


Abstract
Computational identification of ribosomal frameshift sites in genomic sequences is difficult due to their diverse nature, yet it provides useful information for understanding the underlying mechanisms and discovering new genes. We have developed an algorithm that searches entire genomic or mRNA sequences for frameshifting sites, and implements the algorithm as a web-based program called FSFinder (Frameshift Signal Finder). The current version of FSFinder is capable of finding −1 frameshift sites on heptamer sequences X XXY YYZ, and +1 frameshift sites for two genes: protein chain release factor B (prfB) and ornithine decarboxylase antizyme (oaz). We tested FSFinder on ∼190 genomic and partial DNA sequences from a number of organisms and found that it predicted frameshift sites efficiently and with greater sensitivity and specificity than existing approaches. It has improved sensitivity because it considers many known components of a frameshifting cassette and searches these components on both + and − strands, and its specificity is increased because it focuses on overlapping regions of open reading frames and prioritizes candidate frameshift sites. FSFinder is useful for discovering unknown genes that utilize alternative decoding, as well as for analyzing frameshift sites. It is freely accessible at http://wilab.inha.ac.kr/FSFinder/.
INTRODUCTION
Programmed ribosomal frameshifting is involved in the expression of certain genes in a wide range of organisms such as viruses, bacteria and eukaryotes including humans (1–5). In this process, the ribosome switches to an alternative frame at a specific site in response to special signals in the messenger RNA (4). Programmed frameshifting plays a significant role in morphogenesis, autogenous control and in producing alternative enzymatic activities (6).
The most common frameshift is a −1 frameshift, in which the ribosome slips a single nucleotide in the upstream direction. The major elements of −1 frameshifting consist of a slippery site, where the ribosome changes reading frames, and a stimulatory RNA structure such as a pseudoknot or a stem–loop located a few nucleotides downstream (4,6–9). It is generally accepted that ribosomes pause at −1 frameshifts, but Kontos et al. (7) report that pausing is not sufficient to mediate frameshifting. Most slippery sites consist of a heptameric sequence of the form X XXY YYZ in the incoming 0-frame (10), but there are other slippery sequences that do not conform to this motif (5). The slippery heptamer is separated from the stimulatory structure by a sequence of 5–9 nt, the so-called spacer (3,8). The length of the spacer is known to influence the efficiency of frameshifting. Frameshifts typically produce fusion proteins in which the N- and C-terminal domains are encoded by overlapping open reading frames (ORFs) (9), as shown in Figure 1.
Figure 1.
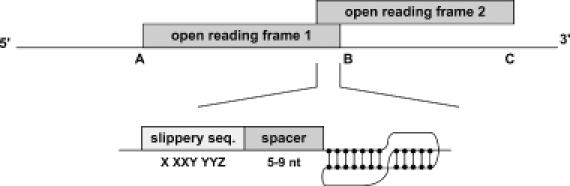
The three components of −1 frameshift signals in the overlap between two ORFs: slippery sequence, spacer and pseudoknot (or stem–loop). When a frameshift takes place, protein synthesis terminates at C rather than at B.
+1 frameshifts are much less common than −1 frameshifts but have been observed in diverse organisms (6). Escherichia coli prfB encoding release factor 2 (RF2) is a well-known gene that utilizes +1 frameshifting (11,12). In RF2 frameshifting, a Shine–Dalgarno (SD) sequence is often observed upstream of a slippery sequence, normally CUU UGA C and in a single known case CUU UAA C (12). Several +1 frameshift sites have also been recognized in eukaryotic mRNA. For example, the expression of mammalian antizyme 1 (AZ1) requires a +1 frameshift, and the frameshift signal consists of a slippery sequence and two stimulatory elements—a sequence of unknown function, upstream of the slippery sequence, and a pseudoknot (13).
Computational identification of frameshift sites from genomic sequences is difficult since the sequence requirements for frameshifting cassettes are diverse and highly dependent on the organism. Several computational approaches have been attempted, but only a few are publicly available. The model for eukaryotic −1 frameshifting developed by Bekaert et al. (8) only considers H-type pseudoknots as stimulatory structures and misses many frameshift sites with other stimulatory structures. Hammell et al. (9) developed a program to identify −1 frameshift sites in prokaryotic and eukaryotic DNA sequences, but the sensitivity of their approach is low; it misses many frameshift sites because it only considers downstream pseudoknots, and its definition of a pseudoknot is too restrictive. For example, their approach does not locate the frameshift sites in Rous sarcoma virus (RSV), because loops 1 and 2 of the pseudoknot are larger than permitted by their approach. FreqAnalysis developed by Shah et al. (14) is usable to identify simple novel slippery sequences, but it does not take in consideration existence of stimulators. A semi-automated approach by Ivanov et al. (13) finds a gene where antizyme frameshifting is expected to occur and then identifies the frameshift. While this approach has been shown to be successful for identifying ornithine decarboxylase antizyme (oaz) frameshifting, it omits universality. There are also computational approaches that identify frameshifting errors in sequencing when the reference protein sequences are available (15–17).
In this paper, we present an algorithm for locating −1 and +1 frameshift sites of certain types in genomic or mRNA sequences. The algorithm is intended to find −1 frameshift sites of X XXY YYZ type in viruses, bacteria and eukaryotes, and considers pseudoknots as well as simple stem–loops as downstream stimulatory structures. It also allows the user to change the stem and loop sizes from their default values. +1 frameshift signals are too diverse among different organisms. Therefore, the algorithm currently finds only those frameshift sites that are conserved among many species, namely frameshift sites used in genes encoding protein chain release factor B (prfB) and ornithine decarboxylase antizyme (oaz). The algorithm has been implemented as a web-based application program called FSFinder (Frameshift Signal Finder), and is accessible at http://wilab.inha.ac.kr/FSFinder/.
COMPUTATIONAL MODEL
Components of frameshift signals
We have modified the computational model for −1 frameshift signals of Hammell et al. (9) to improve its sensitivity and selectivity. Sequences of three codons (9 nt) in a genomic sequence are first examined for possible slippery sequences of the form X XXY YYZ. In this sequence X and Z can be any nucleotide, and Y can be A or U (in Hammell's model, Z is either A, U or C). If a slippery sequence is identified, FSFinder searches for a downstream structure by sliding 4–11 nt along the spacer. Figure 2 shows a programmed −1 frameshift site with a pseudoknot as stimulatory structure. The pseudoknot is of the H-type, in which stem 1 has ≤13 bp, stem 2 has ≤6 bp, and both loops of the pseudoknot have ≤6 nt. The first 4 bp of stem 1 include at least 2 G–C pairs. Some programmed −1 frameshift signals have a simple stem–loop as stimulatory structure. As explained in Figure 3, we examine the sequence in both directions from every pivot nucleotide for possible base pairing. The pivot nucleotide can be either included in, or excluded from, the base pairing.
Figure 2.
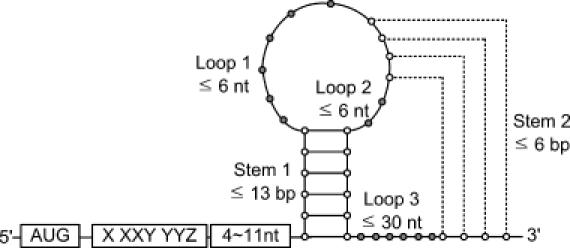
A programmed −1 ribosomal frameshift signal with an H-type pseudoknot.
Figure 3.
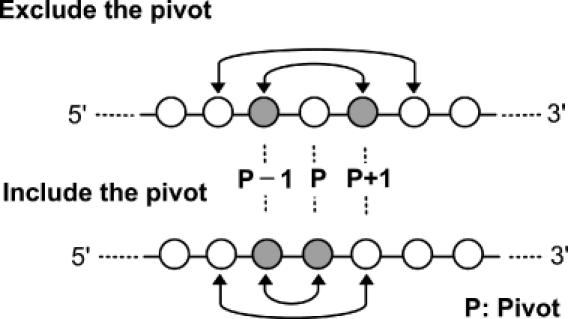
Finding a simple stem–loop structure downstream of a slippery sequence. Nucleotides in both directions from each pivot nucleotide are examined for possible base pairing.
Frameshifting can produce longer or shorter proteins than those resulting from standard decoding (4), as shown in Figure 4. FSFinder currently finds frameshift sites that result in longer products (Figure 4A), and ignores those resulting in shorter products (Figure 4B), since it focuses on frameshift sites in the overlapping region of ORFs. An exception to this is the E.coli dnaX gene. Although dnaX −1 frameshifting results in a shorter product, FSFinder finds its frameshift site using information about the upstream SD-like sequence (18). The SD-like sequence is simplified to GGRG or RGGR in the sequence located 9 nt upstream of the slippery sequence.
Figure 4.

Frameshifting may result in a long (A) or short product (B).
Since +1 frameshift signals are too diverse to model, we focus on +1 frameshift signals in two of the most common genes known to utilize frameshifting: protein chain release factor B (prfB) encoding release factor 2 (RF2), in prokaryota (12), and ornithine decarboxylase antizyme (ODC antizyme, oaz), in eukaryota (13). To detect prfB signals, FSFinder first searches for CUU UGA C or CUU UAA C slippery motifs. It then searches for an SD sequence 3 nt upstream and this sequence is simplified to 5 nt with RGG in the sequence. To detect oaz signals, FSFinder searches for UUU, UCC or CCC codons together with a UGA termination codon, a 3′ RNA pseudoknot, or both. Figure 5 shows a model of +1 frameshift signals. AUU codon that occurs upstream of UGA in Dugesia japonica antizyme frameshift site was not taken into account since it is the only known case where such frameshift site is utilized (19).
Figure 5.
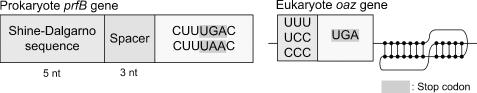
Programmed +1 ribosomal frameshift signals for eukaryotic oaz and prokaryotic prfB genes.
Algorithms for predicting frameshift sites
Algorithms 1 and 2 search for stem–loops and canonical base pairs, respectively. When bases of a single-stranded loop pair with complementary bases outside the loop, they are considered to form a pseudoknot (20). Algorithm 3 finds an overlap of ORFs. This is found as follows: suppose that a pair of ORFs is identified in frame 0 and frame −1, respectively (see Figure 6); the start positions of the ORFs are extended from their original start codons to upstream stop codons (positions A and C in Figure 6). The extended regions A–B and C–D of the two ORFs partially overlap at their termini if position A of frame −1 is to the left of position D of frame 0 and there exists a start codon in frame 0. FSFinder focuses on frameshift sites in the region of overlap (region E in Figure 6).
Figure 6.
The reading frame A–B (region that starts at A and ends at B) and the reading frame C–D partially overlap at their termini. FSFinder focuses on finding frameshift sites in the overlap region E.
Implementation
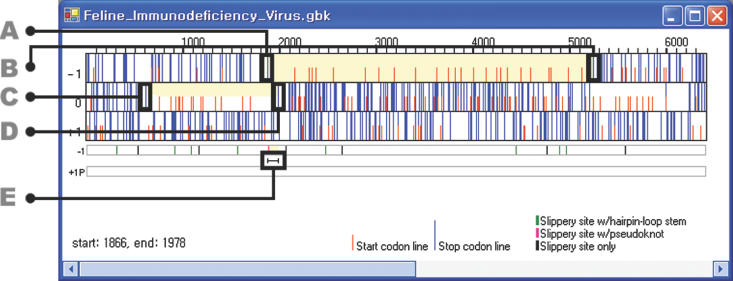
FSFinder has been implemented as a web-based application program using Microsoft C#. It can be executed on a Windows NT/2000/XP system with Microsoft .NET framework installed. Given a DNA or mRNA sequence in GenBank or FASTA format, it shows three frames (−1, 0 and +1 frames) in the upper left window (Figure 7). It considers one start codon, AUG, and three stop codons, UAA, UAG and UGA, for the three frames. Users are asked to choose from a list of available types of frameshifting (e.g. dnaX type, oaz type, etc.), the sequence size, and whether the search should be performed in the + or − strand during the file open operation. This information is used to determine the method of finding genes in the given sequence. For a bacterial genome with the prfB gene or sequence with the oaz gene, FSFinder first finds a gene in a manner similar to Glimmer (21). For a full genomic sequence specified as − strand by a user, frameshift sites are found in the reverse complementary sequence. Candidate −1 and +1 frameshift sites are shown below in the three frame views. +1 frameshift signals are set to prfB signals by default, but can be switched to oaz signals using the run menu. If a user specifies a region for detailed examination by the drag and drop operation, the specified region is enlarged in the lower left window.
Figure 7.
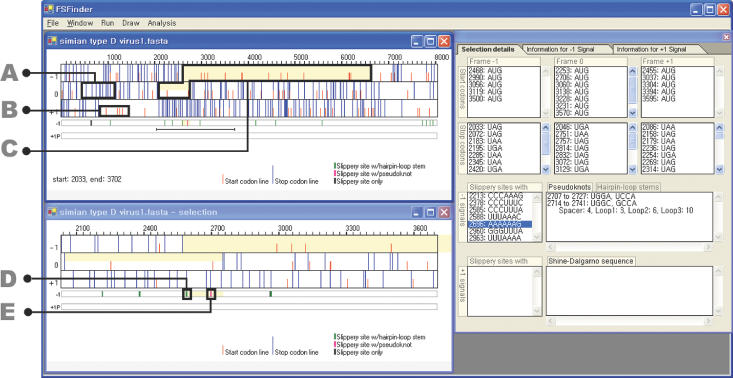
Graphical user interface of FSFinder. (A) Stop codons (long, blue lines). (B) Start codons (short, red lines). (C) Frameshift signal with the highest probability (light yellow). (D) Frameshift signal with a stem–loop (green bar). (E) Frameshift signal with a pseudoknot (pink bar).
The right window of FSFinder consists of three panels (Figure 7) for selection details, −1 signals, and +1 signals. The panel for selection details shows the start and stop codons, slippery sequences, pseudoknots and stem–loops (Figure 7). The panels for −1 and +1 signal panels show the total number of signals detected in overlapping and non-overlapping regions of the frames, as well as the positions of the signals.
Users can also choose the range of a view using the draw option in the draw menu, and change the stem and loop sizes of a stem–loop or pseudoknot using the find option in the run menu. They can also alternate frames to find frameshift sites in different overlapping frames using the analysis menu. Overlapping frames with the largest ORF (light grey) have the highest probability of containing frameshift sites, and overlapping frames with the second largest ORF (dark grey) have the second highest probability of having frameshift sites (see Figure 8).
Figure 8.
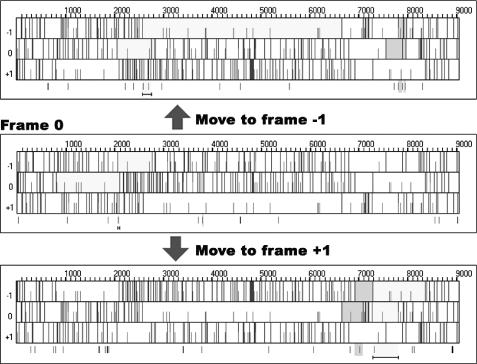
Alternating ORFs.
RESULTS AND DISCUSSION
We tested FSFinder on 71 organisms with known programmed −1 frameshift mutations obtained from the databases PseudoBase (22) and RECODE (23). At the moment when this work has been performed, PseudoBase contained 20 eukaryotic viruses, while RECODE had 65 prokaryotes, eukaryotic viruses, bacteriophages, eukaryotic transposable elements and bacterial insertion sequences. The two databases share 14 frameshifts. Each of these organisms and elements has one or two authentic programmed −1 frameshift sites for 27 genes in total.
FSFinder identifies more potential frameshift sites than the approach of Hammell et al. (9) because both pseudoknots and simple stem–loops are considered as downstream secondary structures and because the conditions for slippery motifs and pseudoknots are relaxed. On the other hand, it finds fewer candidates for non-programmed frameshift sites than the approach of Bekaert et al. (8) because it only searches for frameshift sites in the overlapping regions of ORFs, and prioritizes candidate frameshift signals. Existence of frameshift site in the overlap of two ORFs increases likelihood of frameshift site to be utilized for gene expression purposes.
In total, 26 frameshift sites in RECODE have simple stem–loops as downstream secondary structures, but 5 of these were excluded because PseudoBase assigns them different stimulatory structures or sequences. Eighteen of the remaining 21 frameshift sites were detected by FSFinder while 3 could not be found because their slippery sequences do not conform to the motif X XXY YYZ (Table 1). It turns out that most of bacterial frameshift sites have the slippery motif X XXY YYG. FSFinder identified 13 such sequences, and these can be classified into two types: A AAA AAG and G GGA AAG.
Table 1. Frameshift sites in RECODE with downstream stem–loops and X XXY YYG slippery sequences.
| RECODE ID | Organisms | ||
|---|---|---|---|
| Frameshift signals with X XXY YYZ (Z ≠ G) and a downstream stem | Frameshift signals with X XXY YYG and a downstream stem | Frameshift signals with X XXY YYG and other downstream structures | |
| 71 | Escherichia coli | ||
| 82 | HIV type 1 | ||
| 83 | HIV type 2 | ||
| 84 | Human T-cell lympotrophic virus type 1 | ||
| 85 | Human T-cell lympotrophic virus type 2 | ||
| 92 | Red clover necrotic mosaic virusa | ||
| 97 | Simian T-cell lymphosropic virus type 1 | ||
| 104 | Bacteriophage lambda | ||
| 106 | Drosophila buzzatii Ossvaldo retrotransposon | ||
| 237 | IS2 | ||
| 238 | IS911 | ||
| 251 | IS150 | ||
| 252 | IS1221A | ||
| 257 | Carrot mottle mimic virusa | ||
| 258 | Groundnut rosette virus | ||
| 260 | Pea enation mosaic virus RNA 2a | ||
| 360 | Salmonella typhi | ||
| 361 | Salmonella typhimurium | ||
| 362 | Vibrio cholerae | ||
| 363 | Neisseria meningtidis | ||
| 364 | Neisseria gonorrhoeae | ||
| 365 | Neisseria meningitides | ||
| 392 | Yersinia pestis |
aIndicates a frameshift site that was not identified by FSFinder because the slippery sequence did not conform to the motif X XXY YYZ.
Searching for frameshift signals in the overlapping region of ORFs is effective in predicting strong candidates for programmed frameshift sites. For example, a total of 582 potential −1 frameshift sites were found in the sequences of the test cases in PseudoBase. Only 40 of these were in overlapping ORFs, and only 21 of the 40 proved to be genuine frameshift sites. FSFinder also identifies frameshift sites in alternative frames. For example, simian type D virus 1 has two slippery sequences G GGA AAC and A AAU UUU in different frames at positions 2058 and 2585, respectively. FSFinder detected two different sites in each of six viruses in RECODE: human T-cell lymphotropic virus type 2, mouse mammary tumor virus, simian type D virus 1, simian retrovirus type 2, simian T-cell lymphotropic virus type 1 and visna virus. Only one alternative site (in mouse mammary tumor virus) could not be identified as it had a different motif (G GAU UUA). FSFinder could not detect the nine frameshift sites marked with ‘a’ in Table 2. As mentioned earlier, it only considers frameshift sites resulting in a long product, and those missed are associated with a short product.
Table 2. Predictions for −1 frameshift sites in PseudoBase and RECODE.
| ID | Organism | TP | FN | FP | TN | ID | Organism | TP | FN | FP | TN |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKB1 | BLV | 1 | 0 | 4 | 40 | RECODE96 | Simian retrovirus 2 | 1 | 0 | 1 | 33 |
| PKB2 | BWYV | 1 | 0 | 3 | 16 | RECODE97 | Siman T cell lympotropic virus 1 | 2 | 0 | 3 | 25 |
| PKB3 | EIAV | 1 | 0 | 2 | 41 | RECODE98 | Visna virus | 2 | 0 | 0 | 31 |
| PKB4 | FIV | 1 | 0 | 1 | 41 | RECODE99 | Bacteriophage T7a | 0 | 1 | 0 | 0 |
| PKB42 | PLRV-W | 1 | 0 | 1 | 13 | RECODE104 | Bacteriophage lambda | 1 | 0 | 0 | 0 |
| PKB43 | PLRV-S | 1 | 0 | 0 | 13 | RECODE105 | Cocksfoot mottle virus | 1 | 0 | 0 | 5 |
| PKB44 | CABYV | 1 | 0 | 0 | 10 | RECODE106 | D.buzzatii ossvaldo retrotransposone | 1 | 0 | 1 | 4 |
| PKB45 | PEMV | 1 | 0 | 2 | 12 | RECODE107 | D.ananassae Tom retrotransposone | 1 | 0 | 0 | 33 |
| PKB46 | BYDV-NY_RPV | 1 | 0 | 1 | 12 | RECODE108 | Gill-associated virus | 1 | 0 | 0 | 16 |
| PKB80 | MMTV | 2 | 0 | 0 | 34 | RECODE110 | T.vaginalis virus 2a | 0 | 1 | 0 | 6 |
| PKB106 | IBV | 1 | 0 | 0 | 65 | RECODE114 | B.subtilisa | 0 | 1 | 0 | 3 |
| PKB107 | SRV1_gag/pro | 2 | 0 | 0 | 33 | RECODE115 | D.melanogaster telo-meric retrotransposon Het-Aa | 0 | 1 | 0 | 22 |
| PKB127 | EAVa | 0 | 1 | 1 | 41 | ||||||
| PKB128 | BEV | 1 | 0 | 1 | 53 | RECODE118 | Enzootic nasal tumor V. | 1 | 0 | 1 | 15 |
| PKB171 | HCV_229E | 1 | 0 | 0 | 55 | RECODE233 | Potato leafrol V. | 1 | 0 | 1 | 9 |
| PKB174 | RSV | 1 | 0 | 0 | 17 | RECODE235 | IS1 | 1 | 0 | 1 | 2 |
| PKB217 | LDV-C | 1 | 0 | 0 | 36 | RECODE236 | IS3a | 0 | 1 | 0 | 3 |
| PKB218 | PRRSV-16244B | 1 | 0 | 1 | 43 | RECODE237 | IS2 | 1 | 0 | 0 | 1 |
| PKB233 | PRRSV-LV | 1 | 0 | 0 | 32 | RECODE238 | IS911 | 1 | 0 | 1 | 6 |
| PKB240 | BChV | 1 | 0 | 2 | 17 | RECODE249 | Cereal yellow dwarf V. RPV-NY | 1 | 0 | 1 | 9 |
| RECODE71 | E.coli | 1 | 0 | 0 | 4 | RECODE250 | Cereal yellow dwarf V. RPV-Mex | 1 | 0 | 0 | 3 |
| RECODE72 | Drosophila TE | 1 | 0 | 0 | 33 | RECODE251 | IS150 | 1 | 0 | 0 | 3 |
| RECODE73 | Human astrovirus | 1 | 0 | 1 | 7 | RECODE252 | IS1221A | 1 | 0 | 0 | 30 |
| RECODE79 | Giardiavirus | 1 | 0 | 0 | 7 | RECODE257 | Carrot mottle mimic V.a | 0 | 1 | 0 | 6 |
| RECODE80 | D.melanogaster gypsy TE | 1 | 0 | 0 | 21 | RECODE258 | Groundnut rosette V. | 1 | 0 | 0 | 14 |
| RECODE82 | HIV type 1 | 1 | 0 | 0 | 40 | RECODE260 | PEMV2a | 0 | 1 | 0 | 13 |
| RECODE83 | HIV type 2 | 1 | 0 | 0 | 13 | RECODE360 | S.typhi | 1 | 0 | 0 | 6 |
| RECODE84 | Human T-cell lympotrophic 1 | 1 | 0 | 5 | 22 | RECODE361 | S.typhimurium | 1 | 0 | 0 | 6 |
| RECODE85 | Human T-cell lympotrophic 2 | 2 | 0 | 0 | 16 | RECODE362 | V.cholerae | 1 | 0 | 0 | 5 |
| RECODE86 | IAP | 1 | 0 | 1 | 16 | RECODE363 | N.meningitides | 1 | 0 | 0 | 7 |
| RECODE88 | S.cerevisiae L-A | 1 | 0 | 0 | 15 | RECODE364 | N.gonorrhoeae | 1 | 0 | 0 | 8 |
| RECODE89 | Murine hepatitis V. | 1 | 0 | 0 | 49 | RECODE365 | N.meningitides | 1 | 0 | 0 | 9 |
| RECODE91 | Mason-pfizer monkey V. | 2 | 0 | 0 | 33 | RECODE375 | M.musculus | 1 | 0 | 0 | 19 |
| RECODE92 | Red clover necrotic mosaic V.a | 0 | 1 | 0 | 13 | RECODE376 | H.sapiens | 1 | 0 | 0 | 28 |
| RECODE94 | SIV | 1 | 0 | 2 | 18 | RECODE392 | Y.pestis | 1 | 0 | 0 | 7 |
| RECODE95 | Simian type D V. 1 | 2 | 0 | 0 | 30 | RECODE393 | SARS coronavirus | 1 | 0 | 1 | 62 |
aIndicates a frameshift site missed by FSFinder because a slippery sequence did not conform to the motif X XXY YYZ. TE: transposable element. TP: true positives, TN: true negatives, FP: false positives, FN: false negatives.
We also tested FSFinder on 75 organisms in RECODE with known +1 frameshift cassettes in the prfB gene and oaz genes, and successfully detected 62 out of 75. The reasons FSFinder missed 13 of the sites were as follows. Nine (RECODE19, RECODE34, RECODE35, RECODE37, RECODE44, RECODE52, RECODE64, RECODE67, RECODE369) of the 13 sequences were partial DNA sequences that have a truncated ORF (entire genomic sequences were not available in GenBank), and FSFinder could not find an overlap of ORFs. In three (RECODE9, RECODE14, RECODE21 in Table 3) of the 13 sequences, there was no pair of overlapping ORFs since one of the ORFs has no start codon. One (RECODE43 in Table 3) of the 13 sequences has a different SD sequence (GGUG) from FSFinder definition of a SD signal, and could not be detected.
Table 3. Predictions for +1 frameshift sites in RECODE.
| ID | Organism | TP | FN | FP | TN | ID | Organism | TP | FN | FP | TN |
|---|---|---|---|---|---|---|---|---|---|---|---|
| RECODE1 | B.mori | 1 | 0 | 0 | 1 | RECODE40 | C.pneumoniae | 1 | 0 | 0 | 0 |
| RECODE2 | B.fuckeliana | 1 | 0 | 0 | 0 | RECODE41 | C.acetobutylicum | 1 | 0 | 0 | 1 |
| RECODE3 | C.elegans | 1 | 0 | 0 | 2 | RECODE42 | C.difficile | 1 | 0 | 0 | 0 |
| RECODE4 | D.rerio (long form) | 1 | 0 | 0 | 1 | RECODE43 | D.ethenogenes | 0 | 0 | 0 | 1 |
| RECODE5 | D.rerio (short form) | 1 | 0 | 0 | 1 | RECODE44 | D.radiodurans | 0 | 0 | 0 | 1 |
| RECODE6 | D.melanogaster | 1 | 0 | 1 | 3 | RECODE45 | D.vulgaris | 1 | 0 | 1 | 0 |
| RECODE7 | A.nidulellus | 1 | 0 | 0 | 0 | RECODE46 | E.faecalis | 1 | 0 | 0 | 0 |
| RECODE8 | G.gallus | 1 | 0 | 0 | 1 | RECODE47 | E.coli | 1 | 0 | 0 | 0 |
| RECODE9 | G.pallida | 0 | 0 | 0 | 1 | RECODE48 | H.ducreyi | 1 | 0 | 0 | 0 |
| RECODE10 | H.contortus | 1 | 0 | 0 | 0 | RECODE49 | H.influenzae | 1 | 0 | 0 | 0 |
| RECODE11 | H.sapiens | 1 | 0 | 0 | 1 | RECODE50 | P.multocida | 1 | 0 | 0 | 0 |
| RECODE12 | H.sapiens | 1 | 0 | 0 | 4 | RECODE51 | P.gingivalis | 1 | 0 | 0 | 0 |
| RECODE13 | H.sapiens | 1 | 0 | 0 | 0 | RECODE52 | P.aeruginosa | 0 | 0 | 0 | 1 |
| RECODE14 | H.sapiens | 0 | 0 | 0 | 2 | RECODE53 | P.putida | 1 | 0 | 0 | 0 |
| RECODE15 | M.auratus | 1 | 0 | 0 | 2 | RECODE54 | R.prowazekii | 1 | 0 | 0 | 0 |
| RECODE16 | M.musculus | 1 | 0 | 0 | 2 | RECODE55 | S.typhimurium | 1 | 0 | 0 | 0 |
| RECODE17 | M.musculus | 1 | 0 | 0 | 2 | RECODE56 | S.typhi | 1 | 0 | 0 | 0 |
| RECODE18 | M.musculus | 1 | 0 | 0 | 0 | RECODE57 | S.putrefaciens | 1 | 0 | 0 | 0 |
| RECODE19 | N.americanus | 0 | 0 | 0 | 2 | RECODE58 | S.mutans | 1 | 0 | 0 | 0 |
| RECODE20 | O.volvulus | 1 | 0 | 0 | 1 | RECODE59 | S.aureus | 1 | 0 | 0 | 0 |
| RECODE21 | P.carinii | 0 | 0 | 0 | 1 | RECODE61 | S.pneumoniae | 1 | 0 | 0 | 0 |
| RECODE22 | P.pacificus | 1 | 0 | 0 | 0 | RECODE62 | S.pyogenes | 1 | 0 | 0 | 0 |
| RECODE23 | R.norvegicus | 1 | 0 | 0 | 2 | RECODE63 | S.PCC6803 | 1 | 0 | 0 | 1 |
| RECODE24 | S.pombe | 1 | 0 | 0 | 2 | RECODE64 | T.pallidum | 0 | 1 | 0 | 1 |
| RECODE25 | S.japonicus | 1 | 0 | 0 | 0 | RECODE65 | V.cholerae | 1 | 0 | 0 | 0 |
| RECODE26 | S.octosporus | 1 | 0 | 0 | 2 | RECODE66 | X.campestris pv. campestris | 1 | 0 | 0 | 0 |
| RECODE27 | T.marmorata | 1 | 0 | 0 | 2 | ||||||
| RECODE28 | X.laevis | 1 | 0 | 0 | 2 | RECODE67 | X.fastidiosa | 1 | 0 | 0 | 0 |
| RECODE29 | A.ferrooxidans | 1 | 0 | 0 | 0 | RECODE68 | N.meningitidis | 1 | 0 | 0 | 0 |
| RECODE30 | A.actinomycetemcomitans | 1 | 0 | 0 | 0 | RECODE69 | L.monocytogenes | 1 | 0 | 0 | 0 |
| RECODE32 | B.firmus | 1 | 0 | 0 | 0 | RECODE366 | B.halodurans | 1 | 0 | 0 | 0 |
| RECODE33 | B.subtilis | 1 | 0 | 0 | 0 | RECODE367 | B.parapertussis | 0 | 1 | 0 | 1 |
| RECODE34 | B.bronchiseptica | 0 | 1 | 0 | 2 | RECODE368 | B.sp.APS | 1 | 0 | 0 | 0 |
| RECODE35 | B.pertussis | 0 | 1 | 0 | 0 | RECODE369 | C.psittaci | 0 | 1 | 0 | 1 |
| RECODE36 | B.burgdorferi | 1 | 0 | 0 | 0 | RECODE370 | C.psittaci | 1 | 0 | 0 | 0 |
| RECODE37 | C.crescentus | 0 | 1 | 0 | 1 | RECODE371 | C.tepidum | 1 | 0 | 0 | 0 |
| RECODE38 | C.trachomatis | 1 | 0 | 1 | 0 | RECODE372 | D.hafniense | 1 | 0 | 0 | 0 |
| RECODE39 | C.muridarum | 1 | 0 | 0 | 1 | RECODE373 | M.loti | 1 | 0 | 0 | 0 |
Tables 2 and 3 summarize the predictions for −1 and +1 frameshift sites, respectively. A total of 68 −1 frameshift sites for 21 genes were predicted correctly, and 10 −1 frameshift sites for six genes were missed. The average sensitivity and specificity of prediction for −1 frameshift sites were 0.88 and 0.97, respectively, using Equations 1 and 2. For +1 frameshifts, FSFinder was intended for two genes. A total of 62 +1 frameshift sites were predicted correctly, and six were missed. The average sensitivity and specificity of prediction for +1 frameshift sites were 0.91 and 0.94, respectively, using Equations 3 and 4. It has higher specificity than sensitivity for both types of frameshifting.
![]()
![]()
![]()
![]()
where TP, TN, FP and FN are true positives, true negatives, false positives and false negatives, respectively. TPs are those cases where FSFinder found frameshifts that are annotated in the databases. FPs are those cases where FSFinder reported frameshifts that do not exist. TNs are those frameshifts that conform to the frameshift signal model but were rejected by FSFinder as candidate frameshifts because they exist outside the overlapping regions of ORFs. They are not annotated in databases, either. FNs are actual frameshifts that were missed by FSFinder.
Frameshift signals in microbial genomes
Escherichia coli release factor 2 (RF2) is a well-known example that utilizes +1 frameshifting (11,12), and the role of this frameshifting is widely acknowledged. We extracted 38 bacterial genomes with RF2 genes from GenBank that are not present in the RECODE database and tested FSFinder on them (Table 4). FSFinder missed 11 frameshift sites in the 38 organisms since their slippery sequences were of the form CUU URA C. The average sensitivity and specificity of prediction were 0.72 and 0.92, respectively (Equations 5 and 6). The sensitivity was lower than that for the RECODE data on +1 frameshifts.
Table 4. Predictions for +1 frameshift sites in the RF2 gene in bacterial genomes.
| ID | TP | FN | FP | TN |
|---|---|---|---|---|
| NC_002663 | 1 | 0 | 1 | 8 |
| NC_002737 | 1 | 0 | 0 | 18 |
| NC_002952 | 1 | 0 | 0 | 10 |
| NC_002971 | 1 | 0 | 0 | 5 |
| NC_003062 | 1 | 0 | 1 | 8 |
| NC_003197 | 1 | 0 | 2 | 20 |
| NC_003295 | 1 | 0 | 5 | 5 |
| NC_003304 | 1 | 0 | 1 | 8 |
| NC_003317 | 1 | 0 | 1 | 4 |
| NC_003454 | 1 | 0 | 0 | 11 |
| NC_003869 | 0 | 1 | 0 | 16 |
| NC_003909 | 1 | 0 | 0 | 20 |
| NC_004193 | 1 | 0 | 2 | 15 |
| NC_004307 | 0 | 1 | 0 | 2 |
| NC_004310 | 1 | 0 | 1 | 4 |
| NC_004342 | 0 | 1 | 0 | 18 |
| NC_004344 | 0 | 1 | 0 | 2 |
| NC_004350 | 1 | 0 | 1 | 23 |
| NC_004463 | 1 | 0 | 5 | 20 |
| NC_004551 | 0 | 1 | 0 | 5 |
| NC_004572 | 0 | 1 | 1 | 9 |
| NC_004663 | 1 | 0 | 2 | 17 |
| NC_004722 | 1 | 0 | 0 | 25 |
| NC_004757 | 1 | 0 | 8 | 3 |
| NC_005027 | 1 | 0 | 6 | 21 |
| NC_005042 | 1 | 0 | 0 | 15 |
| NC_005061 | 0 | 1 | 0 | 1 |
| NC_005071 | 1 | 0 | 1 | 14 |
| NC_005072 | 1 | 0 | 0 | 7 |
| NC_005085 | 1 | 0 | 1 | 5 |
| NC_005090 | 0 | 1 | 0 | 31 |
| NC_005126 | 1 | 0 | 1 | 20 |
| NC_005296 | 1 | 0 | 0 | 7 |
| NC_005303 | 0 | 1 | 0 | 3 |
| NC_005363 | 1 | 0 | 1 | 36 |
| NC_005823 | 0 | 1 | 0 | 18 |
| NC_005835 | 1 | 0 | 2 | 50 |
| NC_005861 | 1 | 1 | 1 | 17 |
TP: true positives, TN: true negatives, FP: false positives, FN: false negatives.
![]()
![]()
In Borrelia burgdorferi B31 (gi:15594346, 910 724 bp), FSFinder predicted a CUUUGAC heptameric sequence in the overlapping region of the ORFs (at position 70 196 in the +1 strand of B.burgdorferi), which corresponds to a known +1 frameshift site in prfB (23). It also predicted a new −1 frameshift site in the overlap region (at position 428 613). Biochemical experiments to confirm this are in progress.
We compared these predictions with those using randomly generated sequences in which the number of As and Ts were equal to those of Gs and Cs. FSFinder was tested on 10 random sequences of the same length as B.burgdorferi B31. On average, no −1 frameshift site and 0.9 +1 frameshift sites were detected in the overlapping regions of ORFs. These results indicate that −1 frameshift signals are very unlikely to exist by chance in the overlapping regions of random sequences.
For the purpose of comparison, we tested FreqAnalysis (14) on the ORF regions of the five organisms. FreqAnalysis finds various types of motifs in frameshift sites but does not provide information on motif positions and related RNA structures. It finds all potential frameshift sites in both overlapping and non-overlapping regions. In contrast, FSFinder only finds frameshift sites in overlapping regions and provides detailed information on the frameshift sites.
CONCLUSION
Identifying programmed frameshifts is difficult because of their diverse nature, yet it is important to fully understand the underlying mechanisms and to discover new genes. Existing computational models predict too many false positives, or need reference protein sequences together with DNA sequence data from similar organisms.
We have developed an algorithm and a program called FSFinder for predicting plausible −1 and +1 frameshift sites in long DNA or mRNA sequences. FSFinder was tested on the DNA sequences obtained from different organisms in RECODE, PseudoBase and GenBank, and it predicted both −1 and +1 frameshift signals with higher sensitivity and specificity than other approaches. FSFinder obtains increased sensitivity by considering most of known potentially relevant components and by searching both + and − strands, and has increased specificity because it focuses on the overlapping regions of ORFs and prioritizes candidate signals. We believe FSFinder will be useful to predict frameshift sites.
The development of FSFinder is not yet complete. The current version is capable of finding X XXY YYZ type of −1 frameshifting and prfB and oaz types of +1 frameshifting. Frameshift signals are very diverse and organism-dependent, so that they cannot be modeled in a single, universal way. FSFinder will be extended in future to find any frameshift site modeled by the user.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to John Atkins and Pavel Baranov for their valuable comments on the paper and FSFinder. This work was supported by the Korea Science and Engineering Foundation (KOSEF) under grant R01-2003-000-10461-0.
REFERENCES
- 1.Namy O., Rousset,J., Napthine,S. and Brierley,I. (2004) Reprogrammed genetic decoding in cellular gene expression. Mol. Cell, 13, 157–168. [DOI] [PubMed] [Google Scholar]
- 2.Stahl G., McCarty,G.P. and Farabaugh,P.J. (2002) Ribosome structure: revisiting the connection between translational accuracy and unconventional decoding. Trends Biochem. Sci., 27, 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinman J.D., Icho,T. and Wickner,R.B. (1991) A −1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag-pol fusion protein. Proc. Natl Acad. Sci. USA, 88, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baranov P.V., Gesteland,R.F. and Atkins,J.F. (2002) Recoding: translational bifurcations in gene expression. Gene, 286, 187–201. [DOI] [PubMed] [Google Scholar]
- 5.Licznar P., Mejlhede,N., Prere,M., Wills,N., Gesteland,R.F., Atkins,J.F. and Fayet,O. (2003) Programmed translational −1 frameshifting on hexanucleotide motifs and the wobble properties of tRNAs. EMBO J., 22, 4770–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farabaugh P.J. (1996) Programmed translational frameshifting. Ann. Rev. Genetics, 30, 507–528. [DOI] [PubMed] [Google Scholar]
- 7.Kontos H., Napthine,S. and Brierley,I. (2001) Ribosomal pausing at a frameshifter RNA psuedoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol. Cell. Biol., 21, 8657–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bekaert M., Bidou,L., Denise,A., Duchateau-Nguyen,G., Forest,J., Froidevaux,C., Hatin,I., Rousset,J. and Termier,M. (2003) Towards a computational model for −1 eukaryotic frameshifting sites. Bioinformatics, 19, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammell A.B., Taylor,R.C., Peltz,S.W. and Dinman,J.D. (1999) Identification of putative programmed −1 ribosomal frameshift signals in large DNA databases. Genome Res., 9, 417–427. [PMC free article] [PubMed] [Google Scholar]
- 10.Jacks T. and Varmus,H.E. (1985) Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science, 230, 1237–1242. [DOI] [PubMed] [Google Scholar]
- 11.Weiss R.B., Dunn,D.M., Atkins,J.F. and Gesteland,R.F. (1987) Slippery runs, shifty stops, backward steps, forward hots: −2, −1, +1, +2, +5, and +6 ribosomal frameshifting. Cold Spring Harb. Symp. Quant. Biol., 52, 687–693. [DOI] [PubMed] [Google Scholar]
- 12.Baranov P.V., Gesteland,R.F. and Atkins,J.F. (2002) Release factor 2 frameshifting sites in different bacteria. EMBO Rep., 3, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanov I.P., Gesteland,R.F. and Atkins,J.F. (2000) Antizyme expression: a subversion of triplet decoding, which is remarkably conserved by evolution, is a sensor for an autoregulatory circuit. Nucleic Acids Res., 28, 3185–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah A.A., Giddings,M.C., Parvaz,J.B., Gesteland,R.F., Atkins,J.F. and Ivanov,I.P. (2002) Computational identification of putative programmed translational frameshift sites. Bioinformatics, 18, 1046–1053. [DOI] [PubMed] [Google Scholar]
- 15.Birney E., Thompson,J.D. and Gibson,T.J. (1996) PairWise and SearchWise: finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res., 24, 2730–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halperin E., Faigler,S. and Gill-More,R. (1999) FramePlus: aligning DNA to protein sequences. Bioinformatics, 15, 867–873. [DOI] [PubMed] [Google Scholar]
- 17.Fichant G.A. and Quentin,Y. (1995) A frameshift error detection algorithm for DNA sequencing projects. Nucleic Acids Res., 23, 2900–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen B., Wills,N., Gesteland,R.F. and Atkins,J.F. (1994) rRNA–mRNA base paring stimulates a programmed −1 ribosomal frameshift. J. Bacteriol., 176, 6842–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov I.P., Anderson,C.B., Gesteland,R.F. and Atkins,J.F. (2004) Identification of a new antizyme mRNA +1 framshifting stimulatory pseudoknot in a subset of diverse invertebrates and its apparent absence in intermediate species. J. Mol. Biol., 339, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han K. and Byun,Y. (2003) PseudoViewer2: visualization of RNA pseudoknots of any type. Nucleic Acids Res., 31, 3432–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delcher A.L., Harmon,D., Kasif,S., White,O. and Salzberg,S.L. (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res., 27, 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Batenburg F.H.D., Gultyaev,A.P., Pleij,C.W.A., Ng,J. and Oliehoek,J. (2000) PseudoBase: a database with RNA pseudoknots. Nucleic Acids Res., 28, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baranov P., Gurvich,O.L., Hammer,A.W., Gesteland,R.F. and Atkins,J.F. (2003) RECODE. Nucleic Acids Res., 311, 87–89. [DOI] [PMC free article] [PubMed] [Google Scholar]