts might exist in the human genome and that they could be identified by an alternative prediction method.
A good prediction method should have high sensitivity and specificity. As to sensitivity, our method predicts 22% (5880) of the total 26 741 clusters to form SA pairs, higher than Yelin et al.'s 8.4% (5334 out of the total 63 715 clusters), although the two sets have similar number of pairs. As to specificity, our orientation-specific RT–PCR detected over 90% (23 out of 25) of both sense and antisense transcriptions in brain tissue. In fact, we have also compared our data set with experimental data from other studies (13,14), and found that: (i) of the 25 SA pairs (out of 31 SA pairs with orientation-specific RT–PCR information that was kindly provided by Shendure and Church) appearing in our SA set, 80% of the antisense transcripts were confirmed by their orientation-specific RT–PCR, slightly higher than their overall positive rate (77%: 24 out of 31); and (ii) of the 163 sequences in our SA set that were identical to those studied by Yelin et al.'s (14) micro-array analysis, 52% were verified or indirectly verified by their micro-array analysis, significantly higher (P < 0.05) than their overall positive rate (43%). In addition, only one SA candidate confirmed by our RT–PCR was included in Yelin et al.'s (14) micro-array data set, but was not confirmed by their micro-array; two SA candidates were detected as positive in both RT–PCR analyses of ours and of Shendure and Church's [(13), Figure 2]. The reason that the positive rate of antisense confirmation in micro-array analysis was relative low had been discussed by Yelin et al. (14). Our result also supports the notion that RT–PCR is more sensitive than micro-array analysis. Taken together, these results suggest that our method has high sensitivity and specificity, which would ensure its efficiency in antisense prediction and the validity of our SA set.
In addition, the transcripts selected by our criteria have reliable 3′ ends. This property will facilitate analyses with the massive serial analysis of gene expression (SAGE) expression data (24,32) that has been widely used in the studies at the whole human genome level (23,25,33,34). Although many microarray data are now publicly available, few of them focus on SA pairs. Using available microarray data for genome-wide investigation of SA expression is, therefore, still not feasible. Unlike microarray, the SAGE technique does not require prior knowledge of the sequences to be analyzed, and thus SAGE libraries provide global and unbiased gene expression data that are suitable for SA expression analysis. By taking advantage of the abundant and growing amount of SAGE expression data (e.g. more than 240 SAGE libraries available at the NCBI GEO platform, April 2, 2004) generated from different tissues and various developmental, differentiation, pathological and physiological stages or conditions, one could not only determine the expression levels and patterns of natural human antisense, but also reveal the potential roles and possible functional mechanism(s) played by natural antisense.
Furthermore, in contrast to previous studies on human antisense prediction, our method also predicted 1460 NOB cluster pairs (Figure 1). Such transcription pairs have also been observed in the mouse genome [called ‘non-antisense bi-directional transcription pair’, (15)]. As with SA pairs, the two members of an NOB pair are located at the same genomic locus, but unlike SA pairs, the two members lack exon overlap. We have found that, although both SA and NOB clusters are bi-directional transcription clusters, they have some significant differences with regard to chromosomal distribution (Figure 3 and Table 2), as well as intron length, expression level and pattern and possible involvement in tumorigenesis (J. Chen, M. Sun and J. D. Rowley, unpublished data), suggesting that there might be an intrinsic, biological difference in the nature of the two types of transcription, which in turn implies that they might play different biological roles in the human genome.
Alternative splicing has been demonstrated to be one of the most significant components of the complexity of the human genome (35,36), but the contribution of the natural antisense transcripts to the human genome complexity usually has been underestimated. In this study, we observed that 22% of clusters consisted of SA pairs, and 25% (88 211 out of the total 346 168) human expressed sequences were involved in antisense transcription (Figure 1). These estimates are higher than those from previous analyses (11–14), but are similar to the observations from chromosomes 21 and 22 (29, 37). Moreover, our data set and that of Yelin et al. (14) shared only 38% of the transcripts, indicating that the total number of SA pairs must be more than either of the two data sets. In addition, none of the two data sets included many other kinds of antisense transcripts such as trans-encoded, non-polyadenylated and dsRNAs resulting from bi-directional transcription from repetitive and transposable elements that constitute almost half of the entire human genome (17). Furthermore, the natural antisense transcripts for which expressed sequences are not yet available in the public mRNA/EST databases would not be detected by conventionally computational prediction. Such novel transcripts are likely to be expressed at very low levels (37) and could be identified by combining the use of the SAGE and GLGI methods (38–40). All these data indicate that natural antisense transcripts are much more common in the human genome than estimated previously. The fact that most of our antisense transcripts are non-coding RNAs (Figure 1) and that the majority of novel antisense transcripts may also be non-coding transcripts (37), suggest that the antisense transcripts are unlikely to significantly increase the total number of human protein-coding genes, but they should be one of the major components of the complexity of the human genome.
In GO analysis, we found that SA transcripts have significantly different distribution from non-SA transcripts in some possible molecular functions, involvements in biological process and cellular localizations. Yelin et al. (14) did not observe significant difference in these parameters between SA and other transcripts, probably due to the different strategies used in GO analysis (41), and/or the difference between two SA data sets. The fact that SA transcripts have a significantly higher probability of involvement in ‘translation regulator activity’ and are more frequently located in the ‘nucleus’ and ‘cytoplasm’, might be compatible with their roles related to antisense-mediated gene regulation, because antisense-mediated gene regulation could happen in both nucleus and cytoplasm, and at both transcription and translation levels (5,6). However, systematic experimental analyses should be performed to unravel the actual biological meaning underlying the differences between SA and other transcripts, which will provide new insights into the nature of human antisense transcripts.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Zhijian Qian and Yanming Zhang for critical reading and valuable comments on the manuscript; Galit Rotman for providing published supplementary antisense data; Jay Shendure and George M. Church for providing their published orientation-specific RT–PCR result information; Hehuang Xie and Mervyn G. Marasinghe for help with the statistics analysis and Greg D. Schuler for discussion on UniGene. This work was supported by National Institutes of Health Grant CA84405 (J.D.R.), and the Spastic Paralysis Foundation of the Illinois, Eastern Iowa Branch of Kiwanis International (J.D.R.).
REFERENCES
- 1.Simons R.W. (1988) Naturally occurring antisense RNA control—a brief review. Gene, 72, 35–44. [DOI] [PubMed] [Google Scholar]
- 2.Wagner E.G. and Simons,R.W. (1994) Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol., 48, 713–742. [DOI] [PubMed] [Google Scholar]
- 3.Lacatena R.M. and Cesareni,G. (1981) Base pairing of RNA I with its complementary sequence in the primer precursor inhibits ColE1 replication. Nature, 294, 623–626. [DOI] [PubMed] [Google Scholar]
- 4.Knee R. and Murphy,P.R. (1997) Regulation of gene expression by natural antisense RNA transcripts. Neurochem. Int., 31, 379–392. [DOI] [PubMed] [Google Scholar]
- 5.Kumar M. and Carmichael,G.G. (1998) Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev., 62, 1415–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhee-Brossollet C. and Vaquero,C. (1998) Do natural antisense transcripts make sense in eukaryotes? Gene, 211, 1–9. [DOI] [PubMed] [Google Scholar]
- 7.Terryn N. and Rouze,P. (2000) The sense of naturally transcribed antisense RNAs in plants. Trends Plant Sci., 5, 394–396. [DOI] [PubMed] [Google Scholar]
- 8.Erdmann V.A., Barciszewska,M.Z., Hochberg,A., de Groot,N. and Barciszewski,J. (2001) Regulatory RNAs. Cell Mol. Life Sci., 58, 960–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stolt P. and Zillig,W. (1993) Antisense RNA mediates transcriptional processing in an archaebacterium, indicating a novel kind of RNase activity. Mol. Microbiol., 7, 875–882. [DOI] [PubMed] [Google Scholar]
- 10.Merino E., Balbas,P., Puente,J.L. and Bolivar,F. (1994) Antisense overlapping open reading frames in genes from bacteria to humans. Nucleic Acids Res., 22, 1903–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fahey M.E., Moore,T.F. and Higgins,D.G. (2002) Overlapping antisense transcription in the human genome. Comput. Funct. Genomics, 3, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehner B., Williams,G., Campbell,R.D. and Sanderson,C.M. (2002) Antisense transcripts in the human genome. Trends Genet., 18, 63–65. [DOI] [PubMed] [Google Scholar]
- 13.Shendure J. and Church,G.M. (2002) Computational discovery of sense–antisense transcription in the human and mouse genomes. Genome Biol., 3, research0044.1–research0044.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yelin R., Dahary,D., Sorek,R., Levanon,E.Y., Goldstein,O., Shoshan,A., Diber,A., Biton,S., Tamir,Y., Khosravi,R. et al. (2003) Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol., 21, 379–386. [DOI] [PubMed] [Google Scholar]
- 15.Kiyosawa H., Yamanaka,I., Osato,N., Kondo,S., RIKEN GER Group, GSL Members and Hayashizaki,Y. (2003) Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res., 13, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipman D.J. (1997) Making (anti)sense of non-coding sequence conservation. Nucleic Acids Res., 25, 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmichael G.G. (2003) Antisense starts making more sense. Nat. Biotechnol., 21, 371–372. [DOI] [PubMed] [Google Scholar]
- 18.Adams M.D., Kelley,J.M., Gocayne,J.D., Dubnick,M., Polymeropoulos,M.H., Xiao,H., Merril,C.R., Wu,A., Olde,B., Moreno,R.F. et al. (1991) Complementary DNA sequencing: expressed sequence tags and human genome project. Science, 252, 1651–1656. [DOI] [PubMed] [Google Scholar]
- 19.Boguski M.S., Lowe,T.M. and Tolstoshev,C.M. (1993) dbEST—database for ‘expressed sequence tags’. Nature Genet., 4, 332–333. [DOI] [PubMed] [Google Scholar]
- 20.Schuler G.D., Boguski,M.S., Stewart,E.A., Stein,L.D., Gyapay,G., Rice,K., White,R.E., Rodriguez-Tome,P., Aggarwal,A., Bajorek,E. et al. (1996) A gene map of the human genome. Science, 274, 540–546. [PubMed] [Google Scholar]
- 21.Huang X. and Madan,A. (1999) CAP3: a DNA sequence assembly program. Genome Res., 9, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kent W.J. (2002) BLAT—the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caron H., van Schaik,B., van der Mee,M., Baas,F., Riggins,G., van Sluis,P., Hermus,M.C., van Asperen,R., Boon,K., Voute,P.A. et al. (2001) The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science, 291, 1289–1292. [DOI] [PubMed] [Google Scholar]
- 24.Lash A.E., Tolstoshev,C.M., Wagner,L., Schuler,G.D., Strausberg,R.L., Riggins,G.J. and Altschul,S.F. (2000) SAGEmap: a public gene expression resource. Genome Res., 10, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Versteeg R., van Schaik,B.D., van Batenburg,M.F., Roos,M., Monajemi,R., Caron,H., Bussemaker,H.J. and van Kampen,A.H. (2003) The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res., 13, 1998–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burset M., Seledtsov,I.A. and Solovyev,V.V. (2000) Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res., 28, 4364–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lander E.S., Linton,L.M., Birren,B., Nusbaum,C., Zody,M.C., Baldwin,J., Devon,K., Dewar,K., Doyle,M., et al. and International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 28.Ashburner M., Ball,C.A., Blake,J.A., Botstein,D., Butler,H., Cherry,J.M., Davis,A.P., Dolinski,K., Dwight,S.S., Eppig,J.T. et al. (2000) Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cawley S., Bekiranov,S., Ng,H.H., Kapranov,P., Sekinger,E.A., Kampa,D., Piccolboni,A., Sementchenko,V., Cheng,J., Williams,A.J. et al. (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 116, 499–509. [DOI] [PubMed] [Google Scholar]
- 30.Eddy S.R. (2001) Non-coding RNA genes and the modern RNA world. Nature Rev. Genet., 2, 919–929. [DOI] [PubMed] [Google Scholar]
- 31.Eddy S.R. (2002) Computational genomics of noncoding RNA genes. Cell, 109, 137–140. [DOI] [PubMed] [Google Scholar]
- 32.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Serial analysis of gene expression. Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 33.Lercher M.J., Urrutia,A.O. and Hurst,L.D. (2002) Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nature Genet., 31, 180–183. [DOI] [PubMed] [Google Scholar]
- 34.Hurst L.D., Pal,C. and Lercher,M.J. (2004) The evolutionary dynamics of eukaryotic gene order. Nature Rev. Genet., 5, 299–310. [DOI] [PubMed] [Google Scholar]
- 35.Modrek B. and Lee,C. (2002) A genomic view of alternative splicing. Nature Genet., 30, 13–19. [DOI] [PubMed] [Google Scholar]
- 36.Johnson J.M., Castle,J., Garrett-Engele,P., Kan,Z., Loerch,P.M., Armour,C.D., Santos,R., Schadt,E.E., Stoughton,R. and Shoemaker,D.D. (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science, 302, 2141–2144. [DOI] [PubMed] [Google Scholar]
- 37.Kampa D., Cheng,J., Kapranov,P., Yamanaka,M., Brubaker,S., Cawley,S., Drenkow,J., Piccolboni,A., Bekiranov,S., Helt,G. et al. (2004) Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res., 14, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J., Rowley,J.D. and Wang,S.M. (2000) Generation of longer cDNA fragments from serial analysis of gene expression tags for gene identification. Proc. Natl Acad. Sci. USA, 97, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J., Lee,S., Zhou,G. and Wang,S.M. (2002) High-throughput GLGI procedure for converting a large number of serial analysis of gene expression tag sequences into 3′ complementary DNAs. Genes Chromosomes Cancer, 33, 252–261. [DOI] [PubMed] [Google Scholar]
- 40.Chen J., Sun,M., Lee,S., Zhou,G., Rowley,J.D. and Wang,S.M. (2002) Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc. Natl Acad. Sci. USA, 99, 12257–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie H. (2003) Gene ontology-facilitated genome analysis. Curr. Genomics, 4, 569–574. [Google Scholar]
- 42.Furey T.S. and Haussler,D. (2003) Integration of the cytogenetic map with the draft human genome sequence. Hum. Mol. Genet., 12, 1037–1044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.