

Abstract
Altered methylation patterns have been found to play a role in developmental disorders, cancer and aging. Increasingly, changes in DNA methylation are used as molecular markers of disease. Therefore, there is a need for reliable and easy to use techniques to detect and measure DNA methylation in research and routine diagnostics. We have established a novel quantitative analysis of methylated alleles (QAMA) which is essentially a major improvement over a previous method based on real-time PCR (MethyLight). This method is based on real-time PCR on bisulfite-treated DNA. A significant advantage over conventional MethyLight is gained by the use of TaqMan probes based on minor groove binder (MGB) technology. Their improved sequence specificity facilitates relative quantification of methylated and unmethylated alleles that are simultaneously amplified in single tube. This improvement allows precise measurement of the ratio of methylated versus unmethylated alleles and cuts down potential sources of inter-assay variation. Therefore, fewer control assays are required. We have used this novel technical approach to identify hypermethylation of the CpG island located in the promoter region of the retinoblastoma (RB1) gene and found that QAMA facilitates reliable and fast measurement of the relative quantity of methylated alleles and improves handling of diagnostic methylation analysis. Moreover, the simplified reaction setup and robustness inherent to the single tube assay facilitates high-throughput methylation analysis. Because the high sequence specificity inherent to the MGB technology is widely used to discriminate single nucleotide polymorphisms, QAMA potentially can be used to discriminate the methylation status of single CpG dinucleotides.
INTRODUCTION
In the genomic DNA of vertebrates, cytosine-5 methylation only occurs in CpG dinucleotides and is associated with transcriptional repression. Regions that are rich in CpG dinucleotides, also known as CpG islands, are usually maintained free of methylation. Exceptions are CpG islands associated with imprinted genes or with genes located on the X chromosome. The methylation patterns are established during germ cell development or during early mammalian ontogenesis and are maintained during subsequent mitotic cell divisions. Erroneously established methylation patterns have been found to be associated with human diseases including Prader–Willi syndrome, Angelman syndrome and Fragile X syndrome. In these disorders, diagnosis can be confirmed by identification of specific aberrant methylation patterns (1–3).
Altered methylation patterns can also arise in somatic cells of adult organisms. Somatic epimutations are thought to contribute to aging and tumourigenesis (4,5). In tumor cells, global hypomethylation and regional hypermethylation have been found (6). Specifically, hypermethylation of 5′ regulatory regions of tumor suppressor genes can contribute to gene inactivation and, subsequently, tumor development and progression (7–9). Methylation analysis of several CpG islands in parallel have shown non-random alterations of methylation patterns that can be used to discover tumor classes and predict prognosis (10–12). However, current methods for parallel analysis of multiple CpG islands are either not commercially available (12) or are too laborious for application in routine testing (11).
Methylation-specific PCR (MSP) is another technique frequently used for methylation analysis (13). In this method, the target DNA that has been treated with bisulfite is used. This procedure modifies unmethylated cytosines to uracil while leaving methylated cytosines unaltered (14,15). Therefore, the methylation status can be discriminated by PCR with sequence-specific primers. An improvement over the original MSP is MethyLight (16). This real-time PCR-based assay determines the methylation status of a selected CpG island and has been proposed for use in high-throughput methylation analysis.
The novel assay presented here is a quantitative version of MethyLight. This assay was originally discussed by Eads et al. (16), but was not tested by these authors because they had expected that cross hybridization of probes would interfere with discrimination of methylated versus unmethylated target sequences. Recently, TaqMan probes based on minor groove binder (MGB) technology have been introduced (17). The improved sequence specificity provided by these probes allows reliable discrimination even of single base pair changes (18,19). Here, we make use of this new probe technology to establish quantitative analysis of methylated alleles (QAMA) for the detection of methylated alleles at the promoter of the RB1 gene. In this assay we use one primer set to amplify both methylated and unmethylated alleles starting from bisulfite-treated DNA. The methylation status is discriminated at the level of probe hybridization with two MGB TaqMan probes that have distinct fluorescent labels and that are specific for sequences obtained after bisulfite treatment of methylated and unmethylated DNA.
MATERIALS AND METHODS
Tumor DNA samples
We analyzed 49 unilateral sporadic retinoblastomas and one tumor (Z175AMM) from a bilateral case (Tables 1 and 2). The DNA was extracted from frozen samples using standard methods. All tumors had been assessed for loss of heterozygosity (LOH) at intragenic polymorphic markers Rbi2 and RB1.20 (20). Twenty tumor DNA samples (Table 1), which had been previously analyzed by MSP, were used to establish the methylation assay. To generate a standard curve, we prepared different ratios of methylated versus unmethylated target sequences by mixing universal methylated DNA (Serologicals Corporation, USA) and blood cell DNA of a healthy donor prior to bisulfite modification. The following ratios were prepared (methylated/unmethylated): 0/100, 10/90, 25/75, 50/50, 75/25, 90/10 and 100/0.
Table 1. Tumor samples used to establish the assay.
| Tumor ID | MSP | QAMA | RB1 status |
|---|---|---|---|
| M6733 | Methylated | 47 | Heterozygous |
| M8311 | Methylated | 44 | Heterozygous |
| M20009 | Methylated | 42 | Heterozygous |
| Z137CK | Methylated | 42 | Heterozygous |
| Z088SD | Methylated | 35 | Heterozygous |
| Z054KG | Methylated | 35 | Heterozygous |
| Z157BR | Methylated | 29 | Heterozygous |
| Z075AK | Methylated | 100 | LOH |
| Z177CT | Methylated | 100 | LOH |
| Z153AR | Methylated | 81 | LOH |
| Z064JC | Methylated | 77 | Hemizygous |
| Z112EE | Unmethylated | 0 | Heterozygous |
| Z078HG | Unmethylated | 0 | Heterozygous |
| Z146RR | Unmethylated | 0 | Heterozygous |
| Z106SL | Unmethylated | 0 | Heterozygous |
| Z037NSE | Unmethylated | 0 | Heterozygous |
| Z175AMM | Unmethylated | 0 | Heterozygous |
| Z169CS | Unmethylated | 0 | LOH |
| Z154IH | Unmethylated | 0 | LOH |
| M7395 | Unmethylated | 0 | LOH |
Tumor samples as analyzed by QAMA. RB1 status was assessed using polymorphic markers Rbi2 and RB1.20. Relative prevalence of methylated RB1 alleles as determined by QAMA is given. All samples had previously been analyzed by MSP (21). The methylation ratio is deduced from a standard curve that was generated in an independent experiment.
Table 2. Second set of tumor sample of unknown methylation status.
| Tumour ID | QAMA | RB1 status |
|---|---|---|
| M2087 | 45 | Heterozygous |
| M13074 | 43 | Heterozygous |
| M14893 | 41 | Heterozygous |
| M22808 | 42 | Heterozygous |
| M6734 | 38 | Heterozygous |
| M15295 | 100 | LOH |
| M22088 | 100 | LOH |
| M22058 | 88 | LOH |
| M21414 | 84 | LOH |
| M14382 | 0 | Hemizygous |
| M14687 | 0 | Heterozygous |
| M20882 | 0 | Heterozygous |
| M21951 | 0 | Heterozygous |
| M22049 | 0 | Heterozygous |
| M22455 | 0 | Heterozygous |
| M20060 | 0 | Heterozygous |
| M23115 | 0 | Heterozygous |
| M3625 | 0 | Heterozygous |
| M21549 | 0 | Heterozygous |
| M18916 | 0 | Heterozygous |
| M21371 | 0 | Heterozygous |
| M20168 | 0 | Heterozygous |
| M20945 | 0 | Heterozygous |
| M22507 | 0 | LOH |
| M21851 | 0 | LOH |
| M21111 | 0 | LOH |
| M21681 | 0 | LOH |
| M10492 | 0 | LOH |
| M10493 | 0 | LOH |
| M22724 | 0 | LOH |
Tumor samples as analyzed by QAMA. RB1 status was assessed using polymorphic markers Rbi2 and RB1.20. Relative prevalence of methylated RB1 alleles as determined by QAMA is given.
Bisulfite treatment
The procedure was modified from established protocols (1,21). Genomic DNA (1 μg in 30 μl) was denatured by adding 3 μl of freshly prepared 3 M NaOH and incubating the solution at 37°C for 15 min. For complete denaturation, the samples were incubated at 95°C for 1 min and immediately cooled on ice. The bisulfite solution was prepared by dissolving 8.5 g of sodium bisulfite in 15 ml degassed water, adding 1 ml of a 40 mM hydroquinone solution and adjusting the pH to 5.0 with 600 μl of 10 M NaOH. The bisulfite solution (0.5 ml) was added to the denatured DNA, mixed and incubated at 55°C for 16 h in the dark. The DNA was recovered by using the Wizard DNA Clean-Up System (Promega) followed by elution in 100 μl of water. Subsequently, 11 μl of 3 M NaOH was added, and the samples were incubated for 15 min at 37°C. The solution was then neutralized by adding 110 μl of 6 M NH4OAc pH 7.0. The DNA was ethanol precipitated, washed in 70% ethanol, dried and resuspended in 20 μl of water.
PCR
The PCR was performed using a 96-well optical tray with caps at a final reaction volume of 20 μl. Samples contained 10 μl of TaqMan® Universal PCR Master Mix, No AmpErase® UNG (uracil-N-glycosylase), 2 μl of bisulfite-treated DNA, an additional 2.5 U of AmpliTaq Gold (Perkin Elmer), 2.5 μM each of the primers RBfw and RBrev and 150 nM each of the fluorescently labeled probes RBmet and RBunmet. Initial denaturation at 95°C for 10 min to activate the AmpliTaq Gold DNA polymerase was followed by 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min (ABI Prism, 7000, Sequence Detection System). Primer and probe sequences were selected with the help of the probe and primer test document included with the Primer Express software (ABI). PCR primers were designed to amplify the bisulfite-converted antisense strand of the RB1 promoter sequence lacking any known nucleotide polymorphisms. The software designs primers with a melting temperature (Tm) of 58–60°C and probes with a Tm value of 68°C. The Tm of both primers should be equal. The amplicon size ideally should be 50–150 bp.
Primer sequences: RBfw, 5′-CCACAATCACCCACCAAACTC-3′ (position −303 to −283); RBrev, 5′-GAGGAATTAAATTGGGAAATTTGGA-3′ (position −178 to −154); RBmet, 5′-VIC-TCCGAACCGCGCCGA-MGB-3′ (position −222 to −208); RBunmet, 5′-6FAM-ACATCCAAACCACACCAA-MGB-3′ (position −219 to −208).
The amount of FAM and VIC fluorescence released in each tube was measured as a function of the PCR cycle number at the end of each cycle using an ABI 7000 sequence detection system. The cycle number at which the fluorescence signal crosses a detection threshold is referred to as CT and the difference of both CT values within a sample (ΔCT) is calculated (ΔCT = CT−FAM − CT−VIC). All samples were measured in duplicate using the mean for further analysis. Given that the percentage of methylated DNA molecules in a real-time PCR experiment is given by c, the resulting ΔCT equals Log2[c/(1 − c)]. To account for the differential efficacy of the PCR (methylated/unmethylated) and probe activity, we restate the model as ΔCT = a + b Log2[c/(1 − c)], with a and b representing the additional effects. These nuisance parameters are fitted by means of a linear regression from data of a control experiment. The following equation was deduced from the results generated by the standard curve: c = 100/[1 + 2[(1.33−ΔCT)/−2.27)]. The relative prevalence of either the methylated or the unmethylated allele was set to 100% in the case that only one fluorescence signal crossed the threshold, indicating a relative absence of the opposite target (for detailed derivation of the equation please see the Supplementary Material.).
RESULTS
Rationale
Bisulfite treatment of denatured DNA converts all unmethylated cytosines to uracil, leaving methylated cytosines unaltered (15). After bisulfite modification, a methylated allele differs from the unmethylated allele at all CpG positions within the nucleotide sequence. For RB1-QAMA, we designed primers to amplify the bisulfite-converted antisense strand of the RB1 promoter. The primer binding sites lack CpG dinucleotides and, therefore, the nucleotide sequences in methylated and unmethylated DNA are identical after bisulfite treatment. Consequently, it is possible to amplify both alleles in the same reaction tube with one primer pair. Methylation discrimination occurs during probe hybridization by the use of two differently labeled internal MGB TaqMan® probes (Figure 1) (17,18). Probes bound to their respective target sequence are cleaved by the 5′ nuclease activity of Taq DNA polymerase in the course of PCR (22). Therefore, the amplification of the methylated and unmethylated alleles is monitored independently in the same tube. The binding site of the two MGB probes covers four differently methylated CpG dinucleotides. We used a VIC-labeled MGB probe (RBmet) that specifically hybridizes to the sequence derived from the methylated RB1 allele, and a FAM-labeled probe (RBunmet) that binds to the sequence generated from the unmethylated allele. The amount of fluorescent dye released during PCR is measured by a real-time PCR system and is directly proportional to the amount of PCR product generated. For precise quantification of the ratio of methylated to unmethylated alleles, the ΔCT (=CT−FAM − CT−VIC) value is determined and compared to a standard curve.
Figure 1.
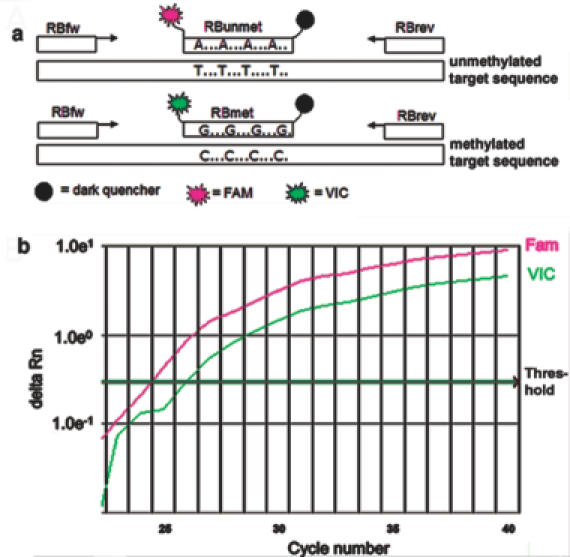
(a) Schematic of the QAMA. Bisulfite-treated target sequence is amplified with the same primer set (RBfw, RBrev) irrespective of its methylation status. Two differently labeled internal MGB TaqMan® probes (RBmet, RBunmet) bind their respective target and are cleaved by the 5′ nuclease activity of Taq DNA polymerase. The amount of fluorescence dyes VIC and FAM released during PCR is directly proportional to the amount of PCR product generated from the methylated or unmethylated allele, respectively. (b) Methylation analysis of a premixed sample with a methylation ratio of 50%. The relative fluorescence of VIC and FAM (delta Rn) is plotted as a function of the cycle number.
To set up the RB1 methylation assay, we mixed methylated control DNA with DNA prepared from peripheral blood cells of a healthy donor with no methylation of the RB1 promoter to generated DNA samples with defined ratios of unmethylated versus methylated RB1 target sequences. After bisulfite modification, each sample was examined by real-time PCR analysis in duplicate. We correlated the ΔCT values with the predefined prevalence of methylated alleles. The curve exhibits a sigmoid shape with a linear part in the range of 25–75% of methylated DNA (Figure 2). From this we deduced an algorithm to calculate the methylation ratio of an unknown sample from its ΔCT value (see Materials and Methods).
Figure 2.
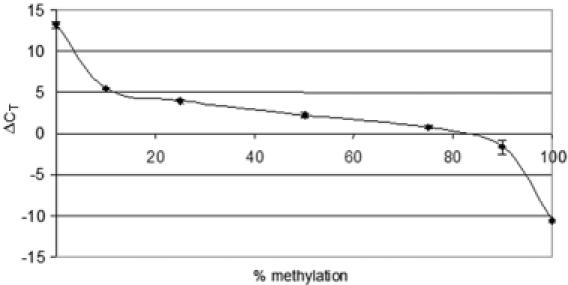
Standard curve as obtained by plotting the ΔCT values against the predefined methylation ratio of each sample. The standard deviation for each sample analyzed in duplicate is indicated by error bars. The CT value was set to 40 in the case that the respective fluorescence signal did not cross the threshold.
We first analyzed the RB1 methylation status of DNA samples of 20 retinoblastomas (Table 1), which had previously been tested by MSP (21). The ΔCT value for each sample was calculated and the methylation ratio deduced from the equation given above. In seven samples without LOH at polymorphic loci within the RB1 gene, the methylation ratios ranged from 29 to 47% (mean 39%). In four samples with LOH, the methylation ratios ranged from 77 to 100% (mean 90%). In each of the remaining tumor samples, the VIC signal representing the methylated RB1 allele did not cross the threshold indicating complete absence of methylated alleles. All results were concordant with the methylation status determined by MSP previously (Table 1). Control experiments showed that unmodified genomic DNA was not amplified under the conditions used here (data not shown).
Next we applied QAMA to analyze another set of 30 DNA samples of unknown methylation status or suspected to be methylated. In nine of these samples, methylation at the RB1 promoter was identified (Table 2). The mean methylation ratio for tumors without LOH was 42%, which likely represents methylation of one of the two RB1 alleles. In two tumors with LOH, only methylated alleles were detected. This is consistent with complete methylation of the remaining allele. Two other tumors with LOH at RB1 showed methylation ratios of 84 and 88%, suggesting the presence of minor amounts of unmethylated alleles, possibly contributed by normal cells that are contained in the tumor sample that was used for DNA preparation. Methylation ratios of less than the expected 50% in tumors without LOH at the RB1 locus may also be caused by this kind of contamination (e.g. Z157BR, Table 1).
To test inter-assay variability, we analyzed 10 tumor samples that had shown methylated alleles in previous experiments (Table 1). To obtain maximum precision, a standard curve was run along with each of the three assays and the resulting equation was used to deduce the methylation ratio of each tumor sample. We found a maximum standard deviation of ±7.2% methylation in the results obtained for the same sample in three different assays (Figure 3 and Table 3).
Figure 3.
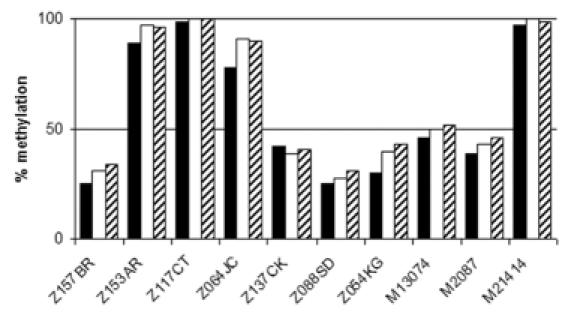
Test for inter-assay variability. Three independent assays, each represented by full, open or hatched bars were performed at different days. The methylation ratio of each sample was deduced from a standard curve running along with each assay.
Table 3. Tumor samples used to test inter-assay variability.
| Tumor ID | QAMA assay | Mean | SD | ||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| Z157BR | 25 | 31 | 34 | 30 | 4.4 |
| Z153AR | 89 | 97 | 96 | 94 | 4.5 |
| Z075AK | 99 | 100 | 100 | 100 | 0.5 |
| Z064JC | 78 | 91 | 90 | 87 | 7.2 |
| Z137CK | 42 | 39 | 41 | 41 | 1.8 |
| Z088SD | 25 | 28 | 31 | 28 | 3.0 |
| Z054KG | 30 | 40 | 43 | 37 | 6.8 |
| M21414 | 97 | 100 | 99 | 99 | 1.6 |
| M13074 | 46 | 50 | 52 | 49 | 3.1 |
| M2087 | 39 | 43 | 46 | 43 | 3.1 |
The mean of three independent experiments and the standard deviation is given. In each of the three assays a standard curve was included.
We also tested if the results are influenced by varying amounts of target DNA. In analyses of serial dilutions (100, 50, 10 and 2.5 ng) of DNA from a tumor sample with a methylated allele and retention of heterozygosity (M20009), the ΔCT value and, consequently, the methylation ratio was almost unchanged (49 ± 2.2%) (data not shown). This indicates that minor variations in the amount of target DNA have only little influence on the results of the assay.
DISCUSSION
Here, we present QAMA, which is a real-time PCR-based methodology for the relative quantification of methylated alleles. When a standard curve of different ratios of methylated versus unmethylated target DNA is included in each assay and each sample is analyzed in duplicate, this assay shows a surprisingly high precision. Testing retinoblastoma samples we found that such precision is not needed to identify tumors with methylated alleles present in a homozygous or heterozygous state. Therefore, a simplified assay may be performed without a standard curve and a smaller number of replicates. However, in individuals in whom methylation changes are present in a mosaic state, the precision provided by QAMA will help in quantifying the ratios of methylated to unmethylated alleles. Interestingly, we found that minor variations in the amount of target DNA do not substantially influence the results. This helps simplify handling as it obviates the need for precise quantification of the target DNA added to the reaction. We would like to point out that, because of its inherent robustness and simplicity, this assay is ideally suited for high-throughput analysis.
However, two caveats must be noted. For one, the results obtained by QAMA could be misinterpreted if nucleotide polymorphisms or mutations affect the probe or primer binding sites. This is not a limitation specific for the assay presented here, but applies to all PCR-based assays and also to methods involving methylation-sensitive restriction enzymes. Second, the detection principle of the assay presented here is based on the assumption that CpG islands are either fully methylated or fully unmethylated. If methylation of the target site for binding of the TaqMan probe is incomplete, the sequence that is derived after bisulfite treatment is recognized by neither of the two probes. If such a situation is present, the relative proportion of methylated alleles will be underestimated. However, the high sequence specificity provided by the MGB technology might be exploited to design probes specific for the methylation status of single CpG dinucleotides.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Bernhard Horsthemke for helpful discussion and continuous support. This work has been supported by the DFG (KFO109, Klinische Forschergruppe Ophthalmologische Onkologie und Genetik, Teilprojekt II-1).
REFERENCES
- 1.Zeschnigk M., Lich,C., Buiting,K., Doerfler,W. and Horsthemke,B. (1997) A single-tube PCR test for the diagnosis of Angelman and Prader–Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur. J. Hum. Genet., 5, 94–98. [PubMed] [Google Scholar]
- 2.Oberle I., Rousseau,F., Heitz,D., Kretz,C., Devys,D., Hanauer,A., Boue,J., Bertheas,M.F. and Mandel,J.L. (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science, 252, 1097–1102. [DOI] [PubMed] [Google Scholar]
- 3.Dittrich B., Robinson,W.P., Knoblauch,H., Buiting,K., Schmidt,K., Gillessen-Kaesbach,G. and Horsthemke,B. (1992) Molecular diagnosis of the Prader–Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11–13. Hum. Genet., 90, 313–315. [DOI] [PubMed] [Google Scholar]
- 4.Issa J.P. (2003) Age-related epigenetic changes and the immune system. Clin. Immunol., 109, 103–108. [DOI] [PubMed] [Google Scholar]
- 5.Jaenisch R. and Bird,A. (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genet., 33 (Suppl), 245–254. [DOI] [PubMed] [Google Scholar]
- 6.Feinberg A.P. and Vogelstein,B. (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301, 89–92. [DOI] [PubMed] [Google Scholar]
- 7.Dobrovic A. and Simpfendorfer,D. (1997) Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res., 57, 3347–3350. [PubMed] [Google Scholar]
- 8.Greger V., Debus,N., Lohmann,D., Hopping,W., Passarge,E. and Horsthemke,B. (1994) Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum. Genet., 94, 491–496. [DOI] [PubMed] [Google Scholar]
- 9.Greger V., Passarge,E., Hopping,W., Messmer,E. and Horsthemke,B. (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet., 83, 155–158. [DOI] [PubMed] [Google Scholar]
- 10.Cui H., Cruz-Correa,M., Giardiello,F.M., Hutcheon,D.F., Kafonek,D.R., Brandenburg,S., Wu,Y., He,X., Powe,N.R. and Feinberg,A.P. (2003) Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science, 299, 1753–1755. [DOI] [PubMed] [Google Scholar]
- 11.Costello J.F., Fruhwald,M.C., Smiraglia,D.J., Rush,L.J., Robertson,G.P., Gao,X., Wright,F.A., Feramisco,J.D., Peltomaki,P., Lang,J.C. et al. (2000) Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nature Genet., 24, 132–138. [DOI] [PubMed] [Google Scholar]
- 12.Adorjan P., Distler,J., Lipscher,E., Model,F., Muller,J., Pelet,C., Braun,A., Florl,A.R., Gutig,D., Grabs,G. et al. (2002) Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res., 30, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herman J.G., Graff,J.R., Myohanen,S., Nelkin,B.D. and Baylin,S.B. (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl Acad. Sci. USA, 93, 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark S.J., Harrison,J., Paul,C.L. and Frommer,M. (1994) High sensitivity mapping of methylated cytosines. Nucleic Acids Res., 22, 2990–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frommer M., McDonald,L.E., Millar,D.S., Collis,C.M., Watt,F., Grigg,G.W., Molloy,P.L. and Paul,C.L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eads C.A., Danenberg,K.D., Kawakami,K., Saltz,L.B., Blake,C., Shibata,D., Danenberg,P.V. and Laird,P.W. (2000) MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res., 28, E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Afonina I., Zivarts,M., Kutyavin,I., Lukhtanov,E., Gamper,H. and Meyer,R.B. (1997) Efficient priming of PCR with short oligonucleotides conjugated to a minor groove binder. Nucleic Acids Res., 25, 2657–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak K.J. (1999) Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Gene Anal., 14, 143–149. [DOI] [PubMed] [Google Scholar]
- 19.de Kok J.B., Wiegerinck,E.T., Giesendorf,B.A. and Swinkels,D.W. (2002) Rapid genotyping of single nucleotide polymorphisms using novel minor groove binding DNA oligonucleotides (MGB probes). Hum. Mutat., 19, 554–559. [DOI] [PubMed] [Google Scholar]
- 20.Lohmann D.R., Gerick,M., Brandt,B., Oelschlager,U., Lorenz,B., Passarge,E. and Horsthemke,B. (1997) Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am. J. Hum. Genet., 61, 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeschnigk M., Lohmann,D. and Horsthemke,B. (1999) A PCR test for the detection of hypermethylated alleles at the retinoblastoma locus. J. Med. Genet., 36, 793–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak K.J., Flood,S.J., Marmaro,J., Giusti,W. and Deetz,K. (1995) Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl., 4, 357–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.