


Abstract
We describe a novel multiplexing technology using a library of small fluorescent molecules, termed eTag molecules, to code and quantify mRNA targets. eTag molecules, which have the same fluorometric property, but distinct charge-to-mass ratios possess pre-defined electrophoretic characteristics and can be resolved using capillary electrophoresis. Coupled with primary Invader® mRNA assay, eTag molecules were applied to simultaneously quantify up to 44 mRNA targets. This multiplexing approach was validated by examining a panel of inflammation responsive genes in human umbilical vein endothelial cells stimulated with inflammatory cytokine interleukin 1β. The laser-induced fluorescence detection and electrokinetic sample injection process in capillary electrophoresis allows sensitive quantification of thousands of copies of mRNA molecules in a reaction. The assay is precise, as evaluated by measuring qualified Z′ factor, a dimensionless and simple characteristic for applications in high-throughput screening using mRNA assays. Our data demonstrate the synergy between the multiplexing capability of eTag molecules by sensitive capillary electrophoresis detection and the isothermal linear amplification characteristics of the Invader® assay. eTag multiplex mRNA assay presents a unique platform for sensitive, high sample throughput and multiplex gene expression analysis.
INTRODUCTION
The ability to simultaneously analyze the expression of thousands of genes has become an indispensable part of genomics and integrated biology. Multiplex gene expression analysis has been used for validating existing targets, identifying new targets and detecting the consequences or genetic causes of diseases (1–4). A disease-related (1,5,6) or compound-specific gene expression pattern (2,7–12) is determined by genome-wide analysis of affected and controlled samples. Typically, a ‘signature’ is characterized by a gene expression pattern arising from a set of marker genes with predictive power (10). For example, drug discovery groups are interested in screening tens of thousands of potential lead compounds against a set of informative marker genes to monitor compounds' potential efficacy and safety. DNA microarray technologies are generally unsuited for this task due to inadequate sample throughput to screen a small subset of genes, high cost and relatively poor reproducibility (13). In contrast, traditional high-throughput screening (HTS) mRNA assays were able to screen large numbers of samples in a cost-effective manner, but the read-out is limited to one or two genes of interest (14–17). Therefore, there is a linear relationship between the total number of assays required to screen a compound library and the number of genes in a given marker set. Novel assay technologies that combine sample throughput and multiplex capability need to be developed to enable high-throughput mRNA analysis. Currently, a limited number of such assays are under development. Multiple transcripts were analyzed based on nuclease protection assay via hybridization on a luminescent array in a pre-configured 96-well format (18). The major advantage of this technology is its capability of using crude lysate as direct target, i.e. RNA purification and reverse transcription are not required. However, the intrinsic problems associated with the hybridization approach, such as high background and cross-hybridization between highly homologous sequences present serious challenges in developing a sensitive and specific assay. Another multiplex mRNA assay technology involves RNA isolation and RT–PCR (19). The multiplexing is based on a gel-based size separation of PCR fragments on capillary electrophoresis (CE). RNA isolation and reverse transcription are indispensable steps of the assay. It is not only a costly process, but also an indirect quantification and exponential target amplification method.
Here, we describe a multiplex approach using a library of small fluorescent molecules to code (interrogate) and quantify mRNA targets. These molecules, collectively termed electrophoretic tags, or eTag molecules, have the same fluorometric property. However, each of them is designed and synthesized to have distinct charge-to-mass ratio, resulting in pre-defined electrophoretic characteristics. Using eTag molecules attached to the 5′ terminal bases of signal probes, we have multiplexed the primary Invader® (7) mRNA assay up to 44 different genes per assay. Upon enzymatic cleavage, the released eTag molecules are subsequently separated, detected and quantified by laser-induced fluorescence (LIF) CE. To demonstrate the performance of the eTag multiplex mRNA assay system, the assay sensitivity was measured using known concentrations of in vitro transcripts (IVTs) and the assay reproducibility was assessed using crude cell lysates from human umbilical vascular endothelial cells (HUVECs). The LIF detection coupled with an electrokinetic sample injection process in CE allows sensitive quantification to be as low as 1000–8000 mRNA molecules in a 10 μl reaction. The gene expression profiles in response to IL1β stimulation in HUVEC are validated using real-time RT–PCR (TaqMan® assay). Owing to the high-specific nature of the Cleavase® enzyme employed in the multiplex eTag mRNA assay, this assay system can discriminate highly homologous RNA sequences (discriminating 95% homologous sequence, 1 in ≥1000), which enables specific quantification of families of genes, such as the cytochrome P450 enzymes, or orthologous genes in different species. The quality of the 10-plex gene expression data is further examined and demonstrated using Z′ factor for its potential application in HTS. This study shows that the combination of the multiplexing capability of eTag molecules, sensitive detection property of CE and the isothermal linear signal amplification characteristics of the Invader® assay technology provides a unique multiplexing mRNA assay system. This system, eTag multiplex mRNA assay system, allows sensitive, high sample throughput gene expression analysis in the area of target validation, molecular signature screening (20), in vitro as well as in vivo toxicology studies and high-throughput compound screening to determine the response of chemical perturbation or disease manifestation.
MATERIALS AND METHODS
Synthetic labeling of a DNA probe with an eTag molecule on an ABI 394 DNA synthesizer
An aliquot 0.1 M solution of 6-carboxyfluorescein (6-FAM) phosphoramidite (Glen Research; Sterling, VA) is prepared by the addition of 2.96 ml of anhydrous acetonitrile to a 0.25 g bottle of the fluorescein phosphoramidite. The standard reagents employed for DNA synthesis (Glen Research) are as follows: oxidizer: 0.02 M iodine; DeBlock: 3% trichloracetic acid in dichloromethane; activator: 1H-tetrazole in anhydrous acetonitrile; high-performance liquid chromatography (HPLC) grade acetonitrile (0.002% water); cap A: acetic anhydride; and cap B: N-methyl imidazole. Probes were synthesized with an eTag molecule coupled to the 5′ terminus of the sequences. A modified cycle is chosen such that the desired scale (0.2–1.0 μmol) of DNA is synthesized. Upon completion of the synthesis the column is removed from the synthesizer and the labeled oligonucleotide is purified using standard HPLC reverse phase and ion exchange protocols.
In vitro transcript generation
RT–PCR products were generated using ThermoScirpt™ RT–PCR System (Invitrogen, Carlsbad, CA) on HUVEC total RNA using the gene-specific primers with a T7 promoter sequence attached to the 5′ terminal base of the forward primer. IVTs were generated using a T7-MEGAscript™ High Yield Transcription Kit (Ambion, Austin, TX).
Cell culture and treatment
Pooled human umbilical vein endothelial cells (no. CC-2519; Clonetics, San Diego, CA) were grown in EGM medium (Clonetics). HUVEC was cultured no more than five passages. About 20 000 cells per well were seeded in a 96-well flat bottom cell culture plate the night before treatment with various concentrations of IL1β (R&D Systems, Minneapolis, MN) for different time durations.
Cell lysing
Cell culture medium was removed by gentle aspiration from 96-well plates. An aliquot of 20 μl of cell lysis buffer containing 0.5% (v/v) NP-40 (Sigma, St Louis, MO), 10 mM Tris, pH 7.5 and 10 ng/μl tRNA were dispensed into each well. The lysates were then heated at 85°C for 10 min. The heat-inactivated lysate can also be stored at −80°C for future use.
eTag multiplex mRNA assay
All invader oligonucleotides were purchased from Biosource International (Camarillo, CA) without HPLC purification. All oligonucleotide sequences are listed in Supplementary Table A (available online). The components in a 10 μl volume assay were 10 mM MOPS (pH 7.5; Ambion), 0.05% (v/v) Tween-20 (Promega, Madison, WI), 0.05% NP-40, 12.5 mM MgCl2 (Ambion), 0.5 μM of signal probe, 0.02 μM invader and stacker oligonucleotides and 2 ng/μl Cleavase® (Third Wave Technologies, Madison, WI). An aliquot of 5 μl of target (crude cell lysates, total RNA or IVT) was added to 5 μl of the assay components described above and incubated in a thermal cycler (Tetrad; MJ Research, Waltham, MA) or incubator (Thermolyne; Cole-Parmer, Chicago, IL) at 60°C for 4 h.
Capillary electrophoresis
Upon completion of the eTag multiplex mRNA assay, 10 μl of CE separation reagent, containing a mixture of internal CE markers (ACLARA BioSciences, Mountain View, CA), was added to the reaction. Samples were analyzed using the ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA) or the MegaBACE™ 1000 DNA Sequencer instrument (Amersham Biosciences, Piscataway, NJ). On the ABI PRISM® 3100 system, samples were injected for 30 s at 3 kV and separated using an applied potential of 15 kV at 30°C. On the MegaBACE 1000 system, a 96-capillary coated array was used and samples were injected at 15 kV for 75 s. Separation was performed using a potential of 15 kV at 44°C. On both systems eTag reporters were detected using LIF, with argon-ion laser excitation at 488 nm and fluorescence emission collected at ∼520 nm.
TaqMan® assay
The TaqMan® probes for IL6, IL8 and ABCA1 were purchased from Applied Biosystems. The assays were performed according to the manufacturer's instructions.
Data analysis
The CE electropherograms from each assay well were analyzed using the eTag Informer™ software (ACLARA BioSciences), which is designed for the identification and quantification of eTag reporter peaks. The background signals (signal from no target control) were subtracted as appropriate. For example, the variation in CE injection was controlled by normalizing the peak height of eTag reporters to that of CE marker 2, and the biological sample variation was normalized by ratio to a housekeeping gene of choice, such as GAPD.
RESULTS AND DISCUSSION
Synthesis of eTag molecule-labeled oligonucleotide signal probes
eTag molecules, fluorescent species containing a charge-to-mass modulator, can be separated and quantified by LIF CE. For example, 6-carboxyfluorescein (6-FAM) phosphoramidite can be used as an eTag molecule. There are a variety of methods to couple eTag molecules to DNA probes. One procedure employs a small fluorescent phosphoramidite eTag molecule, which can be attached to the 5′ end of an oligonucleotide on a DNA synthesizer using standard phosphoramidite chemistry. This method utilizes proven solid-phase support chemistries and allows a single-step purification procedure. Alternatively, other electrophilic species such as maleimides can be used to post-synthetically label the DNA probes. Other DNA assembly chemistries including H-phosphonate or phosphotriester approaches can also be employed. The detailed eTag molecule synthesis and electrophoretic characterization are presented elsewhere (US patents 6 322 980, 6 514 700, 6 673 550 and 6 682 887; T. Matray, V. Hernandez, A. Chenna, C. Calacsan and S. Singh, manuscript in preparation).
The eTag reporter consists of the eTag molecule along with the 5′ terminal nucleic acid residue cleaved from a signal probe by specific enzymatic reaction (vide infra). It is well documented that CE is a powerful way to separate fluorescent molecules or DNA fragments, such as DNA fragments from sequencing reactions (21–23), restriction digestions (24,25), multiplex PCR assay (26) or from ligation assays (27). Similarly, CE can also be applied in separating and quantifying eTag reporters, since each reporter possesses a unique electrophoretic mobility.
eTag Multiplex mRNA Assay principle
The primary Invader® mRNA assay was chosen to test the multiplex capability of the eTag technology; it is a homogenous, isothermal and linear signal amplification system for direct quantitative detection of mRNA. The detailed Invader®mRNA assay has been presented previously (17,28,29). Briefly, two target-specific oligonucleotide probes (signal probe, Invader® oligonucleotide) form an invasive structure by hybridizing in tandem to the mRNA target (17) (Figure 1a). Stacker oligonucleotide is used to increase the melting temperature of the signal probe permitting higher reaction temperature (17,30). The signal probe is coded with an eTag molecule at its 5′ terminal base. Cleavase® (Third Wave Technologies), a thermal stable 5′ nuclease, recognizes the invasive structure and cleaves the 5′ base along with the eTag molecule, releasing it as an eTag reporter (eTag molecules with the 5′ base). Multiplexing is achieved by allocating distinct eTag molecules to multiple target-specific signal probes. The migration address (i.e. eTag reporter migration time measured relative to the migration times of CE internal markers) of each eTag reporter codes the identity of the target mRNA (Figure 1b). The relative fluorescence signal intensity, measured by relative peak height (RPH) with respect to a CE marker, is proportional to the initial target concentration. Since the limit of detection (LOD) of CE is typically in subpicomolar range, it provides sufficient sensitivity for the signals amplified in the multiplexed primary Invader® mRNA assay. The secondary Invader® reaction, normally performed after the primary Invader assay, is omitted. No need for the secondary Invader® assay step and direct measure of mRNA in crude lysate (vide infra) has unprecedentedly simplified gene expression analysis in eTag multiplex mRNA assay system (Figure 1c). To demonstrate multiplexing capability of the eTag technology, 44 distinct eTag molecules were attached to 44 specific signal probes coding for 44 different IVTs. Figure 2a shows an electropherogram generated from the 44-plex primary Invader® assay. To test the specificity of the multiplex assay, the targets (IVTs) for IL10, IL8, IL6 IL1B and IL12A were withdrawn individually or together in the reaction, as shown in Figure 2b. The absence of the corresponding signals confirmed the specificity of each probe in the multiplex assay. A single target (IL6) titration was performed in the presence of constant concentrations (1 × 106 copies per assay) of the other 43 targets (Figure 2c). The signal showed a linear response to the target concentration at a range of 8 × 103 to 2 × 106 as tested (R2 of 0.998, the linear plot is not shown). These tests indicate that each individual reaction in the 44-plex format is an independent assay, i.e. the readout of each reaction is only a function of its target and the presence or absence of other targets or probe sets does not interfere with individual specific reactions. This implies that the eTag multiplex mRNA assay is configurable.
Figure 1.
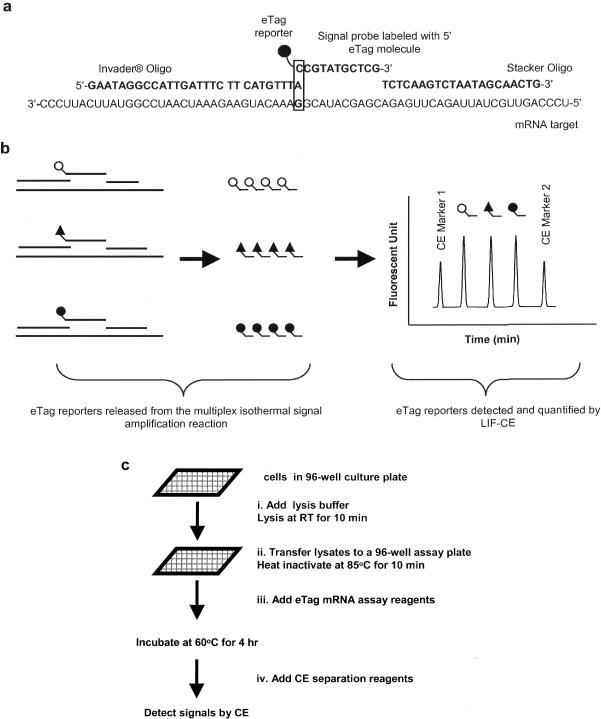
(a) Schematic representation of eTag multiplex mRNA assay coupling the eTag reporter chemistry and the Invader® mRNA reaction. A probe set is shown (from left to right) including Invader® oligonucleotide, signal probe coded with distinct eTag molecule and stacker oligonucleotide specifically designed to anneal to its respective RNA target. The 3′ terminus of Invader® oligonucleotide invades one base (non-complementary to template) into DNA–RNA duplex between signal probe and target, forming an overlapped DNA–RNA triplex structure shown in box. The Cleavase® enzyme, which possesses 5′ nuclease activity, recognizes and cleaves this specific structure, generating an eTag reporter (eTag molecule with 5′ terminal nucleotide). Subsequently, signal probes, in large excess, rapidly undergo association with the mRNA target replacing cleaved signal probes. Multiple signal probes are cleaved per RNA target, resulting in target-specific accumulation of eTag reporters. (b) Multiplex assay is achieved by allocating unique eTag molecules to specific probe sets. Multiple cleaved eTag reporters are separated by CE. (c) eTag multiplex mRNA assay procedure. Assay samples could be total RNA or crude lysate. There are four liquid addition steps starting from sample plate to CE analysis: (i) lysis buffer addition (this step is omitted if the sample is RNA), (ii) transfer the lysate from culture plate to a 96-well assay plate, (iii) reaction mixture (containing probes and enzyme) in addition to the assay plate and (iv) CE marker addition.
Figure 2.
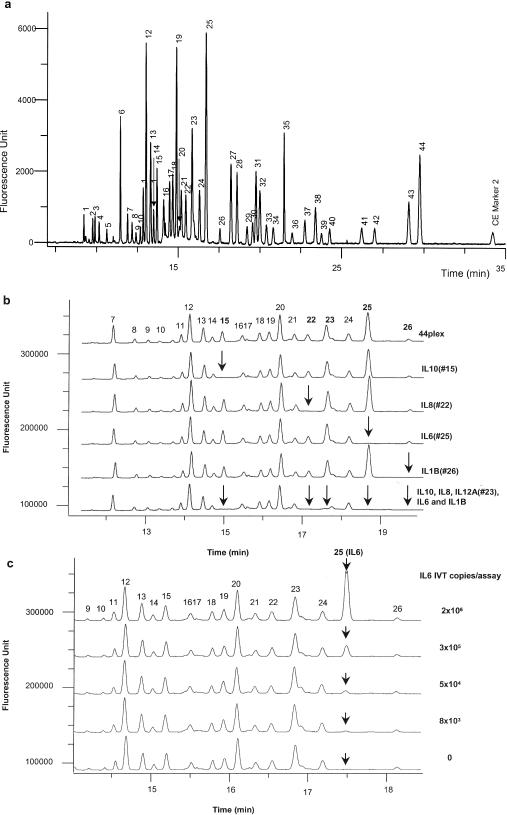
(a) Separation of 44 eTag reporters generated from a single eTag mRNA assay of 44 different in vitro transcription targets at a concentration of 1 × 106 copies per assay. The 44 analytes (based on the order of the electrophoretic mobility) are: CYP1A2 (1), PPIA (2), CYP2B6 (3), CYP2C9 (4), FOS (5), MYC (6), IL1A (7), MMP1 (8), GCS (9), TGFB1 (10), CYP1A1 (11), PPARG (12), C3 (13), TNFAIP3 (14), IL10 (15), GRP78 (16), IL18 (17), SOD (18), VEGFC (19), CCNA1 (20), BCL2 (21), IL8 (22), IL12A (23), JUN (24), IL6 (25), IL1B (26), GMCSF (27), IL12B (28), TNF (29), CCNE1 (30), CYP2C19 (31), GAPD (32), RAD51 (33), CYP3A5 (34), CYP3A4 (35), CCNB1 (36), G6PD (37), VEGF1 (38), ATF3 (39), CREB1 (40), UBQ (41), BAX (42), CDK1 (43) and CDK6 (44). CE marker 2 is an electrophoresis reference control. (b) Probe specificity in a 44-plex assay. The top electropherogram shows signals generated when all the analyte targets (IVTs) were present in the concentration of 1 × 106 copies per assay. As specific target withdrawn from the target mixtures, the corresponding signal was absent (in arrows) as shown in the examples of IL10 (15), IL8 (22), IL6 (25), IL1B (26) and IL12A (23). (c) Signal linearity in the 44-plex assay. The specific signal for IL6 (in arrow) was linearly correlated to the concentration of analyte IL6 (25) tested in the range of 2 × 106 to 8 × 103 copies per assay (R2 = 0.998, the plot is not shown).
Sensitivity and linear dynamic range
A panel of 10 inflammatory responsive genes (shown in Table 1) was selected to evaluate the multiplex assay performance in a model system. Specific probe sets (signal probes, stacker and Invader® oligonucleotides) were designed using Cleavage Site Prediction software (ACLARA BioSciences) and Invader Creator® software (Third Wave Technologies). The sensitivity and linearity of the 10-plex assays were determined using known concentrations of IVTs. Figure 3a shows four representative electropherograms of target titration at concentrations of 1.6 × 107, 1 × 106, 6 × 104 and 4 × 103 molecules per reaction. At low target concentrations of 4 × 103 and 1 × 103 molecules, the signal intensity is displayed with an expanded scale (see the insert). The signal intensity was measured as RPH to the CE marker 2. The linear dynamic range of the assay was observed from 1 × 103 to 6 × 107 molecules per reaction, corresponding to 4.8 orders of magnitude (Figure 3b). The coefficient of correlation (R2) of the detection linearity, reported in Table 1, averaged 0.994. The LOD was determined by Students t-test, which measures the minimum concentration that gives statistically significant response above background (with no target). The limit of quantification (LOQ) is defined as the minimum concentration (no less than LOD) in a linear region of target concentration response. The LOQ of this 10-plex assay varies from probe set to probe set, in a range from 1 × 103 to 8 × 103 molecules per assay. The Cleavase® enzyme cleavage rates (number of cleaved eTag reporters per hour at a fixed enzyme concentration) for each probe set was distinct, as reflected by different slopes in Figure 3b. It is worth mentioning that slope alone is not a sole predictive factor for LOQ, as background signal contributed by impurity of the signal probes would affect both LOD and LOQ. This would explain why the efficient cleavage rate (steep slope) of the IL6 probe set rendered a relatively poor LOQ (8 × 103 molecules per reaction). The high sensitivity of the eTag multiplex mRNA assay system is attributed to two key factors: (i) the separation of eTag reporters from assay background as part of the electrophoresis separation process—the ‘isolation’ of signals (i.e. eTag reporters) from background and (ii) sensitive LIF detection coupled with a electrokinetic injection process that concentrates the eTag reporters in the capillary.
Table 1. The LOQ and coefficient of correlation of linearity assessed in a10-plex eTag multiplex mRNA assay.
| Gene | Accession number | LOQa (molecules per reaction) | Coefficient of correlation (R2) |
|---|---|---|---|
| ICAM 1 | NM_000201 | 1 × 103 | 0.994 |
| IL6 | M 14584 | 8 × 103 | 0.999 |
| TNF | NM_000594 | 4 × 103 | 0.993 |
| IL8 | NM_000584 | 4 × 103 | 0.988 |
| CCL2 (MCP 1) | X14768 | 4 × 103 | 0.979 |
| PTG S2 (COX 2) | NM_000963 | 4 × 103 | 0.995 |
| ABCA 1 | NM_005502 | 4 × 103 | 0.995 |
| SELE | NM_000450 | 4 × 103 | 0.999 |
| VCAM 1 | NM_001078 | 1 × 103 | 0.997 |
| GAPD | AK026525 | 4 × 103 | 0.999 |
aData from five replicates.
Figure 3.
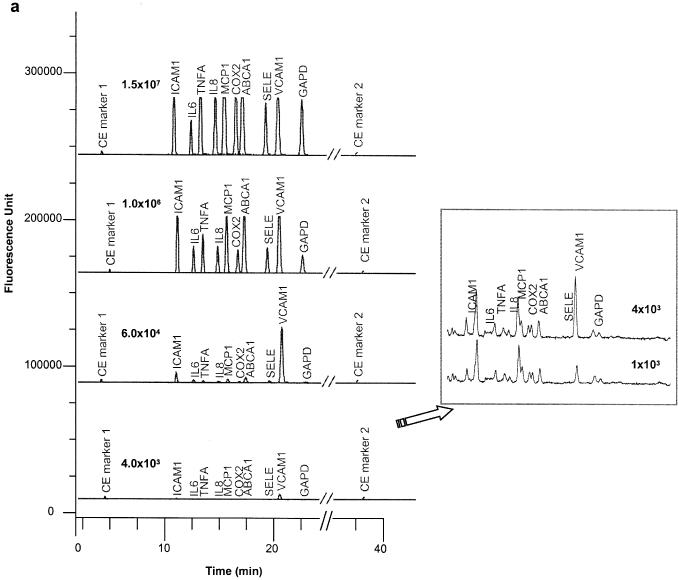
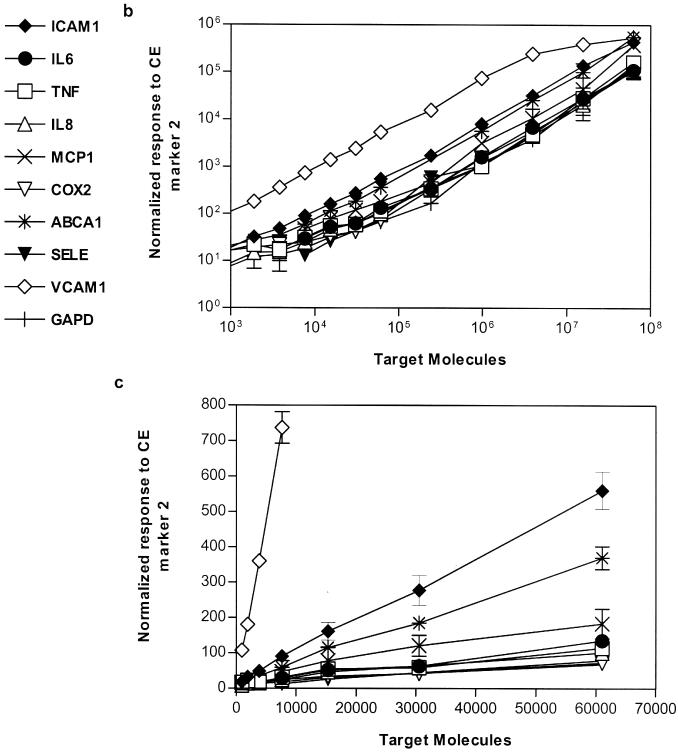
Assay sensitivity and linearity. (a) Electropherograms of 10-plex eTag mRNA assays run on ABI PRISM® 3100 Genetic Analyzer. CE marker 1 and 2 are the electrophoresis reference controls. An expanded view for low concentrations of target at 4 × 103 and 1 × 103 molecules per reaction is shown in the insert. (b) Average RPHs normalized to CE marker 2 in triplicates as a function of target concentration was plotted in double logarithm scale ranging from 1 × 103 to 6.25 × 107. (c) An expanded view for lower target concentration. A linear regression was performed on target concentration response curve for each gene. The LOQ and the coefficient of correlation for signal linearity curves are summarized in Table 1.
Microarray technology, which can monitor the level of gene expression for tens of thousands of genes simultaneously, is one of the most important advances in genomics technology. It is reported that most array-based mRNA measurements require much higher starting amount of total RNA which is equivalent to millions of cells (31) and the typical LOD for microarrays is in the low picomolar range (32). Considering a standard hybridization volume to be 200 μl (31), this LOD corresponds to greater than 106 copies per assay. There are accumulating evidence suggesting that it is possible to perform sensitive array-based measurement using a small amount of RNA (2–20 ng, 100–1000 equivalent mRNA molecules) (33), provided that it has to implement a series of sample preparation steps to couple amplification methods with the array measurements (34–39). The cost on sample preparation is not negligible.
On the other hand, single gene real-time PCR method such as TaqMan® assay has been reported to be able to detect as low as 13 copies per assay (40). Conversely, the least sensitive assays could only detect 104 or more copies (40). In most cases, however, the lower quantification limit is around 50–500 copies of target mRNA (40), depending on both the amplification efficiencies and the sensitivities of the amplicons tested. Interestingly, some single gene (single-plex) eTag assays are able to detect as low as 500 copies (data not shown). Compared to the average lower LOQ reported in real-time RT–PCR assays, the eTag assay LOQ might be 2–20-fold less sensitive. The assay dynamic range for eTag assay is 4.8 orders of magnitude, which could be one to two orders of magnitude less than TaqMan® assay.
The LOQs of TaqMan and eTag assays remain to be carefully compared. Nevertheless, both methods require a small sample input (1000–5000 cells per assay, or equivalent of 3.75–15 ng total RNA per assay), which is already in the range of <1 copy per cell. It is worth noting that eTag gene expression assay may be a choice for certain applications that desire high sample throughput (thousands of samples) and yet measure a set of genes (10–44 genes) simultaneously. The assay chemistry, working directly in crude lysates under isothermal amplification condition, has made it amendable for automation.
Precision
The assay reproducibility was determined by quantifying well-to-well and day-to-day variation. Around 20 000 HUVECs in each well of a 96-well plate were treated with various concentrations of IL1β. Since sample preparation (RNA isolation and reverse transcription) is not required for the eTag mRNA assay (data not shown), the transcripts of 10 endogenous genes were monitored directly in crude lysates in triplicate after 4 h of treatment. Within an assay well, RPHs were measured for all the expressed genes and were normalized to the corresponding housekeeping gene, GAPD (normalized response). The average normalized response of every data point from two identical cell treatment experiments were compared and plotted, as shown in Figure 4a. A linear function of y = 1.01x (correlation coefficient of R2 = 0.997) was observed, demonstrating that the eTag multiplex mRNA assay system is highly reproducible from experiment to experiment. A similar experiment was performed to assess the day-to-day variation. Matched data points assayed on two different days were plotted in Figure 4b. A linear function of y = 0.87x was observed with coefficient of the correlation R2 of 0.962. In order to quantitatively evaluate the overall assay quality, the Z′ factor (41) was calculated for each gene (using the normalized response) in this 10-plex assay, assuming each gene is an independent responsive element in the assay. The positive controls and negative controls refer to data points collected from maximal gene expression induction and no treatment, respectively. Twelve replicates were in each control group. The Z′ factors ranged from 0.5 to 0.8, which passes standard HTS criteria (Z′ > 0.45) (Table 2).
Figure 4.
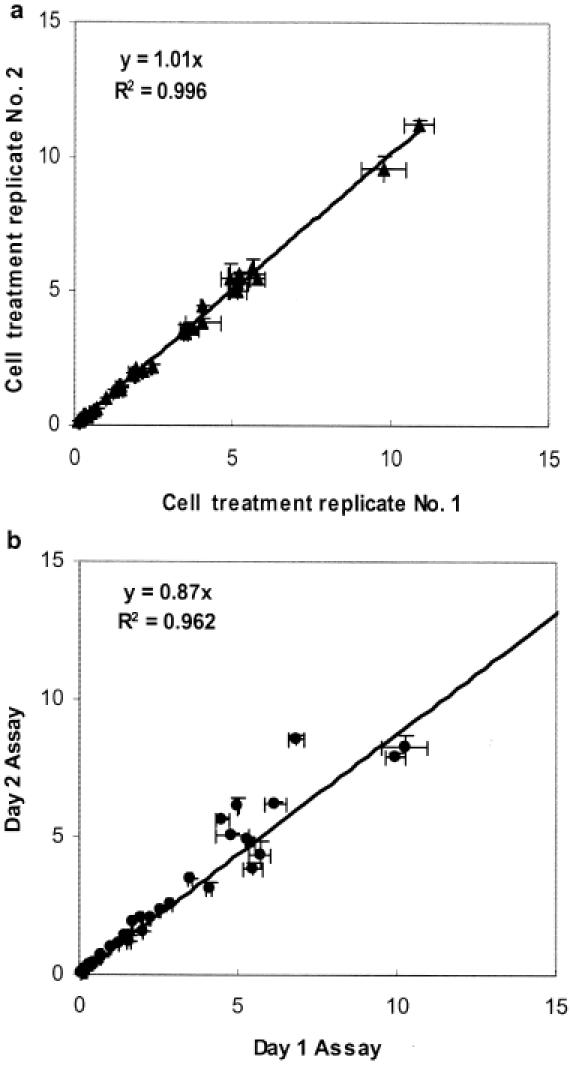
Assay precision. (a) Well-to-well variability for overall assay performance. In two identical experiments, HUVECs were treated with eight doses of IL1β and the assays were performed in triplicates. The expression of the 10 endogenous transcripts from each treatment experiment was compared. The average normalized response to GAPD was calculated and plotted against that of the counterparts. The standard deviations of the triplicate measurement were presented as the X and Y error bars. (b) Day-to-day variability. On two different days, the assays were performed in triplicate on the same sample. The standard deviations of the triplicate measurement are presented as the X and Y error bars.
Table 2. Z′ factor calculated in 10-plex eTag mRNA assay.
| Gene | Normalized responses (to GAPD) | Z′ factor | |||||
|---|---|---|---|---|---|---|---|
| Positive control | Negative control | ||||||
| Mean (N = 12) | SD | CV% | Mean (N = 12) | SD | CV% | ||
| SELE | 5.07 | 0.36 | 7.10 | 0.08 | 0.01 | 12.5 | 0.72 |
| VCAM 1 | 1.82 | 0.19 | 10.61 | 0.08 | 0.01 | 12.5 | 0.69 |
| IL8 | 9.46 | 1.15 | 12.19 | 0.075 | 0.01 | 16.42 | 0.63 |
| COX 2 | 3.59 | 0.39 | 10.82 | 0.20 | 0.043 | 21.36 | 0.62 |
| IL6 | 1.38 | 0.17 | 12.36 | 0.08 | 0.011 | 15.06 | 0.58 |
| ICAM 1 | 5.10 | 0.70 | 13.72 | 0.08 | 0.012 | 16.40 | 0.57 |
| MCP 1 | 4.86 | 0.79 | 16.21 | 0.07 | 0.011 | 14.65 | 0.50 |
| ABCA 1 | 0.19 | 0.03 | 16.39 | 0.14 | 0.038 | 27.74 | NR |
SD, standard deviation; and NR, not reported due to statistically insignificant response.
Specificity
The specificity inherent in the Invader® assay system enables the discrimination of highly homologous sequences. Similar to the Invader® DNA assay for single nucleotide polymorphism detection (28), Invader® mRNA assay also discriminates a single base change (17). Four dual-plex experiment results are summarized in Table 3. In each experiment, the specificity of two pairs of probe sets was tested in the presence of a single IVT at 1 × 108 copies per assay. The discrimination ratio of specific signal (signal generated from the specific IVTs versus the homologous IVTs) was at least 1000-fold. This characteristic is especially important for the analysis of drug targets that arise from close families (such as homologous kinases, matrix metalloproteins and receptors) and drug metabolism enzymes (Cytochrome P450s).
Table 3. Assay specificity assessed in highly homologous transcripts.
| Sequence identity (%) | Target | Normalized responses (to CE Marker 2) in dual-plex eTag mRNA assaya | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CYP3A4 | CYP3A5 | Rn. Cyp2b1 | Rn. Cyp2b2 | Hs. GAPD | Mm. Gapd | Hs. KRAS2 | Mm. Kras2 | ||
| 92 | HS. CYP3A4 | 16.62 | (0.01) | ||||||
| HS. CYP3A5 | (N.D.) | 26.40 | |||||||
| 95 | Rn. Cyp2b1 | 8.82 | (N.D.) | ||||||
| Rn. Cyp2b2 | (0.01) | 22.19 | |||||||
| 97 | HS. GAPD | 26.77 | (0.02) | ||||||
| Mm. Gapd | (0.01) | 10.20 | |||||||
| 99 | Hs. KRAS2 | 45.05 | (0.02) | ||||||
| Mm.Kras2 | (0.02) | 40.66 | |||||||
ND: signal not detected.
aIn a dual-plex assay, specific signal (in boldface) was generated in the presence of 1 × 108 copies of IVTs. The background signal (in parentheses) was determined in the presence of 1 × 108 copies of homologous IVT.
Gene expression profiling—dose–response, time course and EC50 determination
Expression profiles of the 10 genes were characterized upon stimulation of HUVEC with nine different doses of IL1β for 4 h. Among the 10-plex gene panel, ICAM1, IL6, IL8, MCP1, COX2, SELE and VCAM1 were documented previously as being up-regulated by IL1β treatment (42–44). The fold change of gene expression was calculated as the ratio of the normalized response in treated samples versus untreated samples and was plotted as a function of IL1β concentration (Figure 5a). A dose–response curve was fitted for each gene (GraphPad Prism 3.03; GraphPad Software, San Diego, CA). LogEC50 were calculated from the dose–response curves (data not shown). At the dose of 10 ng/ml of IL1β, the mRNA abundance change reached a maximum for ICAM1 (70.3 ± 1.3), IL8 (140.0 ± 2.6), MCP1 (66.3 ± 1.7), COX2 (26.9 ± 7.2), SELE (73.5 ± 3.8) and VCAM1 (26.5 ± 0.1). IL6 was greatly induced upon treatment. The fold change was not obtained due to undetectable basal expression of this gene. TNF was not detected. ABCA1 (1.8 ± 0.6) remained unchanged throughout the doses tested.
Figure 5.
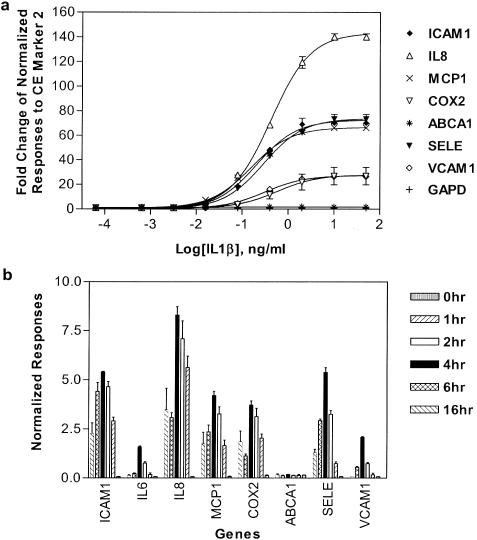
The dose–response and time course on mRNA expression upon IL1β treatment on HUVEC. (a) Dose–response curve. HUVEC received nine doses of IL1β from 50 ng/ml to 0.64 pg/ml (1:5 serial dilutions) for 4 h. A 10-plex eTag mRNA assay was performed in triplicates. The gene expression fold-change was calculated by taking the ratio of normalized response (to CE marker 2) of treatment with respect to that of the untreated controls. The sigmoidal dose–response curve was fitted for eight genes only (as TNF and IL6 which was not expressed in untreated samples) using the following equation: Y = Ylower plateau(Yupper plateau − Y1lower plateau)/[1 + 10(logEC50−X)*Slope], where Ylower plateau and Yupper plateau are the fold changes measured at two plateaus of the curve, and the slope is variable slope at the response region (GraphPad Prism 3.03). (b) The compound treatment time course. HUVECs were treated with 2 ng/ml of IL1β for 1, 2, 4, 6 and 16 h. The normalized responses (to GAPD) were plotted as a function of treatment time. The error bars represent the standard deviation of the average relative normalized responses to GAPD.
A kinetics study was carried out at 2 ng/ml IL1β stimulation for 1, 2, 4, 6 and 16 h time points. The average signal intensity of the normalized response is displayed in Figure 5b. The maximal induction of ICAM1, IL6, IL8, MCP1, COX2, SELE and VCAM1 was achieved after 4 h treatment. ABCA1 was not induced by 2 ng/ml IL1β stimulation. The kinetics of gene expression from this experiment is consistent with previous observation (37,45).
The ability to measure dose- and time-dependent effects of chemical treatment is essential for compound prioritization (46). Multiplex assays exploring multiple factorial responses could reveal critical information about compounds' efficacy and safety. Although only 480 data points (from 48 wells) were generated in the dose–response and kinetics experiments described above, the advantage in sample throughput of eTag multiplex mRNA assay will prevail when sample size increases, compound number increases and experimental conditions vary.
Comparison with real-time RT–PCR method
eTag multiplex mRNA assay was also compared to real-time RT–PCR assay (TaqMan® assay). mRNA abundance was measured upon 4 h treatment with 10 ng/ml IL1β on HUVEC. The TaqMan® assay used 30 ng of total RNA for each gene, while lysates from a total of 1500 cells (30 ng equivalent of total RNA) were used in the same 10-plex eTag mRNA assay. Three representative genes, IL6, IL8 and ABCA1 were measured and compared (Table 4). Relative quantification is used to calculate the fold change (47) for all three genes using the 2[−ΔΔ C(T)] method. IL8 and ABCA1 were in accordance in both methods. IL8 was induced 70-fold after treatment, while ABCA1 remained unchanged. The basal expression level of IL6 was low which was confirmed by an estimated Ct of 34 in the TaqMan® assay. However, after 4 h stimulation, a pronounced induction of IL6 was observed by both eTag multiplex mRNA assay and TaqMan® assay.
Table 4. Comparison of gene expression change with real-time RT–PCR assay.
| Gene | eTag multiplex mRNA assay | TaqMan® assay | ||||
|---|---|---|---|---|---|---|
| Fold change | Normalized responses (N = 3) | Fold changea | CT number (N = 3) | |||
| Untreated | Treated | Untreated | Treated | |||
| IL6 | 276.7 ± 89.3 | 0.002 ± 0.0003 | 0.55 ± 0.11 | 2436.8 ± 1184.5 | 34.0 ± 0.90 | 22.6 ± 0.10 |
| IL8 | 70.40 ± 7.71 | 0.08 ± 0.01 | 5.63 ± 0.47 | 69.3 ± 6.30 | 24.4 ± 0.13 | 18.3 ± 0.04 |
| ABCA1 | 1.32 ± 0.58 | 0.14 ± 0.04 | 0.19 ± 0.03 | 1.29 ± 0.15 | 27.0 ± 0.16 | 26.6 ± 0.29 |
aThe fold change measured in TaqMan® assay (47) was calculated as 2−ΔCt, where ΔCt = Ctuntreated − Cttreated. Calculation of Ct value was determined by interpolation of the RT–PCR progress curve.
CONCLUSIONS
In this report, we assessed several key features of eTag multiplex mRNA assay system: multiplicity, sensitivity, linear dynamic range, precision and specificity. The application of the eTag multiplex mRNA assay in compound dosage and time course studies was demonstrated and the accuracy of gene expression change was compared by a real-time RT–PCR method.
The typical sensitivity of the eTag multiplex mRNA assay is 1000–5000 copies per assay, as demonstrated in this work. This corresponds to <1 copy per cell in 1000–5000 cells (Figure 3). In contrast to conventional gene expression analysis, eTag assays does not require RNA purification and/or reverse transcription. This unprecedentedly simplifies the assay procedure (Figure 1c).
Although the tools for gene expression analysis such as RT–PCR (e.g. TaqMan®) and microarrays are well developed, there is a void in assay technologies for genomics-driven high sample throughput drug development. No sample preparation, multiplex capability, ease of use, amendable to high-throughput and high specificity of the eTag multiplex mRNA assay provides an attractive strategy to fill this void, yet at a comparable or lower cost as compared to either RT–PCR assay or microarray technologies. The data presented here strongly support the notion that eTag multiplex mRNA assay bridges microarray technology (thousands of genes per sample, with limited sample throughput) and real-time RT–PCR, such as TaqMan® technology (single gene at a time, with high sample throughput).
The eTag multiplex mRNA assay system shows promise as a useful tool in large-scale compound screening, structure and activity relationship study, target validation and in vivo as well as in vitro toxicity evaluations.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge Dean Burgi and Jing Wei for technical guidance on CE and data analysis. We thank Kai Nakamura, Carlo Calacsan, Thuy Nguyen, Navneed Mand and Laura Javas for eTag molecule and oligonucleotide synthesis. We thank Robert W. Kwiatkowski, Tsetska Takova and Marilyn Olson at Third Wave Technologies for the fruitful discussions.
REFERENCES
- 1.Golub T.R., Slonim,D.K., Tamayo,P., Huard,C., Gaasenbeek,M., Mesirov,J.P., Coller,H., Loh,M.L., Downing,J.R., Caligiuri,M.A., Bloomfield,C.D. and Lander,E.S. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- 2.Gunther E.C., Stone,D.J., Gerwien,R.W., Bento,P. and Heyes,M.P. (2003) Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc. Natl Acad. Sci. USA, 100, 9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes T.R., Marton,M.J., Jones,A.R., Roberts,C.J., Stoughton,R., Armour,C.D., Bennett,H.A., Coffey,E., Dai,H., He,Y.D., Kidd,M.J., King,A.M., Meyer,M.R., Slade,D., Lum,P.Y., Stepaniants,S.B., Shoemaker,D.D., Gachotte,D., Chakraburtty,K., Simon,J., Bard,M. and Friend,S.H. (2000) Functional discovery via a compendium of expression profiles. Cell, 102, 109–126. [DOI] [PubMed] [Google Scholar]
- 4.Gerhold D.L., Jensen,R.V. and Gullans,S.R. (2002) Better therapeutics through microarrays. Nature Genet., 32 (Suppl.), 547–551. [DOI] [PubMed] [Google Scholar]
- 5.Liotta L. and Petricoin,E. (2000) Molecular profiling of human cancer. Nature Rev. Genet., 1, 48–56. [DOI] [PubMed] [Google Scholar]
- 6.Whitfield M.L., Sherlock,G., Saldanha,A.J., Murray,J.I., Ball,C.A., Alexander,K.E., Matese,J.C., Perou,C.M., Hurt,M.M., Brown,P.O. and Botstein,D. (2002) Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell, 13, 1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson H., Betton,G., Robinson,D., Thomas,K., Monro,A., Kolaja,G., Lilly,P., Sanders,J., Sipes,G., Bracken,W., Dorato,M., Van Deun,K., Smith,P., Berger,B. and Heller,A. (2000) Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol., 32, 56–67. [DOI] [PubMed] [Google Scholar]
- 8.Gerhold D., Lu,M., Xu,J., Austin,C., Caskey,C.T. and Rushmore,T. (2001) Monitoring expression of genes involved in drug metabolism and toxicology using DNA microarrays. Physiol. Genomics, 5, 161–170. [DOI] [PubMed] [Google Scholar]
- 9.Waring J.F., Jolly,R.A., Ciurlionis,R., Lum,P.Y., Praestgaard,J.T., Morfitt,D.C., Buratto,B., Roberts,C., Schadt,E. and Ulrich,R.G. (2001) Clustering of hepatotoxins based on mechanism of toxicity using gene expression profiles. Toxicol. Appl. Pharmacol., 175, 28–42. [DOI] [PubMed] [Google Scholar]
- 10.Thomas R.S., Rank,D.R., Penn,S.G., Zastrow,G.M., Hayes,K.R., Pande,K., Glover,E., Silander,T., Craven,M.W., Reddy,J.K., Jovanovich,S.B. and Bradfield,C.A. (2001) Identification of toxicologically predictive gene sets using cDNA microarrays. Mol. Pharmacol., 60, 1189–1194. [DOI] [PubMed] [Google Scholar]
- 11.Hamadeh H.K., Bushel,P.R., Jayadev,S., DiSorbo,O., Bennett,L., Li,L., Tennant,R., Stoll,R., Barrett,J.C., Paules,R.S., Blanchard,K. and Afshari,C.A. (2002) Prediction of compound signature using high density gene expression profiling. Toxicol. Sci., 67, 232–240. [DOI] [PubMed] [Google Scholar]
- 12.Hamadeh H.K., Bushel,P.R., Jayadev,S., Martin,K., DiSorbo,O., Sieber,S., Bennett,L., Tennant,R., Stoll,R., Barrett,J.C., Blanchard,K., Paules,R.S. and Afshari,C.A. (2002) Gene expression analysis reveals chemical-specific profiles. Toxicol. Sci., 67, 219–231. [DOI] [PubMed] [Google Scholar]
- 13.Huels C., Muellner,S., Meyer,H.E. and Cahill,D.J. (2002) The impact of protein biochips and microarrays on the drug development process. Drug Discov. Today, 7, S119–S124. [DOI] [PubMed] [Google Scholar]
- 14.Warrior U., Fan,Y., David,C.A., Wilkins,J.A., McKeegan,E.M., Kofron,J.L. and Burns,D.J. (2000) Application of QuantiGene nucleic acid quantification technology for high throughput screening. J. Biomol. Screen., 5, 343–352. [DOI] [PubMed] [Google Scholar]
- 15.Liss B. (2002) Improved quantitative real-time RT–PCR for expression profiling of individual cells. Nucleic Acids Res., 30, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storz P., Doppler,H., Horn-Muller,J., Groner,B., Pfizenmaier,K. and Muller,G. (1999) A cellular reporter assay to monitor insulin receptor kinase activity based on STAT 5-dependent luciferase gene expression. Anal. Biochem., 276, 97–104. [DOI] [PubMed] [Google Scholar]
- 17.Eis P.S., Olson,M.C., Takova,T., Curtis,M.L., Olson,S.M., Vener,T.I., Ip,H.S., Vedvik,K.L., Bartholomay,C.T., Allawi,H.T., Ma,W.P., Hall,J.G., Morin,M.D., Rushmore,T.H., Lyamichev,V.I. and Kwiatkowski,R.W. (2001) An invasive cleavage assay for direct quantitation of specific RNAs. Nat. Biotechnol., 19, 673–676. [DOI] [PubMed] [Google Scholar]
- 18.Martel R.R., Bortros,I.W., Rounsevill,M.P., Hinton,J.P., Staples,R.R., Morales,D.A., Farmer,J.B. and Seligmann,B.E. (2002) Multiplexed screening assay for mRNA combining nuclease protection with luminescent array detection. Assay Drug Dev. Technol., 1, 61–71. [DOI] [PubMed] [Google Scholar]
- 19.Johnson P.H., Walker,R.P., Jones,S.W., Stephens,K., Meurer,J., Zajchowski,D.A., Luke,M.M., Eeckman,F., Tan,Y., Wong,L., Parry,G., Morgan,T.K.,Jr, McCarrick,M.A. and Monforte,J. (2002) Multiplex gene expression analysis for high-throughput drug discovery: screening and analysis of compounds affecting genes overexpressed in cancer cells. Mol. Cancer Ther., 1, 1293–1304. [PubMed] [Google Scholar]
- 20.Stegmaier K., Ross,K.N., Colavito,S.A., O'Malley,S., Stockwell,B.R. and Golub,T.R. (2004) Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nature Genet., 36, 257–263. [DOI] [PubMed] [Google Scholar]
- 21.Dovichi N.J. (1997) DNA sequencing by capillary electrophoresis. Electrophoresis, 18, 2393–2399. [DOI] [PubMed] [Google Scholar]
- 22.Dolnik V. (1999) DNA sequencing by capillary electrophoresis (review). J. Biochem. Biophys. Methods, 41, 103–119. [DOI] [PubMed] [Google Scholar]
- 23.Kotler L., He,H., Miller,A.W. and Karger,B.L. (2002) DNA sequencing of close to 1000 bases in 40 minutes by capillary electrophoresis using dimethyl sulfoxide and urea as denaturants in replaceable linear polyacrylamide solutions. Electrophoresis, 23, 3062–3070. [DOI] [PubMed] [Google Scholar]
- 24.McIntosh S.L., Deligeorgiev,T.G., Gadjev,N.I. and McGown,L.B. (2002) Mono- and bis-intercalating dyes for multiplex fluorescence lifetime detection of DNA restriction fragments in capillary electrophoresis. Electrophoresis, 23, 1473–1479. [DOI] [PubMed] [Google Scholar]
- 25.Luo M.C., Thomas,C., You,F.M., Hsiao,J., Ouyang,S., Buell,C.R., Malandro,M., McGuire,P.E., Anderson,O.D. and Dvorak,J. (2003) High-throughput fingerprinting of bacterial artificial chromosomes using the snapshot labeling kit and sizing of restriction fragments by capillary electrophoresis. Genomics, 82, 378–389. [DOI] [PubMed] [Google Scholar]
- 26.Monforte J., Vnsant,G., Pezzoli,P., Ferre,F., Baumhueter,S., Day,G., Dunlea,S., Eynon,B., Fielden,M., Fujimoto,S., Ganter,B., Idury,R., Jarnigan,K., Kolaja,K., Lee,M., Nair,R., Natsoulis,G., Nicholson,S., Pearson,C., Toter,A. and Tolley,A. (2004) Use of expression profiling™ to characterize gene expression biomarker sets for liver toxicity. Proceedings of Society of Toxicology, Baltimore, MD, abstract 653.
- 27.Grossman P.D., Bloch,W., Brinson,E., Chang,C.C., Eggerding,F.A., Fung,S., Iovannisci,D.M., Woo,S., Winn-Deen,E.S. and Iovannisci,D.A. (1994) High-density multiplex detection of nucleic acid sequences: oligonucleotide ligation assay and sequence-coded separation. Nucleic Acids Res., 22, 4527–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyamichev V.I., Kaiser,M.W., Lyamicheva,N.E., Vologodskii,A.V., Hall,J.G., Ma,W.P., Allawi,H.T. and Neri,B.P. (2000) Experimental and theoretical analysis of the invasive signal amplification reaction. Biochemistry, 39, 9523–9532. [DOI] [PubMed] [Google Scholar]
- 29.Ma W.P., Kaiser,M.W., Lyamicheva,N., Schaefer,J.J., Allawi,H.T., Takova,T., Neri,B.P. and Lyamichev,V.I. (2000) RNA template-dependent 5′ nuclease activity of Thermus aquaticus and Thermus thermophilus DNA polymerases. J. Biol. Chem., 275, 24693–24700. [DOI] [PubMed] [Google Scholar]
- 30.Lane M.J., Paner,T., Kashin,I., Faldasz,B.D., Li,B., Gallo,F.J. and Benight,A.S. (1997) The thermodynamic advantage of DNA oligonucleotide ‘stacking hybridization’ reactions: energetics of a DNA nick. Nucleic Acids Res., 25, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lockhart D.J. and Winzeler,E.A. (2000) Genomics, gene expression and DNA arrays. Nature, 405, 827–836. [DOI] [PubMed] [Google Scholar]
- 32.Affymetrix Inc. (2001) Technical Note. The GeneChip arrays provide optimal sensitivity and specificity for microarray expression analysis.
- 33.Lockhart D.J. and Barlow,C. (2001) Expressing what's on your mind: DNA arrays and the brain. Nature Rev. Neurosci., 2, 63–68. [DOI] [PubMed] [Google Scholar]
- 34.Kwoh D.Y., Davis,G.R., Whitfield,K.M., Chappelle,H.L., DiMichele,L.J. and Gingeras,T.R. (1989) Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format. Proc. Natl Acad. Sci. USA, 86, 1173–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Gelder R.N., von Zastrow,M.E., Yool,A., Dement,W.C., Barchas,J.D. and Eberwine,J.H. (1990) Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl Acad. Sci. USA, 87, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eberwine J., Yeh,H., Miyashiro,K., Cao,Y., Nair,S., Finnell,R., Zettel,M. and Coleman,P. (1992) Analysis of gene expression in single live neurons. Proc. Natl Acad. Sci. USA, 89, 3010–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghersa P., Hooft van Huijsduijnen,R., Whelan,J. and DeLamarter,J.F. (1992) Labile proteins play a dual role in the control of endothelial leukocyte adhesion molecule-1 (ELAM-1) gene regulation. J. Biol. Chem., 267, 19226–19232. [PubMed] [Google Scholar]
- 38.Luo L., Salunga,R.C., Guo,H., Bittner,A., Joy,K.C., Galindo,J.E., Xiao,H., Rogers,K.E., Wan,J.S., Jackson,M.R. and Erlander,M.G. (1999) Gene expression profiles of laser-captured adjacent neuronal subtypes. Nature Med., 5, 117–122. [DOI] [PubMed] [Google Scholar]
- 39.Wang E., Miller,L.D., Ohnmacht,G.A., Liu,E.T. and Marincola,F.M. (2000) High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol., 18, 457–459. [DOI] [PubMed] [Google Scholar]
- 40.Bustin S.A. (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol., 25, 169–193. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J.H., Chung,T.D. and Oldenburg,K.R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 42.Bandman O., Coleman,R.T., Loring,J.F., Seilhamer,J.J. and Cocks,B.G. (2002) Complexity of inflammatory responses in endothelial cells and vascular smooth muscle cells determined by microarray analysis. Ann. NY Acad. Sci., 975, 77–90. [DOI] [PubMed] [Google Scholar]
- 43.Mantovani A., Garlanda,C., Introna,M. and Vecchi,A. (1998) Regulation of endothelial cell function by pro- and anti-inflammatory cytokines. Transplant Proc., 30, 4239–4243. [DOI] [PubMed] [Google Scholar]
- 44.Ko Y., Totzke,G., Gouni-Berthold,I., Sachinidis,A. and Vetter,H. (1999) Cytokine-inducible growth factor gene expression in human umbilical endothelial cells. Mol. Cell. Probes, 13, 203–211. [DOI] [PubMed] [Google Scholar]
- 45.Bevilacqua M.P., Stengelin,S., Gimbrone,M.A.,Jr and Seed,B. (1989) Endothelial leukocyte adhesion molecule 1: an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science, 243, 1160–1165. [DOI] [PubMed] [Google Scholar]
- 46.Farr S. and Dunn,R.T.,II (1999) Concise review: gene expression applied to toxicology. Toxicol. Sci., 50, 1–9. [DOI] [PubMed] [Google Scholar]
- 47.Livak K.J. and Schmittgen,T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.