

Abstract
Caveolin-1, a structural protein of caveolae, is cleared unusually slowly from the Golgi apparatus during biosynthetic transport. Furthermore, several caveolin-1 mutant proteins accumulate in the Golgi apparatus. We examined this behavior further in this mutant study. Golgi accumulation probably resulted from loss of Golgi exit information, not exposure of cryptic retention signals, because several deletion mutants accumulated in the Golgi apparatus. Alterations throughout the protein caused Golgi accumulation. Thus, most probably acted indirectly, by affecting overall conformation, rather than by disrupting specific Golgi exit motifs. Consistent with this idea, almost all the Golgi-localized mutant proteins failed to oligomerize normally (even with an intact oligomerization domain), and they showed reduced raft affinity in an in vitro detergent-insolubility assay. A few mutant proteins formed unstable oligomers that migrated unusually slowly on blue native gels. Only one mutant protein, which lacked the first half of the N-terminal hydrophilic domain, accumulated in the Golgi apparatus despite normal oligomerization and raft association. These results suggested that transport of caveolin-1 through the Golgi apparatus is unusually difficult. The conformation of caveolin-1 may be optimized to overcome this difficulty, but remain very sensitive to mutation. Disrupting conformation can coordinately affect oligomerization, raft affinity, and Golgi exit of caveolin-1.
INTRODUCTION
Caveolin-1 is an important component of the membrane-embedded coat that surrounds caveolae (Rothberg et al., 1992; Fra et al., 1994; Smart et al., 1999; Fernandez et al., 2002). Although it is an integral membrane protein, caveolin-1 lacks conventional transmembrane domains. Instead, N- and C-terminal hydrophilic domains flank a central 33-residue hydrophobic domain (Smart et al., 1999). Both hydrophilic domains are located on the cytoplasmic side of the plasma membrane, and the hydrophobic domain has been suggested to form a helical hairpin embedded in the bilayer. A peptide corresponding to the last 20 residues of the N-terminal hydrophilic domain, a region named the caveolin scaffolding domain (CSD), can bind to membranes independently (Schlegel et al., 1999), at least in part because of the high number of aromatic residues in this domain (Arbuzova et al., 2000). The C-terminal domain also can associate with membranes independently, at least in part via three palmitate chains (Luetterforst et al., 1999).
Caveolin-1, alone or with the related protein caveolin-2, forms large (>400 kDa), SDS-stable oligomers (Monier et al., 1995; Sargiacomo et al., 1995; Scheiffele et al., 1998). These oligomers are thought to associate with each other to form the filamentous structure of the caveolar coat (Fernandez et al., 2002). Oligomerization occurs soon after synthesis (Scheiffele et al., 1998). It is not known where along the secretory pathway caveolin oligomers assemble into caveolae, although pits with the distinctive morphology of caveolae have only been reported at the plasma membrane (Rothberg et al., 1992; Denker et al., 1996). Residues 60–100 of caveolin-1 contain the oligomerization domain (Sargiacomo et al., 1995; Schlegel and Lisanti, 2000). However, changes outside this domain can block oligomerization (Machleidt et al., 2000; Lee et al., 2002), presumably through indirect effects.
Caveolin-1 has a high affinity for lipid rafts, membrane microdomains that have a high degree of lipid order and are concentrated in caveolae (Smart et al., 1999). Raft lipids and proteins are resistant to solubilization by nonionic detergents such as Triton X-100 (TX100) and 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate, and can be isolated from cell lysates as detergent-resistant membranes (DRMs; London and Brown, 2000; Schuck et al., 2003; Shogomori and Brown, 2003). DRM association provides a useful tool for identifying proteins that have a high affinity for rafts (London and Brown, 2000; Shogomori and Brown, 2003). Acylation of raft proteins often aids in raft targeting. Caveolin-1 is unusual in that although it is triply acylated, prevention of acylation by mutation does not affect raft affinity (Dietzen et al., 1995). It is not known how caveolin-1 is targeted to rafts, although binding of the protein to cholesterol may play a role (Murata et al., 1995; Thiele et al., 2000).
Unlike most plasma membrane proteins, a significant amount of caveolin-1, estimated at ∼15% by electron microscopy (Denker et al., 1996) is present in the Golgi apparatus at steady state (Rothberg et al., 1992; Dupree et al., 1993; Luetterforst et al., 1999). Although early studies suggested that caveolin-1 cycled constitutively between caveolae and the Golgi apparatus (Conrad et al., 1995), this was later shown not to occur (Nichols, 2002; Thomsen et al., 2002). Instead, as caveolin-1 is selectively cleared from the Golgi apparatus when protein synthesis is inhibited with cycloheximide (Nichols, 2002), the Golgi pool may be a biosynthetic intermediate in transit to the plasma membrane. If so, caveolin-1 exits the Golgi more slowly than most other cargo proteins, which are not detected in the Golgi apparatus at steady state.
Several caveolin-1 mutant proteins accumulate in the Golgi apparatus (Luetterforst et al., 1999; Machleidt et al., 2000; Lee et al., 2002), as does caveolin-2 when expressed without caveolin-1 (Mora et al., 1999; Parolini et al., 1999). Coexpression with caveolin-1 rescues cell-surface expression of caveolin-2. Golgi accumulation of mutant proteins is unusual, because most proteins accumulate in the endoplasmic reticulum (ER) through interaction with chaperone proteins if mutation blocks proper folding. It is not known whether mutation exposes cryptic Golgi retention signals in caveolin-1 (Luetterforst et al., 1999) or whether it destroys information required for efficient Golgi exit (Machleidt et al., 2000). We examined a panel of caveolin-1 mutant proteins to gain more insight into this unusual localization.
MATERIALS AND METHODS
Cells and Antibodies
Culture of COS-1, Fischer rat thyroid (FRT), and mouse melanoma MEB-4 cells has been described previously (Ostermeyer et al., 1999, 2001). The primary antibodies rabbit anti-caveolin-1 and mouse anti-GM130 antibodies were from BD Transduction Laboratories (San Diego, CA); rabbit anti-placental alkaline phosphatase (PLAP) antibodies were from DakoCytomation California (Carpinteria, CA); and rabbit and mouse (12CA5) anti-hemagglutinin (HA) epitope antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The secondary antibodies fluorescein isothiocyanate- and Texas Red-conjugated goat anti-rabbit IgG, Texas Red-conjugated goat anti-mouse Ig(G+M), and horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG were from Jackson ImmunoResearch Laboratories (West Grove, PA).
Plasmids
Plasmids encoding HA- and myc-tagged canine caveolin-1 (Dietzen et al., 1995), Cys- (a triple Cys nonpalmitoylated mutant of this protein; Dietzen et al., 1995), canine caveolin-1 with a C-terminal myc tag followed by the ER retrieval motif KKSL (Ostermeyer et al., 2001), human PLAP, and PLAP-HA, containing the extracellular domain of PLAP fused to the transmembrane and cytoplasmic domains of influenza hemagglutinin (Arreaza and Brown, 1995), have been described. pSAV2M2, encoding canine caveolin-1 with a C-terminal myc tag followed by green fluorescent protein (GFP), was the gift of S. Munro (Medical Research Council, Cambridge, United Kingdom). Plasmids encoding the caveolin-1 mutants 71A75, 76A80, 81A85, 86A90, 91A95, and 96A100 (Machleidt et al., 2000) were gifts of P. Liu and R.G.W. Anderson (University of Texas Southwestern, Dallas, TX). All other caveolin-1 mutants were made by mutagenesis of HA- and myc-tagged canine caveolin-1 (Dietzen et al., 1995) by standard polymerase chain reaction or by using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Amino acids altered in these mutants are referred to based on their position in wild-type canine caveolin-1α (Kurzchalia et al., 1992). Each of these mutants contained an N-terminal HA tag. Each contained a C-terminal myc tag, except the following: Cav-NTD (ending with R101); Y148A5 (Ala substitutions for residues 148–152); H153A5 (Ala substitutions for residues 153–157); the C-terminal truncation mutants Δ13, Δ21, Δ31 (named for the number of residues deleted from the C terminus); and ΔC (lacking the entire C-terminal domain and ending with K135). Cav-ΔN1 lacked residues 3–48, Cav-ΔCSD lacked residues 81–100, and Cav-ΔN1/ΔC lacked residues 3–48 and 136–178. Cav-ΔN2/ΔC lacked residues 46–100 and 136–178. Hydrophobic domain Ala-substitution mutants have been described previously (Ostermeyer et al., 2004). Other mutants are self-explanatory or are described in the figure legends. All mutations were verified by sequencing, and all mutant proteins migrated as expected on SDS-PAGE.
Other Reagents
Dipalmitoyl phosphatidylcholine (DPPC), dioleoyl phosphatidylcholine (DOPC), and cholesterol were from Avanti Polar Lipids (Alabaster, AL). N-Octyl-β-d-glucopyranoside (OG) was from Anatrace (Maumee, OH). Polyvinylidene difluoride (PVDF) membrane was from Millipore (Bedford, MA). X-omat AR film was from Eastman Kodak (Rochester, NY), and Western Lightning chemiluminescence reagent from PerkinElmer Life and Analytical Sciences (Boston, MA). Serva Blue G was from Serva (Heidelberg, Germany). Other reagents were from Sigma-Aldrich (St. Louis, MO).
Transfection and Immunofluorescence
Cells were transfected using Lipofectamine 2000 from Invitrogen (Carlsbad, CA) as described previously (Ostermeyer et al., 2004). All data were from transiently transfected cells, harvested 2 d after transfection, except immunofluorescence (IF) images of Ala-scan mutants in and near the CSD (71A75 and 76A80 shown in Figure 7B), where mixed populations of stably transfected FRT cells expressing each mutant protein were examined.
Figure 7.

Localization, oligomerization, and raft association of caveolin-1 N-terminal domain mutant proteins. (A) Left, schematic diagram of two deletion mutant proteins, ΔCSD (lacking F81-Y100) and ΔN1 (lacking G3-E48). (A) Right, sequence of the last 31 residues of the N-terminal domain is shown, with the start of the hydrophobic domain shown as an open box. The alterations in two substitution mutant proteins are indicated below the N-terminal domain sequence. In WFF/AAA, Ala is substituted for each of W85, F89, and F92. In 97/SASA, the sequence SASA is substituted for residues Y97-Y100. The alterations in five mutant proteins in which sequential groups of five residues were changed to Ala are shown above the wild-type sequence (Machleidt et al., 2000). These proteins are named for the first and last altered residues (i.e., 71A75 contains Ala substituted for each of residues 71–75). In Figure 7C, these proteins are indicated by the first changed residue (i.e., 71A75 is called 71). (B) Localization of the indicated proteins and GM130 in transfected FRT cells is shown. (C) Oligomerization was determined on blue native gels as in Figure 4C. Only the oligomer bands are shown in iii and iv. 71* and 76*, substitution of Ala for residues 71–75 or 76–80, respectively, in the myc and HA-tagged wild-type caveolin-1 shown on the same blot. In iii, the amounts of lysate loaded in each lane were varied to obtain more uniform oligomer bands. (D) TX100-insolubility of the indicated mutant proteins in raft-containing liposomes was determined as in Figure 4D.
IF microscopy on paraformaldehyde-fixed, TX100-permeabilized, coverslip-grown cells was as described previously (Ostermeyer et al., 2001). Where indicated, cells were incubated with 50 μg/ml cycloheximide for the indicated times before fixation. Green and red images, obtained with a 100× objective (numerical aperture 1.3), were captured separately into Adobe Photoshop from a SPOT cooled charge-coupled device 24 bit color digital camera (Diagnostic Instruments, Sterling Heights, MI) mounted on an Axioskop 2 microscope (Carl Zeiss, Oberkochen, Germany).
Reconstitution, Detergent Extraction, and DRM Analysis
For reconstitution of proteins and lipids from cell lysates into mixed liposomes, COS-1 or transfected MEB-4 cells in one 60-mm dish (expected to contain 400 nmol of membrane lipid; Brown and Rose, 1992) were lysed with 0.7 ml of TNE (25 mM Tris-HCl, 150 mM NaCl, and 5 mM EDTA, pH 7.4) containing 120 mM OG and protease inhibitors (0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, and 0.2 mM phenylmethylsulfonyl fluoride). Lysates were clarified by ultracentrifugation at 100,000 × g in a TL-100 tabletop ultracentrifuge (Beckman Coulter, Fullerton, CA) for 10 min. Supernatant (200 μl) was mixed with 0.5 ml of lipids [4 mM, DPPC:cholesterol 2:1 (mol:mol) or DOPC, as indicated] in TNE containing 180 mM OG. Mixtures were subjected to dialysis overnight against 5 L phosphate-buffered saline (PBS; 150 mM NaCl and 20 mM phosphate buffer, pH 7.4) to form mixed proteoliposomes containing cellular proteins and lipids and artificial lipids. Where indicated, the association of caveolin-1 with these liposomes was analyzed by flotation in sucrose gradients. To do this, an aliquot of liposomes was brought to 0.5 ml with PBS, mixed with 1.5 ml of TNE containing 53% sucrose, placed in a 12-ml ultracentrifuge tube, overlaid with 8 ml of TNE/38% sucrose and then 1 ml of TNE/5% sucrose, and subjected to ultracentrifugation in an SW41 rotor at 80,000 × g for 18 h. Three fractions (3.6 ml each) were harvested from the bottom, and any pelleted material was collected and resuspended in 3.6 ml of TNE/1% TX100. Proteins in 0.5-ml aliquots of each fraction were collected by precipitation in trichloroacetic acid in the presence of 10 μg of bovine serum albumin as carrier. Protein pellets were washed once with acetone and once with diethyl ether, dissolved in gel loading buffer, and analyzed by SDS-PAGE and Western blotting.
To analyze DRM association by using sucrose gradients, an aliquot of liposome suspension was adjusted to 1% TX100 in 0.5 ml by using PBS and a 10% TX100 stock and incubated on ice for 30 min. Sucrose step gradients were then formed as described above for detergent-free samples and subjected to sucrose gradient centrifugation, fractionation, SDS-PAGE, and Western blotting as described above. To analyze DRM association without sucrose gradients, after dialysis 0.2 ml of liposome suspension was mixed with 0.9 ml of TNE and collected by centrifugation at top speed in a Microfuge for 5 min. Supernatants were discarded and pellets resuspended in 0.5 ml of ice-cold PBS, adjusted to 1% TX100 with a 10% stock, incubated on ice for 30 min, and then subjected to ultracentrifugation in standard Microfuge tubes at 48,000 rpm for 30 min in a TLA-100 rotor in a TL-100 tabletop ultracentrifuge. Supernatants were collected, and pellets resuspended in 0.5 ml of TNE/1% TX100. Equal volumes of supernatant and pellet fractions were subjected to SDS-PAGE and Western blotting to visualize the indicated proteins.
Gels and Blots
Blue native PAGE was performed as described previously (Schagger et al., 1994), with changes as noted next. Cells were lysed in lysis buffer (500 mM 6-amino caproic acid, 2 mM EDTA, and 25 mM Bistris, pH 7.0) containing 120 mM OG for 30 min at 4°C. Lysates were clarified by centrifugation for 10 min at top speed in a Microfuge. Supernatants were mixed with 1/10 volume sample buffer (5% Serva Blue G, 500 mM ε-amino n-caproic acid, and 100 mM Bistris, pH 7.0) and 1/10 volume glycerol. Proteins were separated on linear 5.5–16% acrylamide gradient gels run at 100 V at 4°C until the dye reached the middle of the gel. Blue cathode buffer (50 mM Tricine, 15 mM Bistris, and 0.02% Serva Blue G, pH 7.0) was then replaced by colorless cathode buffer (lacking Serva Blue G), and gels were run at 200 V until the dye reached the bottom. After soaking gels for 10 min at room temperature in transfer buffer containing 0.1% SDS, proteins were transferred to PVDF membranes and detected by Western blotting. Sizes were roughly estimated by comparison to standards: apoferritin (440 kDa), β-amylase (200 kDa), bovine serum albumin (66 kDa), and carbonic anhydrase (29 kDa).
SDS-PAGE, transfer of proteins to PVDF membranes, and Western blotting were as described previously (Ostermeyer et al., 2004). Caveolin-1 and all mutant proteins were detected on Western blots with anti-caveolin-1 antibodies and HRP-conjugated secondary antibodies, visualizing bands by enhanced chemiluminescence. Detergent-insolubility results of different mutants shown in the same figure did not always come from the same blot. Caveolin-1–positive bands were all aligned for presentation, although they would not necessarily have comigrated if analyzed side by side.
RESULTS
Cellular Localization of Caveolin-1
Caveolin-1 was clearly detectable in the Golgi apparatus, as well as punctate caveolae, in most COS cells (Figure 1A) and after transient expression in FRT cells, which do not express endogenous caveolin-1 (Lipardi et al., 1998). The Golgi apparatus in FRT cells was heterogeneous. It had a normal perinuclear distribution in some cells, but in other cells consisted of dispersed elements (Figure 1, G–L), as seen previously (Luetterforst et al., 1999). None of the mutant caveolin-1 proteins examined here had any obvious effect on Golgi morphology. Golgi staining of caveolin-1, relative to caveolar staining, gradually declined over a few hours of cycloheximide treatment (Figure 1, A–L), as reported previously (Nichols, 2002). By contrast, another exogenously expressed plasma membrane protein, PLAP-HA (Arreaza and Brown, 1995), was usually (>90% of transfected cells) undetectable in the Golgi apparatus at steady state (Figure 1M; GM130 localization in the same cell shown in Figure 1N) and was never detected there after 1-h incubation with cycloheximide. Thus, caveolin-1 was cleared unusually slowly from the Golgi apparatus.
Figure 1.

Selective clearance of caveolin-1 from the Golgi apparatus in cycloheximide-treated cells. COS (A–F) or FRT cells expressing caveolin-1 (G–L) or PLAP-HA (M and N) were left untreated (A, D, G, J, M, and N) or treated with cycloheximide (CHX) for 1 h (B, E, H, and K) or 3 h (C, F, I, and L) before fixation, permeabilization, and detection of caveolin-1 (A–C and G–I) or PLAP-HA (M) and GM130 in the same cells (D–F, J–L, and N) by IF.
Golgi Accumulation of Caveolin-1 Deletion Mutants
Both the C-terminal domain of caveolin-1, expressed without the rest of the protein, and caveolin-1 lacking the C-terminal domain, accumulate in the Golgi apparatus (Luetterforst et al., 1999; Machleidt et al., 2000). These findings suggested that caveolin-1 might contain two Golgi-targeting signals; one in the C-terminal domain and the second in either the N-terminal or hydrophobic domain.
To localize the latter signal more precisely, we expressed the N-terminal domain of caveolin-1 alone (Cav-NTD). This protein was largely cytosolic, but it was concentrated in the Golgi apparatus when membrane association was seen (Figure 2A). To localize the putative signal further, we attempted to express constructs encoding smaller fragments of the N-terminal domain, but we could not detect the proteins. As an alternate approach, we made two new constructs, Cav-ΔN1/ΔC and Cav-ΔN2/ΔC. Each encoded half of the N-terminal domain, linked to the hydrophobic domain (Figure 2). Surprisingly, both proteins accumulated in the Golgi apparatus (Figure 2, D and G). In addition to the Golgi apparatus, Cav-ΔN2/ΔC also accumulated in the ER (Figure 2G), and sometimes in lipid droplets (Figure 2G, arrows), because it lacked a sequence needed for efficient ER exit (Machleidt et al., 2000). Caveolin-1 is partially redirected to lipid droplets when it overaccumulates in the ER (Ostermeyer et al., 2001, 2004). The pool of Cav-ΔN2/ΔC in the Golgi apparatus probably came from molecules that escaped lipid droplet targeting, and slowly left the ER via the secretory pathway. Despite the Golgi accumulation of ΔN1/ΔC, neither it nor any of the caveolin-1 mutant proteins examined here affected transport of a coexpressed plasma membrane protein, PLAP, to the cell surface (Figure 2, J–O).
Figure 2.

Cellular localization of caveolin-1 deletion mutant proteins. Cav-NTD (A), Cav-ΔN1/ΔC (D), or Cav-ΔN2/ΔC (G) were detected together with endogenous GM130 (B, E, and H) in transfected FRT cells by IF. Merged images are shown in C, F, and I. In G, arrows; lipid droplets. In J–O, PLAP was coexpressed with wild-type caveolin-1 (J–L) or with ΔN1/ΔC (M–O) in FRT cells. Caveolin-1 (J) or ΔN1/ΔC (M) was detected with mouse anti-HA antibodies (red), and PLAP in the same cells (K and N, respectively) with anti-PLAP antibodies (green). (L and O); merged images. Schematic diagrams of caveolin-1 and the mutant proteins, indicating the N- and C-terminal hydrophilic domains as open boxes, and the central hydrophobic domain as a shaded box, are shown. The dashed line in the diagram of Cav-ΔN2/ΔC indicates a deleted region.
It is difficult to explain these and earlier findings (Luetterforst et al., 1999; Machleidt et al., 2000) by invoking specific Golgi retention signals in caveolin-1. Such a model would require that caveolin-1 contained three separate retention signals: one in the C-terminal domain, one in the N-terminal domain, and the third either in the N-terminal domain or the hydrophobic domain. All three signals would be cryptic in the intact protein, but they could be exposed by mutation.
An alternate model is that caveolin-1 must achieve the correct conformation for efficient Golgi exit. To determine whether Golgi accumulation might result from conformational defects, we examined a panel of caveolin-1 mutant proteins. In addition to localization, we examined two properties of caveolin-1 that might be sensitive to conformational changes: oligomerization and lipid raft targeting (Luetterforst et al., 1999; Machleidt et al., 2000; Lee et al., 2002).
A Reconstituted System for Assaying DRM Association of Caveolin-1 Mutant Proteins
Raft affinity of membrane proteins is usually examined by determining their association with DRMs after detergent extraction of cells. However, as a protein's lipid environment at the time of extraction affects DRM association (Brown and Rose, 1992; Schroeder et al., 1998), we could not use this method to compare the raft affinity of proteins in different organelles. Instead, we examined DRM association in vitro, by using a liposome reconstitution assay. We mixed lysates of cells expressing the mutant proteins with excess raft-forming lipids, formed liposomes by dialysis, isolated DRMs from the liposomes after TX100 extraction, and determined whether the mutant proteins associated with the DRMs.
To test this system, we first showed that wild-type caveolin-1 derived from both COS and transfected MEB-4 cells associated efficiently with liposomes, as it floated with them in sucrose gradients (Figure 3A, top). MEB-4 cells were chosen because they produce very little endogenous caveolin-1 (Ostermeyer et al., 1999), but they express transfected constructs at high efficiency. About half the wild-type caveolin-1 associated with floating DRMs after Triton X-100 extraction (Figure 3A, middle). Very little caveolin-1 aggregated and pelleted in the gradients. To show that DRM association depended on liposome lipids, and not cellular lipids that remained associated with caveolin-1, we repeated the assay, mixing cell lysates with DOPC, a nonraft forming lipid. Caveolin-1 associated well with these liposomes (our unpublished data), but was efficiently solubilized when these liposomes were extracted with TX100 (Figure 3A, bottom).
Figure 3.

TX100 insolubility of caveolin-1 in mixed proteoliposomes containing cellular proteins and lipids and artificial lipids. OG lysates of COS (left) or transfected MEB-4 cells (right) were mixed with excess OG-solubilized DPPC:cholesterol (2:1) or DOPC as indicated, and proteoliposomes were formed by dialysis. (A) Proteoliposomes were subjected to sucrose density gradient centrifugation (top) or were extracted with TX100 before applying to sucrose gradients (middle and bottom). Gradients were harvested in three fractions from the top. In each case, fraction 1 contained floating membranes (Mem) and fraction 3 contained soluble and detergent-solubilized proteins (Sol). Gradient pellets (Pel) contained any aggregated proteins. (B) Proteoliposomes were extracted with TX100 and subjected to ultracentrifugation, and the TX100-soluble supernatant (S) and TX100-insoluble pellet (P) fractions were separated. (A and B) After resuspension and solubilization of pelleted material, proteins in each fraction were separated by SDS-PAGE and transferred to PVDF membranes. Caveolin-1 was detected by Western blotting.
For convenience, in later experiments, DRMs were collected as pellets rather than by flotation in sucrose gradients. We detected ∼70% of both endogenous COS cell caveolin-1 and wild-type caveolin-1 expressed in MEB4 cells in DRMs by using this method (Figure 3B). We do not know why more caveolin-1 was present in the pellet in this assay than floated with DRMs in sucrose gradients (Figure 3A). A small fraction of the protein may have aggregated and may have pelleted along with DRM-associated protein in the pelleting assay. Aggregates may have been small and may not have pelleted in sucrose gradients. Alternatively, some of the DRM-associated protein may not have withstood the rigors of sucrose gradient analysis, and may have been stripped from DRMs during centrifugation. In any event, as described next, the pelleting assay was useful in distinguishing qualitatively between mutants with high and low DRM association.
Correlation between Localization, Oligomerization, and DRM Association of Hydrophobic Domain Mutant Proteins
We next examined the behavior of a panel of caveolin-1 mutant proteins in the three assays. Results, shown in Figures 4, 5, 6, 7 and summarized in Figure 8, are grouped according to the mutated protein domain. Almost all the mutant proteins showed a striking correlation between behavior in the three assays.
Figure 4.

Localization, oligomerization, and raft association of caveolin-1 hydrophobic domain Ala-scan mutant proteins. (A) The sequence of the hydrophobic domain of caveolin-1, bounded by basic residues, is shown. Sequential groups of five or six residues, together spanning the hydrophobic domain, were changed to Ala as indicated. Each mutant protein was named for the position of the first altered residue and the number of altered residues (i.e., 102A5) or simply by the position of the first altered residue (i.e., 102). (B) Localization of the indicated mutant proteins (green) and GM130 (red) in transfected FRT cells is shown. (C) Proteins in lysates of MEB-4 cells expressing wild-type caveolin-1 or the indicated mutant protein were separated by blue native gel electrophoresis and transferred to PVDF membranes. Caveolin-1 and the mutant proteins were detected by Western blotting. Positions of molecular weight standards (MW), detected by Coomassie Blue staining of the membrane, are shown. Exact molecular weights of membrane proteins cannot be determined by native gel analysis. (D) Mixed proteoliposomes formed from lysates of MEB-4 cells expressing the indicated mutant protein and excess DPPC:cholesterol (2:1) were subjected to TX100 extraction and centrifugation. Supernatant (S) and pellet (P) fractions were analyzed for the presence of the mutants by SDS-PAGE and Western blotting.
Figure 5.

Localization, oligomerization, and raft association of caveolin-1 hydrophobic domain mutant proteins. (A) FRT cells expressing (G108+A112)L (i and iv), P110L (ii and v), or both myc/GFP-tagged caveolin-1 and HA-tagged P132L (iii and vi) were processed for IF. i, ii, iv, and v, (G018+A112)L (i) and P110L (ii) were detected together with GM130 (iv and v, respectively). iii and vi, caveolin-1 was detected by GFP fluorescence (iii), whereas P132L was detected with rabbit anti-HA and Texas Red goat anti-rabbit antibodies (vi). (B) Oligomerization of the indicated proteins [(G108+A112)L, labeled (GA)L; P132L; wild-type caveolin-1; and P110, respectively] was determined on blue native gels as in Figure 4C. The smear migrating more slowly than the (G108+A112)L monomer band was variable. (C) TX100-insolubility of the indicated mutant proteins in raft-containing liposomes was determined as in Figure 4D.
Figure 6.

Localization, oligomerization, and raft association of caveolin-1 C-terminal domain mutant proteins. (A) Sequence of the C-terminal domain of caveolin-1, with the end of the hydrophobic domain indicated as an open box, is shown. Arrows indicate the last residue in each of a series of mutant proteins. All proteins (except ΔC, which lacked the entire C-terminal domain except K135) are named for the number of deleted residues (i.e., Δ13 lacked the last 13 residues of caveolin-1, and ended with K165). Two mutant proteins, Y148A5 and H153A5 (labeled 148 and 153, respectively, in Figure 6C), contained five Ala residues in place of the indicated residues. Of these proteins, only Δ39 contained a C-terminal myc tag. (B) The localization of the indicated mutant proteins and GM130 in transfected FRT cells is shown. (C) Oligomerization was determined on blue native gels as in Figure 4C. -Myc, wild-type caveolin-1 lacking the Myc tag. Cys-, a nonpalmitoylated mutant protein (Dietzen et al., 1995). (D) TX100-insolubility of the indicated mutant proteins in raft-containing liposomes was determined as in Figure 4D.
Figure 8.

Summary of localization, oligomerization, and TX100-insolubility results. The caveolin-1 mutant proteins are diagrammed schematically as in Figure 2A. Mutants are grouped according to the domain altered. Mult., deletions in two domains. HydD, CTD, and NTD, changes in the hydrophobic, C-terminal, or N-terminal domains, respectively. Asterisks indicate substitutions. Four or more substitutions are indicated by a single larger asterisk. Column 3 (PM); efficiency of Golgi-to-plasma membrane transport. ++, wild-type localization; +, slightly enhanced Golgi localization; -, Golgi accumulation. None of these proteins except Cav-ΔN2/ΔC accumulated predominantly in the ER, except in highly expressing cells. Column 4 (Olig); oligomerization on blue native gels. ++, wild-type oligomerization, with very little monomer; +, predominantly oligomeric, but some monomer; +/-, substantial oligomer and monomer; -/+, mostly monomer; -, no oligomer detected; *, oligomer migrated more slowly than wild-type caveolin-1. Column 5 (DRM); association with TX100-resistant membranes after extraction of raft-containing mixed proteoliposomes. +, similar to wild-type caveolin-1 (∼70% insoluble); +/-, about equal amounts soluble and insoluble; -, poor DRM association (∼30% insoluble). ND, not determined.
We first examined six mutant proteins in which sequential groups of five or six residues through the hydrophobic domain were changed to Ala (Figure 4A). Three of these proteins (102A5, 113A5, and 130A5) were similar to wild-type caveolin-1 in all three assays. First, they had similar localization patterns, although they were somewhat more enriched in the Golgi apparatus than the wild-type protein (Figure 4B). We examined oligomerization by using blue native gel electrophoresis. In this method, after gentle cell lysis with OG, detergent is replaced by Coomassie Blue dye, which supplies charge to allow migration in native acrylamide gels, but does not denature proteins and can preserve protein complexes (Schagger et al., 1994). Wild-type caveolin-1 expressed in MEB-4 cells migrated as a high-molecular-weight oligomer on blue native gels (Figure 4C), as expected from previous sucrose velocity gradient analysis (Monier et al., 1995; Sargiacomo et al., 1995; Machleidt et al., 2000). 102A5, 113A5, and 130A5 were largely oligomeric and comigrated with the wild-type proteins, although we also saw a fainter, faster migrating form. This was probably a monomer, because it comigrated with mutant proteins lacking the oligomerization domain that migrate as monomers on velocity gradients (Sargiacomo et al., 1995). The same three proteins also associated well with DRMs isolated from mixed proteoliposomes (Figure 4D).
By contrast, the other three hydrophobic domain Ala-scan mutant proteins, 107A5, 118A5, and 123A6, were defective in all three assays: they were highly concentrated in the Golgi apparatus, migrated as monomers on blue native gels, and associated poorly with DRMs (Figure 4).
We examined three additional hydrophobic domain mutant proteins. We made the first protein (G108+A112)L, after noticing that if the first half of the hydrophobic domain formed an α helix, then the helix face containing G108 and A112 would contain only small residues (Ostermeyer et al., 2004), suggesting that it might pack closely against another helix (Eilers et al., 2002). We changed these residues to Leu to determine whether their small size might be important in structure. (G108+A112)L accumulated in the Golgi apparatus (Figure 5A, i), did not oligomerize properly (Figure 5B, lane 1), and associated poorly with DRMs in the reconstitution assay (Figure 5C), supporting the idea that these residues were structurally important.
P132L, which was detected in 16% of human breast tumors (Hayashi et al., 2001), colocalized with GM130 in the Golgi apparatus in FRT cells (our unpublished data), as reported previously (Lee et al., 2002). Those authors reported that coexpression with P132L caused retention of wild-type caveolin-1 in a perinuclear compartment in COS-7 and human mammary epithelial cells (Lee et al., 2002). By contrast, we saw no effect of coexpression on the localization of caveolin-1-GFP or P132L in FRT cells. Except in very highly expressing cells, wild-type caveolin-1-GFP was present largely in caveolae (Figure 5A, iii), whereas P132L had a Golgi-like distribution (Figure 5A, vi).
P132L migrates as a monomer on sucrose velocity gradients (Lee et al., 2002). We saw two forms of P132L on blue native gels; oligomers that migrated more slowly than wild-type caveolin-1, and monomers (Figure 5B, lane 2). The relative amounts of the two forms varied between experiments. These results suggested that the slowly migrating form of P132L was an abnormal oligomer that dissociated more easily than wild-type caveolin-1. We could not detect P132L in reconstituted liposomes. Changing the second conserved Pro residue in the hydrophobic domain to Leu (P110L) did not affect cellular localization, oligomerization, or detergent insolubility in the reconstitution assay (Figure 5).
Correlation between Localization, Oligomerization, and DRM Association of C-Terminal Domain Mutants
A series of C-terminal truncation mutant proteins (Figure 6A, arrows indicate the last residue in each protein) showed good correlation between behavior in the three assays. Δ13, containing the longest C-terminal domain, had a wild-type distribution in cells (Figure 6B) and oligomerized and associated with DRMs normally (Figure 6, C and D). Δ21 was somewhat more concentrated in the Golgi apparatus than wild-type caveolin-1, although it was easily detectable in caveolae as well (Figure 6B). This protein was mostly oligomeric and associated well with DRMs (Figure 6, C and D). Δ31 was substantially more concentrated in the Golgi apparatus (Figure 6B), although not as completely as some other mutants and did not oligomerize or associate well with DRMs (Figure 6, C and D). Finally, Δ39 and ΔC were concentrated in the Golgi apparatus (ΔC, Figure 6B; Δ39, our unpublished data), as reported previously for a C-terminal deletion mutant in Chinese hamster ovary cells (Machleidt et al., 2000), failed to oligomerize and associated poorly with DRMs (Figure 6, C and D). We sometimes saw a second form of Δ39 and ΔC that migrated slightly more slowly than the monomer, as seen for ΔC in Figure 6C.
Because Δ21 was more severely affected than Δ31 in all three assays, we changed either the first or second five residues in the 10-residue sequence that distinguished these proteins to Ala, in the context of the full-length protein. Both of these mutant proteins (Y148A5 and H153A5, Figure 6A) had a wild-type localization in cells (our unpublished data), oligomerized efficiently (Figure 6C), and associated well with DRMs (Figure 6D). For unknown reasons, oligomers of Y148A5 and H153A5 migrated slightly faster than those of the wild-type protein, an effect that did not depend on their lack of a myc tag (Figure 6C). We conclude that residues 148–157 did not contain a specific motif that directly affected localization, oligomerization, or raft affinity. Finally, Cys-, a nonpalmitoylated caveolin-1 mutant, was indistinguishable from wild-type caveolin-1 in all three assays (localization not shown; oligomerization and detergent-insolubility in Figure 6, C and D), in agreement with previous reports (Dietzen et al., 1995; Monier et al., 1996).
Localization, Oligomerization, and DRM Association of N-Terminal Domain Mutants
The first three N-terminal domain mutants that we examined were defective in all three assays. ΔCSD, which lacked F81-Y100 (Figure 7A, left), accumulated in the Golgi apparatus (Figure 7B), failed to oligomerize, (Figure 7C, i), and did not associate well with DRMs (Figure 7D, left). Aromatic residues, which are abundant in the CSD, were previously shown to aid in membrane association of a CSD peptide (Arbuzova et al., 2000). We made two mutant proteins, WFF/AAA and 97/SASA (Figure 7A, right), to examine the importance of these residues. Both proteins showed the same behavior: they accumulated in the Golgi apparatus (97/SASA, Figure 7B; WFF/AAA, our unpublished data), failed to oligomerize (Figure 7C, i), and associated poorly with DRMs in the reconstitution assay (Figure 7D, left). Thus, the CSD, and especially aromatic residues in that domain, were important in caveolin-1 structure.
We next examined six mutant proteins in which sequential groups of five residues through the CSD and the region just upstream of it were changed to Ala (71A75–96A100; Figure 7A, right). Our results were largely similar to those of Machleidt et al. (2000), who made these constructs. All the proteins were concentrated in the Golgi apparatus, but they were also detectable in punctate structures. 71A75 and 76A80 illustrate the range of localization patterns (Figure 7B). We saw both monomeric and oligomeric forms of all the proteins except 96A100, which was entirely monomeric (Figure 7C, ii). 76A80 and 86A90 were largely oligomeric, whereas a higher fraction of 71A75, 81A85, and 91A95 was monomeric.
Oligomers of these proteins migrated more slowly than wild-type caveolin-1 (Figure 7C, ii). To determine whether this resulted from the respective epitope tags, we introduced the changes present in either 71A75 or 76A80 into our caveolin-1 construct. The new mutants 71* and 76* had the same oligomer:monomer ratio as 71A75 and 76A80, respectively (our unpublished data). Furthermore, they comigrated with the original mutant proteins and migrated more slowly than wild-type caveolin-1, although they migrated faster than P132L (Figure 7C, iii and iv, only oligomer bands shown). 71A75 and 76A80 associated well with DRMs (Figure 7D). 81A85 and 86A90 showed intermediate and variable behavior, and 91A95 and 96A100 were mostly TX100 soluble. By contrast, Machleidt et al. (2000) found that all these mutants were detergent soluble when transfected cells were extracted with Triton X-100, probably because the mutants accumulated in raft-poor Golgi membranes.
Although 91A95 and 96A100 were defective in all three assays, behavior of the other four proteins was less consistent. We do not know the significance of the slow migration of their oligomers. These proteins may have been borderline cases, with slightly altered conformations that affected their behavior in unpredictable ways.
By contrast, ΔN1, which lacked the first half of the N-terminal domain (G3-E48), was the only mutant protein that showed clearly discordant behavior in the three assays. ΔN1 accumulated in the Golgi apparatus (Figure 7B), consistent with previous reports of similar mutant proteins (Luetterforst et al., 1999; Machleidt et al., 2000). Nevertheless, ΔN1 oligomerized and associated with DRMs as well as wild-type caveolin-1 (Figure 7C, v and D). Thus, oligomerization and DRM association were not sufficient for efficient Golgi exit. Results of the behavior of the mutant proteins in the three assays are summarized in Figure 8.
Persistence of Caveolin-1 Mutants in the Golgi Apparatus
We next determined the localization of selected mutant proteins in cycloheximide-treated cells. 97/SASA and 118A5 remained highly concentrated in the Golgi apparatus after prolonged cycloheximide treatment (Figure 9, A–F). Golgi localization of 102A5 and Δ21, which were not as highly concentrated in the Golgi apparatus at steady state, was reduced but still detectable after 3-h cycloheximide treatment (Figure 9, G–L). By contrast, Golgi staining of Δ31 (the C-terminal truncation mutant that was more Golgi-enriched than Δ21 at steady state) was still pronounced in the Golgi after 3-h cycloheximide treatment (Figure 9, M–O). Thus, the concentration of caveolin-1 mutants in the Golgi apparatus at steady state was at least partially explained by their reduced rate of Golgi exit.
Figure 9.

Persistence of caveolin-1 mutant proteins in the Golgi apparatus after cycloheximide treatment. The localization of the indicated proteins (A, D, G, J, and M) and GM130 (B, E, H, J, and N) in transfected FRT cells after cycloheximide treatment for 3 h (except cells expressing 118A5, treated for 5 h) is shown. C, F, I, L, and O; merged anti-caveolin-1 (green) and anti-GM130 (red) images.
Transport through the Secretory Pathway Is Not Required for Oligomerization or Acquisition of Raft Affinity
Transport to the late Golgi apparatus or plasma membrane might be necessary for acquisition of raft affinity, for instance, through a posttranslational modification or conformational change that occurred only in a late compartment. To test this possibility, we examined Cav-KKSL, wild-type caveolin-1 with an appended ER retrieval signal (Ostermeyer et al., 2001). As reported previously (Ostermeyer et al., 2001), Cav-KKSL was only detectable in the ER and in ER-derived lipid droplets (Figure 10, top), presumably through efficient retrieval to the ER in COPI-coated vesicles. Cav-KKSL oligomerized and associated with DRMs as well as wild-type caveolin-1 (Figure 10, middle and bottom). Thus, transport to late compartments of the secretory pathway was not required for oligomerization or acquisition of raft affinity.
Figure 10.
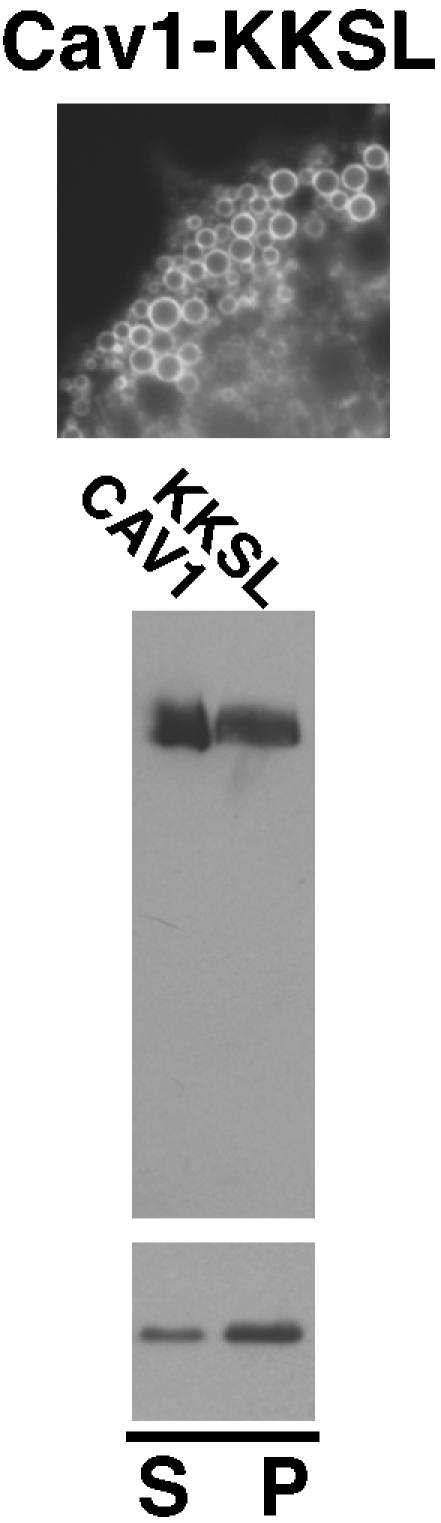
Localization, oligomerization, and raft association of Cav1-KKSL. Top, localization of Cav1-KKSL in FRT cells by IF is shown. Round structures are lipid droplets, as reported previously (Ostermeyer et al., 2001). Middle, oligomerization of wild-type caveolin-1 and Cav-KKSL was determined on blue native gels as in Figure 4C. Bottom, TX100-insolubility of Cav-KKSL in raft-containing liposomes was determined as in Figure 4D.
DISCUSSION
Golgi exit, oligomerization, and raft affinity correlated strikingly among a broad spectrum of caveolin-1 mutant proteins. Of 26 mutant proteins examined, nine were close to normal in all three assays, and 14 were defective in all three. Only two mutant proteins (71A75 and 76A80) showed abnormal oligomerization and Golgi exit, but they retained raft affinity. ΔN1 also convincingly broke the correlation: it oligomerized normally and associated with DRMs, but it was defective for Golgi exit. Based on these data, normal oligomerization and raft affinity might be required for Golgi exit, although ΔN1 showed that neither is sufficient. Alternatively, conformational defects might affect the three properties independently.
The clearest conclusion of this work is that the conformation of caveolin-1 is highly optimized, and extremely sensitive to small changes. Even caveolin-2, which accumulates in the Golgi apparatus and does not oligomerize normally without caveolin-1, cannot achieve this optimized conformation on its own (Mora et al., 1999; Parolini et al., 1999). Sequences in caveolin-1 that were sensitive to mutation were spread among all three domains of the protein. Thus, most of the changes probably acted indirectly, by affecting overall conformation, and not by disrupting specific motifs.
We can speculate on how changes in a few of the mutant proteins might have affected conformation. (G108+A112)L contained bulky Leu residues in place of Gly108 and A112 that may normally lie on a helix face that interacts closely with other protein domains. Similarly, P132L was more severely affected than 130A5, in which P132 was changed to the smaller Ala. Proper hydrophobic domain packing interactions may be required for correct conformation, and preventing this packing may affect surface delivery, oligomerization, and raft targeting indirectly.
Aromatic residues aid in membrane binding of a CSD peptide (Arbuzova et al., 2000) and thus may affect protein conformation by positioning the CSD close to the membrane. WFF/AAA and 97/SASA, in which aromatic residues were specifically targeted, were more severely affected than 81A85, 86A90, or 91A95, Ala scan mutant proteins through the same region. Two Ala-scan mutants just upstream of the CSD, in which no aromatic residues were changed (71A75 and 76A80), associated well with DRMs. By contrast, all the other mutant proteins in this series had changes in at least one aromatic residue and also had reduced DRM association. Thus, aromatic residues in the CSD may play important and additive roles in raft targeting, as well as overall protein conformation.
Cellular Localization
Caveolin-1 mutant proteins that lacked residues 66–70 accumulated in the ER, supporting a previously established role for that domain in ER exit (Machleidt et al., 2000). By contrast, a surprising number of caveolin-1 mutant proteins accumulated in the Golgi apparatus. Previous workers have used a mutagenesis approach in an attempt to identify specific Golgi targeting signals in caveolin-1 (Luetterforst et al., 1999), caveolin-2 (Breuza et al., 2002), or Golgi exit signals in caveolin-1 (Machleidt et al., 2000). Our data suggest that most of these alterations caused Golgi accumulation indirectly, by affecting protein conformation. Golgi accumulation of these proteins probably resulted from inhibition of Golgi exit, rather than exposure of retention signals, as we showed that signal-mediated Golgi retention would require at least three separate signals.
However, ΔN1 accumulated in the Golgi apparatus, even though its oligomerization and raft affinity suggested that its conformation might not be grossly disturbed. Thus, the first half of the N-terminal domain may be especially important in Golgi exit. Consistent with this possibility, caveolin-1 and caveolin-2 (which accumulates in the Golgi apparatus in the absence of caveolin-1) show little sequence similarity in this region.
Golgi Transport of Caveolin-1 Is Unusually Difficult
Cargo proteins are generally transported rapidly and synchronously through the Golgi apparatus. By contrast, Golgi passage of caveolin-1 seems to be unusually difficult. One function of the optimized conformation of caveolin-1 may be to partially overcome this difficulty. Disruption of this optimized conformation by mutation would then lead to Golgi accumulation. One model explaining how this might occur is described next.
Munro and Bretscher proposed a model to explain how newly synthesized plasma membrane proteins in transit through the Golgi apparatus are sorted from Golgi resident proteins (Bretscher and Munro, 1993; Munro, 1995). Transmembrane spans of plasma membrane proteins are longer than those of Golgi proteins. Munro and Bretscher proposed that microdomains of different thickness coexist in the Golgi apparatus. Cholesterol increases bilayer thickness, and they proposed that thick domains contain more cholesterol than thin domains. They also proposed that Golgi enzymes and cargo proteins destined for the plasma membrane partition into the appropriate domains according to transmembrane domain length.
According to the cisternal maturation model of Golgi dynamics, cargo proteins move through the Golgi apparatus passively, as individual cisternae progress through the stack (Pelham and Rothman, 2000; Martínez-Menárguez et al., 2001; Mironov et al., 2001). This model predicts that Golgi resident proteins, as well as any cargo proteins—like caveolin-1—that move through the Golgi more slowly than the rate of cisternal flow, must transiently leave the cisternae, probably in retrograde-directed recycling vesicles. To accommodate these ideas, the model of Munro and Bretscher might predict that recycling vesicles are thinner than bulk cisternae. Thus, long transmembrane spans of plasma membrane proteins would keep these proteins in forward-directed cisternae and prevent incorporation into retrograde-directed carriers.
Caveolin-1 lacks defined transmembrane spans. It may not be able to sense the thickness of cholesterol-rich domains and may not partition as strongly into these domains as other proteins. Instead, at each sorting step, some caveolin-1 may be incorporated into retrograde-directed vesicles. The resulting cycle would slow the net rate of forward transport. However, caveolin-1 is transported through the Golgi apparatus faster than most of the mutants we examined and thus may partition better into thick domains than they do. Caveolin-1 may recognize some other property of these domains, rather than their thickness. This property may be shared with membrane rafts. This property might be the cholesterol-induced increase in acyl-chain packing found in rafts (Xu and London, 2000), or some other feature of the higher cholesterol concentration. In any case, the same structural features that target caveolin-1 to rafts also may increase its affinity for cholesterol-rich Golgi domains. Mutants with reduced raft affinity may have lower affinity for these domains, enhancing their incorporation into retrograde vesicles, and thus their Golgi accumulation.
This model also could explain slow Golgi transport of some other proteins. Like caveolins, peripheral membrane proteins that are transported to the plasma membrane via the secretory pathway cannot be sorted in the Golgi based on transmembrane span length. Palmitoylation of several of these proteins is required for efficient Golgi exit (Choy et al., 1999; Kanaani et al., 2002; Navarro-Lérida et al., 2002; Takimoto et al., 2002; Takida and Wedegaertner, 2003). Palmitoylation can target proteins to rafts (Brown and London, 2000) and thus also might sort these proteins into cholesterol-rich Golgi domains in the Golgi. Palmitoylation also enhances Golgi transport of some transmembrane proteins (van't Hof and Crystal, 2002; Kalinina and Fricker, 2003), possibly through the same mechanism.
Three caveolin-1 mutants, 71A75, 76A80, and ΔN1, did not fit this model. They had apparently normal raft affinity but accumulated in the Golgi apparatus. It is possible that the sensitivity of our DRM assay is not high enough to detect small changes in raft affinity that are important in vivo.
It is also possible that Golgi accumulation of caveolin-1 mutants is related to quality control, a process of recognition and degradation of misfolded proteins. Many of the mutant proteins that we examined were degraded rapidly, as reported previously (Machleidt et al., 2000). Golgi accumulation of misfolded caveolin-1 mutant proteins might occur as part of a post-ER quality control system.
Caveolin-1 Oligomerization
Blue native gel analysis gave some new insights into caveolin-1 oligomerization. We saw remarkably few intermediate sized oligomers. We occasionally saw a band that migrated slightly more slowly than the monomer, when examining mutants that were otherwise entirely monomeric. Some mutants were present in both monomeric and oligomeric forms, suggesting that oligomerization was inefficient or oligomers unstable. However, we never saw any other intermediate-sized forms of these proteins. This suggested that oligomerization of caveolin-1 is highly cooperative.
A few mutant proteins oligomerized, but did not quite comigrate with wild-type caveolin-1 on blue native gels. These complexes may contain different numbers of monomers, or may have a different shape or ability to bind dye. These mutants also may bind differentially to other proteins, although caveolin-1 oligomers are thought to contain no additional proteins except caveolin-2 (Monier et al., 1995; Scheiffele et al., 1998). Oligomers of Y148A5 and H135A5 migrated slightly faster than wild-type caveolin-1. These mutant proteins oligomerized as efficiently as wild-type caveolin-1 and showed normal cellular localization and DRM association. This suggested that their abnormal migration on blue native gels reflected only minor conformational changes.
Two classes of mutant proteins formed oligomers that migrated unusually slowly. P132L was the only member of the first class. The other class included six Ala-scan mutants in and near the CSD (Figure 7), and also a W85A point mutant (our unpublished data). As all these oligomers were similarly affected, changes throughout this region seemed to cause the same structural alteration. P132L oligomers migrated more slowly than those of the CSD mutants. Oligomers of both classes migrated as distinct bands, suggesting that they did not aggregate nonspecifically. We saw significant amounts of monomeric forms of all these mutant proteins, suggesting that oligomerization was inefficient or unstable. All these mutant proteins accumulated in the Golgi apparatus. Thus, structural changes that reduced oligomer mobility seemed to affect caveolin-1 conformation more severely than those that increased migration.
The correlation between in vitro DRM association and oligomerization suggested that wild-type caveolin-1 and oligomerization-competent mutant proteins were probably incorporated into liposomes as oligomers. Blue native gel analysis showed that the wild-type protein remained oligomeric after reconstitution (our unpublished data), and we expect that this was true of the mutant proteins as well.
Further work will clearly be required to fully understand how so many different mutations can alter the conformation of caveolin-1 and how these changes can have such profound effects on Golgi exit, oligomerization, and raft affinity of the protein.
Acknowledgments
We thank S. Munro (Medical Research Council) and P. Liu and R.G.W. Anderson (University Texas Southwestern Medical College) for plasmids. This work was supported by grants GM-47897 (to D.A.B.) and GM-41297 (to D.M.L.) from the National Institutes of Health.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04–06–0480. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–06–0480.
Abbreviations used: CSD, caveolin scaffolding domain; DOPC, dioleoyl phosphatidylcholine; DPPC, dipalmitoyl phosphatidylcholine; DRM, detergent-resistant membrane; FRT, Fischer rat thyroid; HRP, horseradish peroxidase; IF, immunofluorescence; OG, N-octyl-β-D-glucopyranoside; PBS, phosphate-buffered saline; PLAP, placental alkaline phosphatase; TX100, Triton X-100.
References
- Arbuzova, A., Wang, L., Wang, J., Hangyas-Mihalyne, G., Murray, D., Honig, B., and McLaughlin, S. (2000). Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39, 10330-10339. [DOI] [PubMed] [Google Scholar]
- Arreaza, G., and Brown, D.A. (1995). Sorting and intracellular trafficking of a glycosylphosphatidylinositol-anchored protein and two hybrid proteins with the same ectodomain in MDCK kidney epithelial cells. J. Biol. Chem. 270, 23641-23647. [DOI] [PubMed] [Google Scholar]
- Bretscher, M.S., and Munro, S. (1993). Cholesterol and the Golgi apparatus. Science 261, 1280-1281. [DOI] [PubMed] [Google Scholar]
- Breuza, L., Corby, S., Arsanto, J.-P., Delgrossi, M.-H., Scheiffele, P., and Le Bivic, A. (2002). The scaffolding domain of caveolin 2 is responsible for its Golgi localization in Caco-2 cells. J. Cell Sci. 115, 4457-4467. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and London, E. (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221-17224. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and Rose, J.K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533-544. [DOI] [PubMed] [Google Scholar]
- Choy, E., Chiu, V.K., Silletti, J., Feoktistov, M., Morimoto, T., Michaelson, D., Ivanov, I.E., and Philips, M.R. (1999). Endomembrane trafficking of Ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98, 69-80. [DOI] [PubMed] [Google Scholar]
- Conrad, P.A., Smart, E.J., Ying, Y.-S., Anderson, R.G.W., and Bloom, G.S. (1995). Caveolin cycles between plasma membrane caveolae and the Golgi complex by microtubule-dependent and microtubule-independent steps. J. Cell Biol. 131, 1421-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker, S.P., McCaffrey, J.M., Palade, G.E., Insel, P.A., and Farquhar, M.G. (1996). Differential distribution of α subunits and βγ subunits of heterotrimeric G proteins on Golgi membranes of the exocrine pancreas. J. Cell Biol. 133, 1027-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzen, D.J., Hastings, W.R., and Lublin, D.M. (1995). Caveolin is palmitoylated on multiple cysteine residues: palmitoylation is not necessary for localization of caveolin to caveolae. J. Biol. Chem. 270, 6838-6842. [DOI] [PubMed] [Google Scholar]
- Dupree, P., Parton, R.G., Raposo, G., Kurzchalia, T.V., and Simons, K. (1993). Caveolae and sorting in the trans-Golgi network of epithelial cells. EMBO J. 12, 1597-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers, M., Patel, A.B., Liu, W., and Smith, S.O. (2002). Comparison of helix interactions in membrane and soluble alpha-bundle proteins. Biophys. J. 82, 2720-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, I., Ying, Y., Albanesi, J., and Anderson, R.G.W. (2002). Mechanism of caveolin filament assembly. Proc. Natl. Acad. Sci. USA 99, 11193-11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fra, A.M., Williamson, E., Simons, K., and Parton, R.G. (1994). Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J. Biol. Chem. 269, 30745-30748. [PubMed] [Google Scholar]
- Hayashi, K., Matsuda, S., Machida, K., Yamamoto, T., Fukuda, Y., Nimura, Y., Hayakawa, T., and Hamaguchi, M. (2001). Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res. 61, 2361-2364. [PubMed] [Google Scholar]
- Kalinina, E.V., and Fricker, L.D. (2003). Palmitoylation of carboxypeptidase D. Implications for intracellular trafficking. J. Biol. Chem. 278, 9244-9249. [DOI] [PubMed] [Google Scholar]
- Kanaani, J., el-Husseini Ael, D., Aguilera-Moreno, A., Diacovo, J.M., Bredt, D.S., and Baekkeskov, S. (2002). A combination of three distinct trafficking signals mediates axonal targeting and presynaptic clustering of GAD65. J. Cell Biol. 158, 1229-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzchalia, T.V., Dupree, P., Parton, R.G., Kellner, R., Virta, H., Lehnert, M., and Simons, K. (1992). VIP21, a 21-kD membrane protein, is an integral component of trans-Golgi-network-derived transport vesicles. J. Cell Biol. 118, 1003-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H., Park, D.S., Razani, B., Russell, R.G., Pestell, R.G., and Lisanti, M.P. (2002). Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (-/-) null mice show mammary epithelial cell hyperplasia. Am. J. Pathol. 161, 1357-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipardi, C., Mora, R., Colomer, V., Paladino, S., Nitsch, L., Rodriguez-Boulan, E., and Zurzolo, C. (1998). Caveolin transfection results in caveolae formation but not apical sorting of glycosylphosphatidylinositol (GPI)-anchored proteins in epithelial cells. J. Cell Biol. 140, 617-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London, E., and Brown, D.A. (2000). Insolubility of lipids in Triton X-100. Physical origin and relationship to sphingolipid/cholesterol domains (rafts). Biochim. Biophys. Acta 1508, 182-195. [DOI] [PubMed] [Google Scholar]
- Luetterforst, R., Stang, E., Zorzi, N., Carozzi, A., Way, M., and Parton, R.G. (1999). Molecular characterization of caveolin association with the Golgi complex: identification of a cis-Golgi targeting domain in the caveolin molecule. J. Cell Biol. 145, 1443-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleidt, T., Li, W.-P., Liu, P., and Anderson, R.G.W. (2000). Multiple domains in caveolin-1 control its intracellular traffic. J. Cell Biol. 148, 17-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Menárguez, J.A., Prekeris, R., Oorschot, V.M.J., Scheller, R., Slot, J.W., Geuze, H.J., and Klumperman, J. (2001). Peri-Golgi vesicles contain retrograde but not anterograde proteins consistent with the cisternal progression model of intra-Golgi transport. J. Cell Biol. 155, 1213-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov, A.A., et al. (2001). Small cargo proteins and large aggregates can traverse the Golgi by a common mechanism without leaving the lumen of cisternae. J. Cell Biol. 155, 1225-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier, S., Dietzen, D.J., Hastings, W.R., Lublin, D.M., and Kurzchalia, T.V. (1996). Oligomerization of VIP21-caveolin in vitro is stabilized by long chain fatty acylation or cholesterol. FEBS Lett. 388, 143-149. [DOI] [PubMed] [Google Scholar]
- Monier, S., Parton, R.G., Vogel, F., Behlke, J., Henske, A., and Kurzchalia, T.V. (1995). VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol. Biol. Cell 6, 911-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora, R., Bonilha, V.L., Marmorstein, A., Scherer, P.E., Brown, D., Lisanti, M.P., and Rodriguez-Boulan, E. (1999). Caveolin-2 localizes to the Golgi complex but redistributes to plasma membrane, caveolae, and rafts when co-expressed with caveolin-1. J. Biol. Chem. 274, 25708-25717. [DOI] [PubMed] [Google Scholar]
- Munro, S. (1995). An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 14, 4695-4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata, M., Peränen, J., Schreiner, R., Wieland, F., Kurzchalia, T.V., and Simons, K. (1995). VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 92, 10339-10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Lérida, I., Alberto Álvarez-Barrientos, F.G., and Ignacio, and Rodriguez-Crespo, I. (2002). Distance-dependent cellular palmitoylation of denovo-designed sequences and their translocation to plasma membrane subdomains. J. Cell Sci. 115, 3119-3130. [DOI] [PubMed] [Google Scholar]
- Nichols, B.J. (2002). A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 4, 374-378. [DOI] [PubMed] [Google Scholar]
- Ostermeyer, A.G., Beckrich, B.T., Ivarson, K.A., Grove, K.E., and Brown, D.A. (1999). Glycosphingolipids are not essential for formation of detergent-resistant membrane rafts in melanoma cells. Methyl-beta-cyclodextrin does not affect cell-surface transport of a GPI-anchored protein. J. Biol. Chem. 274, 34459-34466. [DOI] [PubMed] [Google Scholar]
- Ostermeyer, A.G., Paci, J.M., Zeng, Y., Lublin, D.M., Munro, S., and Brown, D.A. (2001). Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J. Cell Biol. 152, 1071-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeyer, A.G., Ramcharan, L.T., Zeng, Y., Lublin, D.M., and Brown, D.A. (2004). Role of the hydrophobic domain in targeting caveolin-1 to lipid droplets. J. Cell Biol. 164, 69-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolini, I., et al. (1999). Expression of caveolin-1 is required for the transport of caveolin-2 to the plasma membrane. Retention of caveolin-2 at the level of the Golgi complex. J. Biol. Chem. 274, 25718-25725. [DOI] [PubMed] [Google Scholar]
- Pelham, H.R.B., and Rothman, J.E. (2000). The debate about transport in the Golgi—two sides of the same coin? Cell 102, 713-719. [DOI] [PubMed] [Google Scholar]
- Rothberg, K.G., Heuser, J.E., Donzell, W.C., Ying, Y.-S., Glenney, J.R., and Anderson, R.G.W. (1992). Caveolin, a protein component of caveolae membrane coats. Cell 68, 673-682. [DOI] [PubMed] [Google Scholar]
- Sargiacomo, M., Scherer, P.E., Tang, Z.L., Kübler, E., Song, K.S., Sanders, M.C., and Lisanti, M.P. (1995). Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc. Natl. Acad. Sci. USA 92, 9407-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger, H., Cramer, W.A., and von Jagow, G. (1994). Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217, 220-230. [DOI] [PubMed] [Google Scholar]
- Scheiffele, P., Verkade, P., Fra, A.M., Virta, H., Simons, K., and Ikonen, E. (1998). Caveolin-1 and -2 in the exocytic pathway of MDCK cells. J. Cell Biol. 140, 795-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel, A., and Lisanti, M.P. (2000). A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J. Biol. Chem. 275, 21605-21617. [DOI] [PubMed] [Google Scholar]
- Schlegel, A., Schwab, R.B., Scherer, P.E., and Lisanti, M.P. (1999). A role for the caveolin scaffolding domain in mediating the membrane attachment of caveolin-1. The caveolin scaffolding domain is both necessary and sufficient for membrane binding in vitro. J. Biol. Chem. 274, 22660-22667. [DOI] [PubMed] [Google Scholar]
- Schroeder, R.J., Ahmed, S.N., Zhu, Y., London, E., and Brown, D.A. (1998). Cholesterol and sphingolipid enhance the Triton X-100-insolubility of GPI-anchored proteins by promoting the formation of detergent-insoluble ordered membrane domains. J. Biol. Chem. 273, 1150-1157. [DOI] [PubMed] [Google Scholar]
- Schuck, S., Honsho, M., Ekroos, K., Shevchenko, A., and Simons, K. (2003). Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 100, 5795-5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogomori, H., and Brown, D.A. (2003). Use of detergents to study membrane rafts: The good, the bad, and the ugly. Biol. Chem. 384, 1259-1263. [DOI] [PubMed] [Google Scholar]
- Smart, E.J., Graf, G.A., McNiven, M.A., Sessa, W.C., Engelman, J.A., Scherer, P.E., Okamoto, T., and Lisanti, M.P. (1999). Caveolins, liquid-ordered domains, and signal transduction. Mol. Cell. Biol. 19, 7289-7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takida, S., and Wedegaertner, P.B. (2003). Heterotrimer formation, together with isoprenylation, is required for plasma membrane targeting of Gβγ. J. Biol. Chem. 278, 17284-17290. [DOI] [PubMed] [Google Scholar]
- Takimoto, K., Yang, E.-K., and Conforti, L. (2002). Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J. Biol. Chem. 277, 26904-26911. [DOI] [PubMed] [Google Scholar]
- Thiele, C., Hannah, M.J., Fahrenholz, F., and Huttner, W.B. (2000). Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 2, 42-49. [DOI] [PubMed] [Google Scholar]
- Thomsen, P., Roepstorff, K., Stahlhut, M., and van Deurs, B. (2002). Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol. Biol. Cell 13, 238-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Hof, W., and Crystal, R.G. (2002). Fatty acid modification of the cox-sackievirus and adenovirus receptor. J. Virol. 76, 6382-6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., and London, E. (2000). The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 39, 843-849. [DOI] [PubMed] [Google Scholar]