

Abstract
Developmental geneticists continue to make substantial jumps in our understanding of the genetic pathways that regulate development. This understanding stems predominantly from analyses of genetically tractable model organisms developing in lab environments. This environment is vastly different from that in which human development occurs. As such, most causes of developmental defects in humans are thought to involve multifactorial gene-gene and gene-environment interactions. In this review, we discuss how gene-environment interactions with environmental teratogens may predispose embryos to structural malformations. We elaborate on the growing number of gene-ethanol interactions that might underlie susceptibility to Fetal Alcohol Spectrum Disorders.
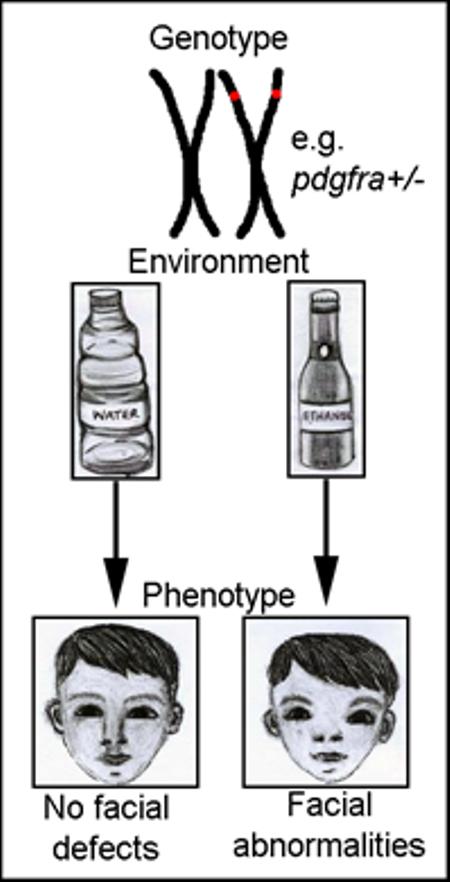
Graphical abstract
Introduction
Nature versus nurture, or the relative contributions of genetics versus the environment to traits, has long been an area of great debate. Given that all organisms evolved within an environment that can be variable, it comes as no surprise that “versus” is a fairly contrived term in many instances. A classic example of this is the disease progression of Phenylketonuria, which requires both the mutation of PHENYLALANINE HYDROXYLASE (PAH) and the presence of dietary phenylalanine. The maintenance of mutant alleles of PAH in human populations is also likely due to gene-environment interactions. In some populations, such as Irish, where the mutant PAH allele is at a relatively high frequency, maternal heterozygosity associates with a reduced risk for miscarriage1. This is thought to be due to higher levels of phenylalanine in the blood of heterozygotes protecting against the mycotoxin, Ochratoxin A, which competes with phenylalanine for PAH and is toxic to embryos1. Such gene-environment interactions abound in psychological and biological systems, yet we still understand relatively little about these interactions.
This lack of understanding can confound many of the studies examining therapeutic approaches to diseases and disorders. An example is the reduction of neural tube defects by folic acid supplementation. Studies showing that folic acid supplementation reduces neural tube defects prompted the US medical community to mandate folic acid fortification of grains2. However, subsequent work in mice showed that the protective benefits of folic acid is dependent on genetic background and that, in varying genetic contexts, folic acid supplementation can be detrimental3-5. Collectively, these findings with PAH and folic acid demonstrate that neither a genotype nor an environmental factor can readily be a priori declared deleterious or beneficial. Instead, development is highly context dependent and requires not only an understanding of genetic variation but also of the environmental context in which the genotype is functioning.
Understanding the mechanism and breadth of gene-environment interactions requires careful analysis of both the genetics of development and the preponderance of environmental influences. Recent work published in the journal Nature has shown that extrinsic, or environmental, factors have a greater influence on cancer risk than intrinsic factors6. Thus, genetic risk is only one part of a complicated equation determining total cancer risk. Cancer can be thought of as a disease of development as many of the pathways necessary for proper development have been implicated in many types of cancer. Ultimately, this work shows that the interplay of both genetics and the environment is critical in understanding the etiology of cancer and, by extension, development.
Teratogens are environmental factors that can disrupt normal development, causing birth defects. While the timing (Figure 1) and dosage of teratogen exposure are critical variables that determine phenotypic outcomes, genetic predisposition is also an important variable that is, in most instances, poorly understood. In this review, we focus our discussion on gene-environment interactions with teratogenic agents that disrupt early development. We first discuss a set of environmental agents that have been associated with gene-environment interactions and elaborate on progress made on understanding gene-environment interactions with the most common teratogen, ethanol.
Figure 1.
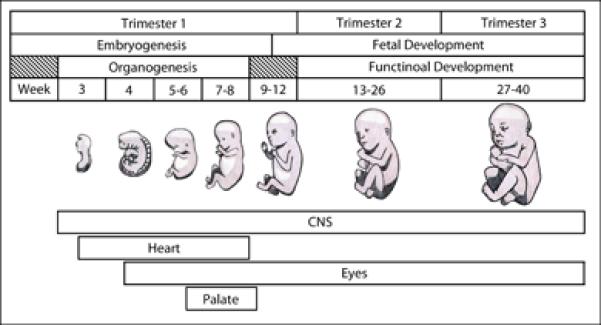
Timing of teratogen exposure can dictate disrupted organ systems. The timing of development of several organ systems discussed in this review is listed. The long development of the central nervous system (CNS) makes it particularly susceptible to teratogenic insult.
Environmental influences: the dark side of progress
Due to human activity, the environment has changed drastically in the last 50 years. Currently, there are more than 80,000 synthetic chemicals, and of these, close to 3,000 are produced in amounts at or exceeding 1 million kg per year7. These 3,000 chemicals are readily found in the environment and are in measureable quantities in the blood of individuals7. In addition, there are 631 different pharmaceutical agents found at measurable concentrations in the water worldwide and this number is likely an underestimate due to a lack of testing8. Adding to these large numbers, there may be many more unknown metabolites of these chemicals with unknown effects. Alarmingly, very few of these chemicals have been examined in a developmental context7, 8. These industrial and pharmaceutical chemicals add to an already complex environment (Figure 2), which includes disease, maternal factors (e.g. diet), natural factors (e.g. oxygen levels) and drugs (e.g. alcohol). These facts demonstrate an ever-increasing likelihood for gene-environment interactions but also have important implications, particularly in human studies that rely on self-reported exposures, given that many exposures may be unknown to an individual.
Figure 2.
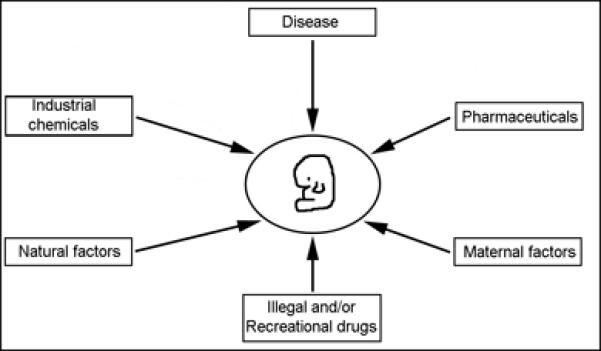
Environmental influences on development. A developing embryo or fetus (center) can be exposed to numerous environmental factors. These factors can interact with the genetic susceptibility of the developing embryo or fetus to alter the outcome of development.
There is growing evidence that chemicals originally considered safe can cause developmental defects. Thalidomide is arguably the clearest example of this. Thalidomide was originally developed in the early 1950's in Germany as a sedative and was actually prescribed to pregnant women as a treatment for morning sickness in over 46 countries9. This resulted in severe birth defects, principally phocomelia, consisting primarily of dramatically shortened limbs, though various other birth defects can and do present9. Due to the complex chemistry and actions of the drug, determining a mechanism of action has been difficult. Multiple mechanisms have been proposed, including oxidative stress and anti-angiogenic actions, though the direct thalidomide targets and how they mediate teratogenesis are still not known10. Nitric Oxide (NO) is a promising as a target of thalidomide activity and exogenous NO has been shown to rescue thalidomide-induced limb and eye deformities via reducing oxidative stress and increasing angiogenesis in both chicken and zebrafish11. While no specific loci are known, a genetic susceptibility to thalidomide-induced birth defects is possible as not all exposed embryos developed birth defects12. Understanding this potential genetic susceptibility is critical because there is a resurgence of thalidomide use as an anti-cancer therapy13.
Recent work has shown that common pharmaceuticals may also increase the risk for developmental disorders and that there is genetic susceptibility to these effects. Schill and colleagues14 demonstrated that ibuprofen inhibits migration and colonization of the bowel by enteric neural crest cells in zebrafish, chicken and mouse. Mice lacking a single copy of Ret have increased sensitivity to ibuprofen. In humans, mutation of RET is a risk factor for Hirschsprung Disease, in which the bowel is not adequately colonized by enteric neural crest cells15. Overall, this work suggests that, in sensitive genetic backgrounds, ibuprofen could increase the risk for Hirschsprung Disease. It should be of concern that this type of research is lacking for most of the 3,000 pharmaceutical agents currently produced.
Similar concerns exist in the large numbers of other naturally occurring and synthetic chemicals. Chemicals such as lead, methylmercury, toluene and ethanol can all cause teratogenesis. Lead, used by humans for thousands of years, has spread widely throughout the environment and human exposure can result in severe neurotoxicity. Recent events in Flint, Michigan demonstrate the impact that environmental lead still has on our society today. Prenatally, lead is able to pass the placental barrier and result in reduced cognitive development and decreased IQ scores16. Postnatally, young children are especially sensitive where exposure can lead to cognitive impairments, decreased IQ scores and behavioral problems, and at later ages may cause a host of diseases including Alzeimer's and Parkinson's17, 18. The differential progressive nature of lead-induced neurological damage across individuals suggests that permanent changes in the CNS may be mediated by genetic background. APOE epsilon 4 (ε4) associates with poorer prognosis following neural trauma19, implicating it in neural repair and potentially lead-induced CNS defects. A study from 2002 has shown that APOEε4 may result in an increased sensitivity to lead toxicity20. However, this study looked at adult exposures rather than in utero exposure and did not hypothesize a potential mechanism for this gene-environmental interaction.
The APOEε4-environment interaction is also observed in mercury toxicity. Methylmercury is formed in fresh water environments from both natural and human-made sources of inorganic mercury. Methylmercury enters the aquatic food chain where it accumulates in fishes. There is a long history of the toxic effects of methylmercury dating back to 186521. However, it wasn't until the 1950's that methylmercury was identified as a teratogen, leading primarily to neurodevelopmental alterations, but also affecting overall growth and limb development22, 23. Recent work has identified gene-mercury interactions with APOE ε4 resulting in increased risk for neurodevelopmental deficits and maladaptive behavioral outcomes24, 25. Additionally, work has shown that polymorphisms in other genes (ABC transporters and glutathione processing enzymes) involved in the processing and elimination of methylmercury may lead to accumulation of methylmercury in utero resulting in reduced birth weight26, 27. Thus, the genetic capacity of an embryo (or potentially the mother) to clear an environmental contaminant may be critical in the risk of teratogenesis.
In addition to environmental contaminants, “life style” can negatively impact development. While United States smoking rates are declining, as of 2014, 16.8% of adults still smoke cigarettes (http://www.cdc.gov/tobacco/data_statistics/fact_sheets/adult_data/cig_smoking/). Smoking is a known risk factor for birth defects, such as orofacial clefting28. Gene-smoking interactions associated with risk for orofacial clefting have been identified predominantly by candidate approaches in humans. In a large study for gene-smoking interactions underlying orofacial clefting, null and hypomorphic alleles for the detoxifying enzymes GSTT1 and NAT2, respectively, were found to associate with orofacial clefting29. Several studies using candidate gene approaches have also identified genetic variants mediating risk to smoking-induced orofacial clefting. These include genes associated with nicotine dependence (DDC)30, DNA repair (RAD51)31 and orofacial clefting (MSX1 and TGFB3)32. It is of interest that other, similar, studies failed to associate either MSX1 or TGFB3 and smoking in the risk for orofacial clefting33, 34. Many possible reasons for such discrepancies exist and experiments in animal models where potential confounds can be controlled would be of great assistance. Such animal models for the effects of smoking on development are being generated35 and should greatly increase our understanding of genetic risk to smoking-induced birth defects as such models have done for our understanding of gene-ethanol interactions.
Ethanol: the emperor of all teratogens
Humans have been consuming ethanol for millennia and alcohol consumption is socially acceptable in most cultures. While the first evidence that ethanol exposure could damage developing embryos was published more than a century ago36, it was not clinically appreciated that prenatal alcohol exposure could cause human birth defects until 196837. In 1973, the term Fetal Alcohol Syndrome (FAS) was coined in reference to a set of severe birth defects in individuals with prenatal alcohol exposure38. An FAS diagnosis requires the presence of characteristic facial defects, such as a smooth philtrum, thin upper lip and short palpebral fissures or eye openings (Figure 3). Additionally, reduced growth and CNS deficits are present in FAS38. While FAS requires the presence of this set of characteristic phenotypes, it is clear that much variability exists in the phenotypic outcomes of ethanol exposure.
Figure 3.
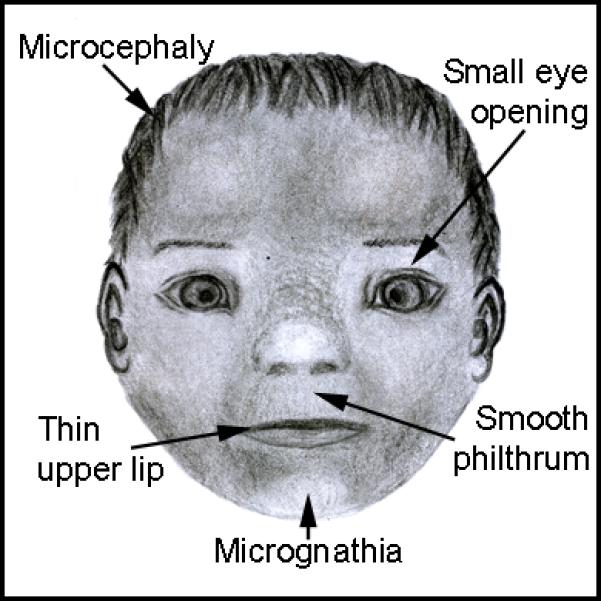
Facial features characteristic of Fetal Alcohol Syndrome.
It is now well appreciated that ethanol can cause a wide range of structural, neural and neurological impairments. A more complete discussion of these ethanol-induced defects can be found elsewhere39, but briefly prenatal alcohol exposure is a risk factor for orofacial clefting as well as cardiac and eye defects40. Additionally, numerous structural defects of the brain are found in ethanol-exposed children, including reduced size of the cerebellum and structural changes to the corpus callosum39, 41. Subsequently, ethanol-exposed individuals may have learning and memory impairments42. These individuals frequently lack the appropriate initiative to form and maintain friendships, leading to a lack of social relationships43, 44. These deficits in social skills can result in employment problems, trouble with the law, inappropriate sexual behaviour, suicide and depression45, 46. This full range of ethanol-induced phenotypes, with FAS at the severe end, is collectively referred to as Fetal Alcohol Spectrum Disorders (FASD)39.
Despite our understanding of FASD, significant numbers of individuals are exposed to at least some alcohol prenatally. In the US the numbers vary between studies, ranging as low as 12% to as high as 25%47, 48. However these numbers may be underestimates as more than 50% of women of childbearing age consume ethanol and nearly half of pregnancies are unplanned47. Estimates of the prevalence of FASD are as high as 1 in 100 live births in the US49. More recent estimates give US prevalence rates of 2-5%50. Recent studies in Italy and South Africa have shown even higher rates of FASD, 3.6% and 7.2%, respectively50. These rates may well be underestimates because pediatricians frequently fail to recognize FASD51. Collectively research shows that FASD is strikingly common and has no single set of phenotypes that define it. Instead, FASD is a highly complex disorder suggesting its genesis is multifactorial.
The variability of FASD and the comorbidity of other negative environments such as poor nutrition, tobacco or drug use can confound human studies of FASD52, 53. Thus, animal models have been crucial in developing our understanding of the pathogenesis of FASD. Indeed, it was work in animal models that definitively showed ethanol was a clear teratogen54. Work across animal models has shown that ethanol was capable of disrupting development of organ systems commonly disrupted in FASD, including the brain, face, heart and eyes36, 55-58. While development can be disrupted by ethanol at any developmental time point, some of the most severe phenotypes are generated when exposure occurs during gastrulation, when the progenitors of the CNS and the face are being generated. Therefore, disrupting embryonic development during these developmental time windows can lead to a wide range of ethanol-induced phenotypes, including growth retardation, facial dysmorphologies and CNS abnormalities55. There is also interest in animal studies of the behavioral outcomes of FASD and these studies have been extensively reviewed elsewhere59. For the purpose of this review, we will focus primarily on the genesis of ethanol-induced structural defects.
Gene-ethanol interactions: A tale of two inputs
Timing, dosage, pattern, and duration of ethanol exposure all impact the phenotypes in FASD 60, 61. Furthermore, multiple studies demonstrate that genetic predisposition also plays a role in FASD. Human twin studies show there is 100% concordance for FAS in monozygotic twins while only 64% concordance in dizygotic twins62. In every animal model system studied, zebrafish, chicken, mice and rat, different inbred strains show different sensitivity to ethanol-induced defects63. Thus, across species there is substantial evidence that the risk for ethanol-induced developmental defects is genetically modulated.
Some of the insight into the genetic risk for FASD comes from phenotypes in individuals with FASD. While FASD has an extremely wide spectrum of phenotypes, some of these mirror holoprosencephaly, which is also highly phenotypically variable. Prenatal ethanol exposure is a risk factor for holoprosencephaly64. Mouse studies have shown that ethanol exposure during days 7 and 8 of pregnancy results in a range of holoprosencephaly-like phenotypes55. The genetics behind holoprosencephaly are complex, but most genes known to be involved in the genesis of holoprosencephaly function in the Sonic Hedgehog (Shh) pathway65. Collectively, these findings initially suggested that mutations in the Shh pathway could enhance the teratogenicity of ethanol.
Work in multiple animal model systems has demonstrated that mutations disrupting Shh signaling predispose to ethanol teratogenesis. Work from Hong and Krauss66 demonstrated that the Shh co-receptor Cdon interacted with ethanol resulting in an increased incident of holoprosencephaly-like phenotypes in mutant mice. Recent work has shown that heterozygosity for either Shh or Gli2 enhanced the facial and neural defects caused by ethanol67. Additionally, work in zebrafish, using morpholinos, revealed that ethanol interacts with shha leading to disrupted GABAergic and glutamatergic neural development68. Morpholinos against agrin, which mediates Shh signaling, also interact with ethanol resulting in defects to ocular development69. Thus, a substantial body of evidence exists suggesting that genetic attenuation of the Shh pathway is a risk factor for FASD.
Several possibilities may explain these interactions of members of the Shh pathway with ethanol. First, ethanol has been shown to disrupt lipid modification of Shh that is required for proper signaling70. Second, it is possible that a source of Shh is undergoing apoptosis following ethanol treatment71. Third is that ethanol disrupts Retinoic acid levels. Retinoic acid is critical in inducing Shh expression in the notochord and neural plate. The timing of ethanol exposure needed to phenocopy holoprosencephaly is just prior to the induction of Shh expression. It has been proposed that ethanol is a competitive inhibitor of retinoic acid synthesis72, 73, although this model remains contentious74. Numerous studies have examined if retinoic acid supplementation can rescue ethanol-induced defects. Most relevant to whether retinoic acid is involved in interactions between ethanol and the Shh pathway is the finding that retinoic acid supplementation can rescue mid-hindbrain defects in ethanol-treated, shha morpholino-injected embryos75. The same study found that retinoic acid did not rescue ocular defects under these same conditions. An inability of retinoic acid to rescue ethanol-induced eye defects was independently demonstrated by Kashyap and colleagues76. These findings, among others detailed more extensively elsewhere77suggest that the involvement of retinoic acid in interactions between ethanol and the Shh pathway is likely to be context dependent. Given that Shh and retinoic acid only explain a portion of the phenotypic spectrum in FASD many other gene-ethanol interactions must exist.
As with the teratogens discussed above, genes mediating clearance of ethanol are likely candidates to modulate FASD risk. Early work in human populations focused on allelic differences in ethanol metabolizing enzymes. Across animal species, degradation of ethanol is a multi-step process. Ethanol is metabolized initially to acetaldehyde, primarily through the action of ALCOHOL DEHYDROGENASE (ADH), formed as a complex of ADH1A, ADH1B and ADH1C. Acetaldehyde is highly reactive and also potentially teratogenic. It is converted to acetate via ALDEHYDE DEHYDROGENASE (ALDH). In several human studies, alleles of ADH1B that are predicted to metabolize ethanol more quickly are underrepresented in children with FASD78-82. Similarly, a slow metabolizing variant of ADH1C associates with orofacial clefting in ethanol-exposed children 83.
While ethanol metabolism may be protective against FASD, it also generates by-products that can be deleterious to cells, making clearance of such by-products another potential level of gene-ethanol interactions. Ethanol processing leads to reactive oxygen species production, disrupting the balance between prooxidants and antioxidants leading to increased oxidative damage, including DNA damage84, 85. Disrupting endogenous antioxidant production can lead to sensitivity to ethanol, which can be mitigated by supplementation with the antioxdiant vitamin E 84. These approaches have been partially successful but may be dependent on a range of factors including timing, dosage, cellular and tissue context and genetic background84, 86. In mice, maternal loss of Superoxide dismutase, responsible for clearing reactive oxygen species, predisposes to ethanol teratogenesis87-89. Work in mouse has shown that combined loss of Aldh2 and the Fanconi Anemia DNA repair enzyme, Fancd2, results in ethanol-induced exencephaly and eye defects90. Thus, both reactive oxygen clearance and DNA damage repair are promising pathways to mediate susceptibility to FASD.
Similar concerns are observed in reactive nitrogen species, in particular nitric oxide. Changes in nitric oxide have been shown to play a role in ethanol teratogenesis with nitric oxide production being protective at low ethanol concentrations and toxic at higher ethanol concentrations85, 91-93. However, attenuation of nitric oxide levels, in Nitric oxide synthase 1 mutants, predisposes embryos to ethanol-induced neural defects91-93. These studies used a third trimester model of exposure, demonstrating that deleterious gene-ethanol interactions are not limited to early development. Collectively, these studies demonstrate that genes involved in clearing ethanol and reversing potential deleterious consequences of ethanol and its metabolism are likely involved in the genetic risk for FASD.
Other studies have taken broader approaches and demonstrated that the genetic susceptibility to ethanol is likely more complex than what would be predicted based on overt phenotypes or ethanol metabolism. The ease of performing genetic screens in zebrafish makes it an appealing model organism to understand genetic risk for FASD. In an initial screen of five craniofacial mutants housed in our lab using doses of ethanol that did not disrupt development in wild-type embryos, we found that pdgfra interacted with ethanol and this interaction was highly synergistic94. Loss of Pdgfra results in orofacial clefting in zebrafish, mice and human95-98. Ethanol-treated pdgfra mutant zebrafish lose the entire palate94. In addition, haploinsufficiency was observed in the majority of pdgfra heterozygous embryos. Pdgfra acts through the PI3K/mTOR pathway to regulate cell survival, proliferation and growth99, 100. We found that this pathway mediates the pdgfra-ethanol interaction and elevating PI3K and mTOR signaling could partially rescue the ethanol-treated mutants94. In humans, we identified single nucleotide polymorphisms (SNPs) in PDGFRA and PDGFRB that associate with in changes in outer canthal width and midfacial depth, respectively, in ethanol-exposed individuals94. In a follow up screen of 20 mutants available from the Zebrafish International Resource Center (ZIRC), we found that mars, hinfp, plk1, foxi1 and vangl2 all genetically interacted with ethanol101. The nature of these genetic interactions is of ongoing interest. These results demonstrate the strength of genetic screens to identify risk factors and we are currently performing a forward genetic screen to identify and characterize new ethanol-sensitive loci.
With the advent, and ever decreasing cost, of deep sequencing, whole genome association studies in humans are becoming more and more feasible. Aside from metabolic enzymes (discussed above), previous candidate based approaches of identifying risk factors in humans have had mixed results. Using a set of genes implicated in human orofacial clefting, one study found an association between the Bmp target, MSX1, and ethanol34. However, two other studies failed to find a similar association32, 33. Recently, a genome-wide association study found two loci, MLLT3 and SMC2, which associated with ethanol-exposure and orofacial clefting102. Future studies will be essential to understand these interactions as neither gene has been implicated in ethanol teratogenesis previously. Overall, this work along with the genetic screens described above demonstrates that gene-ethanol interactions are not readily predicted. The phenotypes from gene-ethanol interactions can be synergistic in nature, requiring a methodical approach to their identification.
Identifying genetic risk factors to one teratogen may help us understand other teratogens. Toluene is an aromatic hydrocarbon used extensively as a solvent in the production of many industrial products, including paint, varnish, lacquer and glue. It is also used as a recreational drug via ‘sniffing or huffing,’ which can lead to neurotoxic events103. The teratogenic effects of toluene (methylbenzene) exposure result in microcephaly, craniofacial abnormalities and neurological impairments103, strikingly similar to prenatal ethanol exposure. “Fetal solvents syndrome” was proposed to describe these features104. However this description is controversial because in some cases clinicians could not rule out concomitant exposure to other teratogens103, 105. The phenotypic similarity between ethanol and toluene suggests that they may share similar mechanisms of teratogenesis103, 106. In addition, the degradation of toluene uses several of the same enzymatic steps as ethanol105. Genetic risk for toluene-induced birth defects is unknown, but it will be of great interest to determine if our understanding of gene-ethanol interactions can inform the study of gene-toluene interactions.
Mechanisms of Gene-environment interactions
The interplay between genetic background and the environment plays a key role in a multitude of diseases and disorders. Even among “simple” Mendelian diseases there is substantial phenotypic variability that could be due to gene-gene, gene-environment or even more complicated multifactorial interactions. These more complicated interactions that include environmental inputs probably abound in more complex disorders. A significant problem remains though in identifying and then characterizing gene-environment interactions that underlie disease as well as healthy development. A key observation across all of the teratogens discussed here is that clearance of a teratogen is critical for healthy development and those genotypes that are slower in this clearance are more susceptible to harm. For most teratogens, we know little more than this regarding genetic risk.
With ethanol as a model, we see that genetic risk for teratogenesis is vastly more complicated than simply clearing the substance from our system. A teratogenic insult must set off a cascade of deleterious events, be that cellular damage or altered signaling. Sometimes, we can predict gene-environment interactions based on mechanisms used to repair such damage (such as DNA damage repair) or based on similar phenotypes of genetic mutants and teratogen-exposed embryos (such as holoprosencephaly-like phenotypes). However, sometimes these interactions appear truly synergistic. For instance, it is only in embryos with both attenuated Pdgf signaling, via mutation of pdgfra, and exposure to ethanol that exhibit a substantial elevation in the death of facial progenitor cells. It is likely that casting a broad net to capture all possible gene-environment interactions will serve us best in understanding this complex problem.
Once identified, understanding the nature of gene-environment interactions may represent a substantial hurdle. We point readers to a recent manuscript detailing how some individual gene-environment interaction researchers conceptualize these interactions and the challenges therein107. Even with gene-gene interactions, there are several possible causes of a different phenotype occurring in a double mutant versus either single mutant. Gene-environment interactions are no exception and if we consider teratogens there are several possibilities.
The first is a direct, physical, interaction with a gene product. This is almost assuredly the case with gene-environment interactions between loci encoding the enzymatic machinery needed to clear a teratogen. Physical interactions between environmental contaminants and other gene products are difficult to detect, although some examples exist. One example with ethanol is the L1 cell adhesion molecule (L1CAM). L1CAM is a membrane bound immunoglobulin-like protein that has multiple functions during neural development108. The extracellular domain of L1CAM has an ethanol-binding pocket that when bound reduces L1CAM function leading to neurodevelopmental defects109. While it is unknown if L1CAM itself is an ethanol-sensitive locus, ERK-dependent phosphorylation of L1CAM alters the ethanol-binding pocket and modulates ethanol sensitivity in a genetic background-dependent manner110. Thus, the physical interaction between ethanol and a protein may also be genetically modulated by activity of signaling pathways.
The direct disruption of a signaling pathway is a second mechanism by which teratogens may interact with genetic risk factors. In this case, ethanol attenuates, or potentially elevates, a signaling pathway upon which the gene product impinges. This is likely to be the case for the pdgfra-ethanol interaction discussed above. It is unlikely that ethanol is interacting directly with the Pdgfra protein because PI3K signaling immediately downstream of the receptor, determined by phosphor-AKT levels, is actually elevated in the presence of ethanol. It is only downstream of mTOR, phospho-Eif4b, where the pathway is attenuated94. In this scenario, it is the combined genetic and environmental insults that result in a failure of the embryo to regulate development.
A third likely mechanism is epigenetics. Teratogens such as ethanol have been shown to alter the levels of small noncoding RNAs, microRNAs (miRNAs). Because each individual miRNA is capable of modulating the levels of translation of many different genes, these small RNAs may well be an important target for ethanol teratogenesis111. Ethanol also effects DNA methylation as well as histone methylation and acetylation112. These types of modifications can give rise to transgenerational epigenetic inheritance, which can be carried through both the male and female germline112.
There is a growing body of work in model organisms demonstrating transgenerational inheritance following teratogen exposure. Recent work suggests that prenatal lead exposure can lead to heritable epigenetic modifications in genes regulating immune response (e.g. APOA5) and neural development and function (e.g. NDRG4 and NINJ2) 113. However, how these epigenetic modifications contribute to lead-induced development disorders is currently not known. In zebrafish, failure of the embryo (or mother) to properly clear methylmercury can lead to a build up to methylmercury in the developing embryo 114. This accumulation of methylmercury can alter DNA methylation patterns. If these modifications occur in the germ line cells of the embryo, then subsequent generations may exhibit developmental impairments in the absence of the environmental insult115. This work showed that F2 and F3 embryos from parents exposed embryonically to methylmercury had persistent learning impairments even without a methylmercury exposure. Similar transgenerational effects have been observed for other environmental toxins including, dioxin, bisphenol A, 17α-ethinylestradiol and polyaromatic hydrocarbons116-119. Overall, this suggests that embryonic exposure to teratogens can not only disrupt development in an exposed embryo but could lead to developmental defects in subsequent, naïve generations.
Conclusion
Gene-environment interactions are likely to underlie much of the variability and susceptibility to birth defects. No single mechanism fully explains the breadth of phenotypes in these interactions. Rather multiple interactions, all tissue and context dependent, are likely to produce the observed phenotypic spectrum.
The complexity of just what is a gene-environment interaction and how these impact human development are major hurdles that must be overcome. Is anything that is not a gene the environment? For instance, it seems straightforward that a bottle of beer or a shot of whiskey is environmental, but what about blood alcohol concentration (BAC)? BAC is a critical variable for ethanol teratogenicity, but it is, in itself, governed by gene-environment interactions: ethanol metabolizing enzymes and the amount and rate of ethanol consumed (which itself has a genetic component modulated by environmental conditions). Thus, even in a “simple” model of gene-environment interactions, it becomes clear that our resulting phenotype is driven by interwoven, layered, and potentially interdependent gene-environment interactions (Fig. 4).
Figure 4.
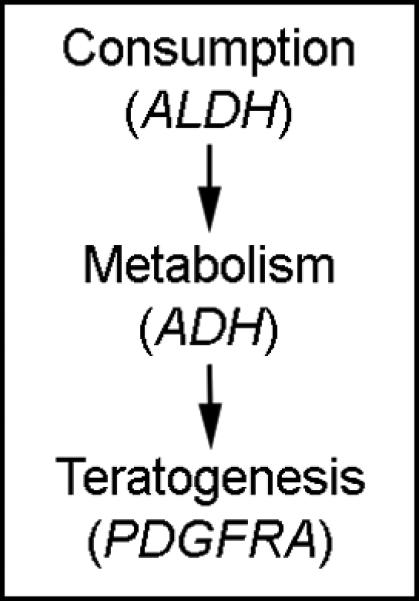
Complexity of gene-environment interactions. Multiple gene-environment interactions are involved in even the simplest model of ethanol teratogenesis. Prenatal alcohol exposure is required for the development of FASD, but is, itself, regulating by gene- environment interactions that mediate both consumption patterns and ethanol metabolism. Prenatal development, then, can be thought of as a set of complex, hierarchical and often interrelated gene-environment interactions.
Further Reading.
The following recent reviews will be of interest to readers wishing to understand our current knowledge of the roles of gene-environment interactions in complex psychological disorders.
Mandy, W. and Lai, M.-C. (2016) Annual Research Review: The role of the environment in the developmental psychopathology of autism spectrum condition. The Journal of Child Psychology and Psychiatry 57:3, 271-292.
Ayhan, Y., McFarland, R. and Pletnikov, M.V. (2016) Animal models of gene-environment interaction in schizophrenia: A dimensional perspective. Progress in Neurobiology 136, 1-27.
Klengel, T. and Binder, E.B. (2015) Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron 86:6, 1343-1357.
Caspi, A. and Moffitt, T.E. (2006) Gene-environment interactions in psychiatry: joining forces with neuroscience. Nature Reviews Neuroscience 7(7) 583-590.
Acknowledgments
This work was supported by grants from NIH/NIDCR R01DE020884 and NIH/NIAAA R01AA023426 and U24AA014811 (PI, Riley) to JKE and NIH/NIAAA K99AA023560 to CBL. Grant U24AA014811 is done in conjunction with the Collaborative Initiative on Fetal Alcohol Spectrum Disorders (CIFASD) Additional information about CIFASD can be found at www.cifasd.org.
References
- 1.Woolf LI. The heterozygote advantage in phenylketonuria. Am J Hum Genet. 1986;38:773–775. [PMC free article] [PubMed] [Google Scholar]
- 2.Recommendations for the use of folic acid to reduce the number of cases of spina bifida and other neural tube defects. MMWR Recomm Rep. 1992;41:1–7. [PubMed] [Google Scholar]
- 3.Marean A, Graf A, Zhang Y, Niswander L. Folic acid supplementation can adversely affect murine neural tube closure and embryonic survival. Hum Mol Genet. 2011;20:3678–3683. doi: 10.1093/hmg/ddr289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mikael LG, Deng L, Paul L, Selhub J, Rozen R. Moderately high intake of folic acid has a negative impact on mouse embryonic development. Birth Defects Res A Clin Mol Teratol. 2013;97:47–52. doi: 10.1002/bdra.23092. [DOI] [PubMed] [Google Scholar]
- 5.Pickell L, Brown K, Li D, Wang XL, Deng L, Wu Q, Selhub J, Luo L, Jerome-Majewska L, Rozen R. High intake of folic acid disrupts embryonic development in mice. Birth Defects Res A Clin Mol Teratol. 2011;91:8–19. doi: 10.1002/bdra.20754. [DOI] [PubMed] [Google Scholar]
- 6.Wu S, Powers S, Zhu W, Hannun YA. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016;529:43–47. doi: 10.1038/nature16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landrigan PJ, De Garbino JP, Newman B. Framing the future in light of the past: living in a chemical world. Ann N Y Acad Sci. 2006;1076:657–659. doi: 10.1196/annals.1371.030. [DOI] [PubMed] [Google Scholar]
- 8.Aus der Beek T, Weber FA, Bergmann A, Hickmann S, Ebert I, Hein A, Kuster A. Pharmaceuticals in the environment-Global occurrences and perspectives. Environ Toxicol Chem. 2016;35:823–835. doi: 10.1002/etc.3339. [DOI] [PubMed] [Google Scholar]
- 9.Vargesson N. Thalidomide-induced limb defects: resolving a 50-year-old puzzle. Bioessays. 2009;31:1327–1336. doi: 10.1002/bies.200900103. [DOI] [PubMed] [Google Scholar]
- 10.Ito T, Ando H, Handa H. Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci. 2011;68:1569–1579. doi: 10.1007/s00018-010-0619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siamwala JH, Veeriah V, Priya MK, Rajendran S, Saran U, Sinha S, Nagarajan S, Pradeep T, Chatterjee S. Nitric oxide rescues thalidomide mediated teratogenicity. Sci Rep. 2012;2:679. doi: 10.1038/srep00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JH, Scialli AR. Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci. 2011;122:1–6. doi: 10.1093/toxsci/kfr088. [DOI] [PubMed] [Google Scholar]
- 13.Zhou S, Wang F, Hsieh TC, Wu JM, Wu E. Thalidomide-a notorious sedative to a wonder anticancer drug. Curr Med Chem. 2013;20:4102–4108. doi: 10.2174/09298673113209990198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schill EM, Lake JI, Tusheva OA, Nagy N, Bery SK, Foster L, Avetisyan M, Johnson SL, Stenson WF, Goldstein AM, et al. Ibuprofen slows migration and inhibits bowel colonization by enteric nervous system precursors in zebrafish, chick and mouse. Dev Biol. 2016;409:473–488. doi: 10.1016/j.ydbio.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 16.Sanders T, Liu Y, Buchner V, Tchounwou PB. Neurotoxic effects and biomarkers of lead exposure: a review. Rev Environ Health. 2009;24:15–45. doi: 10.1515/reveh.2009.24.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ordemann JM, Austin RN. Lead neurotoxicity: exploring the potential impact of lead substitution in zinc-finger proteins on mental health. Metallomics. 2016 doi: 10.1039/c5mt00300h. [DOI] [PubMed] [Google Scholar]
- 18.Grandjean P, Herz KT. Trace elements as paradigms of developmental neurotoxicants: Lead, methylmercury and arsenic. J Trace Elem Med Biol. 2015;31:130–134. doi: 10.1016/j.jtemb.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nathoo N, Chetty R, van Dellen JR, Barnett GH. Genetic vulnerability following traumatic brain injury: the role of apolipoprotein E. Mol Pathol. 2003;56:132–136. doi: 10.1136/mp.56.3.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart WF, Schwartz BS, Simon D, Kelsey K, Todd AC. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ Health Perspect. 2002;110:501–505. doi: 10.1289/ehp.02110501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards GN. Two cases of poisoning by mercuric methide. Saint Bartholomew's Hosp Rep. 1865;1:141–150. [Google Scholar]
- 22.Harada M. Congenital Minamata disease: intrauterine methylmercury poisoning. Teratology. 1978;18:285–288. doi: 10.1002/tera.1420180216. [DOI] [PubMed] [Google Scholar]
- 23.Ceccatelli S, Bose R, Edoff K, Onishchenko N, Spulber S. Long-lasting neurotoxic effects of exposure to methylmercury during development. J Intern Med. 2013;273:490–497. doi: 10.1111/joim.12045. [DOI] [PubMed] [Google Scholar]
- 24.Ng S, Lin CC, Hwang YH, Hsieh WS, Liao HF, Chen PC. Mercury, APOE, and children's neurodevelopment. Neurotoxicology. 2013;37:85–92. doi: 10.1016/j.neuro.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Ng S, Lin CC, Jeng SF, Hwang YH, Hsieh WS, Chen PC. Mercury, APOE, and child behavior. Chemosphere. 2015;120:123–130. doi: 10.1016/j.chemosphere.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Llop S, Engstrom K, Ballester F, Franforte E, Alhamdow A, Pisa F, Tratnik JS, Mazej D, Murcia M, Rebagliato M, et al. Polymorphisms in ABC transporter genes and concentrations of mercury in newborns--evidence from two Mediterranean birth cohorts. PLoS One. 2014;9:e97172. doi: 10.1371/journal.pone.0097172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee BE, Hong YC, Park H, Ha M, Koo BS, Chang N, Roh YM, Kim BN, Kim YJ, Kim BM, et al. Interaction between GSTM1/GSTT1 polymorphism and blood mercury on birth weight. Environ Health Perspect. 2010;118:437–443. doi: 10.1289/ehp.0900731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung KC, Kowalski CP, Kim HM, Buchman SR. Maternal cigarette smoking during pregnancy and the risk of having a child with cleft lip/palate. Plast Reconstr Surg. 2000;105:485–491. doi: 10.1097/00006534-200002000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Shi M, Christensen K, Weinberg CR, Romitti P, Bathum L, Lozada A, Morris RW, Lovett M, Murray JC. Orofacial cleft risk is increased with maternal smoking and specific detoxification-gene variants. Am J Hum Genet. 2007;80:76–90. doi: 10.1086/510518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jugessur A, Wilcox AJ, Murray JC, Gjessing HK, Nguyen TT, Nilsen RM, Lie RT. Assessing the impact of nicotine dependence genes on the risk of facial clefts: An example of the use of national registry and biobank data. Nor Epidemiol. 2012;21:241–250. doi: 10.5324/nje.v21i2.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Machado RA, Moreira HS, de Aquino SN, Martelli-Junior H, de Almeida Reis SR, Persuhn DC, Wu T, Yuan Y, Coletta RD. Interactions between RAD51 rs1801321 and maternal cigarette smoking as risk factor for nonsyndromic cleft lip with or without cleft palate. Am J Med Genet A. 2016;170:536–539. doi: 10.1002/ajmg.a.37281. [DOI] [PubMed] [Google Scholar]
- 32.Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology. 1999;59:39–50. doi: 10.1002/(SICI)1096-9926(199901)59:1<39::AID-TERA9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 33.Etheredge AJ, Christensen K, Del Junco D, Murray JC, Mitchell LE. Evaluation of two methods for assessing gene-environment interactions using data from the Danish case-control study of facial clefts. Birth Defects Res A Clin Mol Teratol. 2005;73:541–546. doi: 10.1002/bdra.20167. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell LE, Murray JC, O'Brien S, Christensen K. Evaluation of two putative susceptibility loci for oral clefts in the Danish population. Am J Epidemiol. 2001;153:1007–1015. doi: 10.1093/aje/153.10.1007. [DOI] [PubMed] [Google Scholar]
- 35.Esposito ER, Horn KH, Greene RM, Pisano MM. An animal model of cigarette smoke-induced in utero growth retardation. Toxicology. 2008;246:193–202. doi: 10.1016/j.tox.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stockard CR. The influence of alcohol and other ane ae sthetics on embryonic development. American Journal of Anatomy. 1910;10:369–392. [Google Scholar]
- 37.Lemoine P, Harousseau H, Borteyru J-P, Menuet J-C. Les Enfants de parents alcooliques: anomalies observees a propos de 127 cas. Ouest Medical. 1968;21:476–482. [Google Scholar]
- 38.Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;302:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 39.Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev. 2011;21:73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munger RG, Romitti PA, Daack-Hirsch S, Burns TL, Murray JC, Hanson J. Maternal alcohol use and risk of orofacial cleft birth defects. Teratology. 1996;54:27–33. doi: 10.1002/(SICI)1096-9926(199607)54:1<27::AID-TERA4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Nunez CC, Roussotte F, Sowell ER. Focus on: structural and functional brain abnormalities in fetal alcohol spectrum disorders. Alcohol Res Health. 2011;34:121–131. [PMC free article] [PubMed] [Google Scholar]
- 42.Kodituwakku PW. Defining the behavioral phenotype in children with fetal alcohol spectrum disorders: a review. Neurosci Biobehav Rev. 2007;31:192–201. doi: 10.1016/j.neubiorev.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 43.Streissguth AP, Aase JM, Clarren SK, Randels SP, LaDue RA, Smith DF. Fetal alcohol syndrome in adolescents and adults. JAMA. 1991;265:1961–1967. [PubMed] [Google Scholar]
- 44.Roebuck TM, Mattson SN, Riley EP. Behavioral and psychosocial profiles of alcohol-exposed children. Alcohol Clin Exp Res. 1999;23:1070–1076. [PubMed] [Google Scholar]
- 45.Kelly SJ, Day N, Streissguth AP. Effects of prenatal alcohol exposure on social behavior in humans and other species. Neurotoxicol Teratol. 2000;22:143–149. doi: 10.1016/s0892-0362(99)00073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kully-Martens K, Denys K, Treit S, Tamana S, Rasmussen C. A review of social skills deficits in individuals with fetal alcohol spectrum disorders and prenatal alcohol exposure: profiles, mechanisms, and interventions. Alcohol Clin Exp Res. 2012;36:568–576. doi: 10.1111/j.1530-0277.2011.01661.x. [DOI] [PubMed] [Google Scholar]
- 47.Flak AL, Su S, Bertrand J, Denny CH, Kesmodel US, Cogswell ME. The association of mild, moderate, and binge prenatal alcohol exposure and child neuropsychological outcomes: a meta-analysis. Alcohol Clin Exp Res. 2014;38:214–226. doi: 10.1111/acer.12214. [DOI] [PubMed] [Google Scholar]
- 48.Ethen MK, Ramadhani TA, Scheuerle AE, Canfield MA, Wyszynski DF, Druschel CM, Romitti PA. 2009 doi: 10.1007/s10995-008-0328-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM., Jr. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 50.May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev. 2009;15:176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- 51.Rojmahamongkol P, Cheema-Hasan A, Weitzman C. Do Pediatricians Recognize Fetal Alcohol Spectrum Disorders in Children With Developmental and Behavioral Problems? J Dev Behav Pediatr. 2015 doi: 10.1097/DBP.0000000000000146. [DOI] [PubMed] [Google Scholar]
- 52.Schneider ML, Moore CF, Adkins MM. The effects of prenatal alcohol exposure on behavior: rodent and primate studies. Neuropsychol Rev. 2011;21:186–203. doi: 10.1007/s11065-011-9168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelly SJ, Goodlett CR, Hannigan JH. Animal models of fetal alcohol spectrum disorders: impact of the social environment. Dev Disabil Res Rev. 2009;15:200–208. doi: 10.1002/ddrr.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murawski NJ, Moore EM, Thomas JD, Riley EP. Advances in Diagnosis and Treatment of Fetal Alcohol Spectrum Disorders: From Animal Models to Human Studies. Alcohol Res. 2015;37:97–108. [PMC free article] [PubMed] [Google Scholar]
- 55.Sulik KK. Genesis of alcohol-induced craniofacial dysmorphism. Exp Biol Med (Maywood) 2005;230:366–375. doi: 10.1177/15353702-0323006-04. [DOI] [PubMed] [Google Scholar]
- 56.Ponnappa BC, Rubin E. Modeling alcohol's effects on organs in animal models. Alcohol Res Health. 2000;24:93–104. [PMC free article] [PubMed] [Google Scholar]
- 57.Becker HC, Diaz-Granados JL, Randall CL. Teratogenic actions of ethanol in the mouse: a minireview. Pharmacol Biochem Behav. 1996;55:501–513. doi: 10.1016/s0091-3057(96)00255-9. [DOI] [PubMed] [Google Scholar]
- 58.Kiecker C. The chick embryo as a model for the effects of prenatal exposure to alcohol on craniofacial development. Dev Biol. 2016 doi: 10.1016/j.ydbio.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 59.Gerlai R. Fish in behavior research: Unique tools with a great promise! J Neurosci Methods. 2014 doi: 10.1016/j.jneumeth.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 60.Cudd TA. Animal model systems for the study of alcohol teratology. Exp Biol Med (Maywood) 2005;230:389–393. doi: 10.1177/15353702-0323006-06. [DOI] [PubMed] [Google Scholar]
- 61.Williams JF, Smith VC, Committee On Substance A. Fetal Alcohol Spectrum Disorders. Pediatrics. 2015;136:e1395–1406. doi: 10.1542/peds.2015-3113. [DOI] [PubMed] [Google Scholar]
- 62.Streissguth AP, Dehaene P. Fetal alcohol syndrome in twins of alcoholic mothers: concordance of diagnosis and IQ. Am J Med Genet. 1993;47:857–861. doi: 10.1002/ajmg.1320470612. [DOI] [PubMed] [Google Scholar]
- 63.Eberhart JK, Parnell SE. The Genetics of Fetal Alcohol Spectrum Disorders (FASD). Alcoholism: Clinical and Experimental Research. doi: 10.1111/acer.13066. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen MM, Jr., Shiota K. Teratogenesis of holoprosencephaly. Am J Med Genet. 2002;109:1–15. doi: 10.1002/ajmg.10258. [DOI] [PubMed] [Google Scholar]
- 65.Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, et al. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:133–141. doi: 10.1002/ajmg.c.30240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong M, Krauss RS. Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 2012;8:e1002999. doi: 10.1371/journal.pgen.1002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kietzman HW, Everson JL, Sulik KK, Lipinski RJ. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS One. 2014;9:e89448. doi: 10.1371/journal.pone.0089448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang C, Ojiaku P, Cole GJ. Forebrain and hindbrain development in zebrafish is sensitive to ethanol exposure involving agrin, Fgf, and sonic hedgehog function. Birth Defects Res A Clin Mol Teratol. 2013;97:8–27. doi: 10.1002/bdra.23099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang C, Turton QM, Mackinnon S, Sulik KK, Cole GJ. Agrin function associated with ocular development is a target of ethanol exposure in embryonic zebrafish. Birth Defects Res A Clin Mol Teratol. 2011;91:129–141. doi: 10.1002/bdra.20766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li YX, Yang HT, Zdanowicz M, Sicklick JK, Qi Y, Camp TJ, Diehl AM. Fetal alcohol exposure impairs Hedgehog cholesterol modification and signaling. Lab Invest. 2007;87:231–240. doi: 10.1038/labinvest.3700516. [DOI] [PubMed] [Google Scholar]
- 71.Dunty WC, Jr., Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- 72.Duester G. A hypothetical mechanism for fetal alcohol syndrome involving ethanol inhibition of retinoic acid synthesis at the alcohol dehydrogenase step. Alcohol Clin Exp Res. 1991;15:568–572. doi: 10.1111/j.1530-0277.1991.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 73.Pullarkat RK. Hypothesis: prenatal ethanol-induced birth defects and retinoic acid. Alcohol Clin Exp Res. 1991;15:565–567. doi: 10.1111/j.1530-0277.1991.tb00561.x. [DOI] [PubMed] [Google Scholar]
- 74.Napoli JL. Effects of ethanol on physiological retinoic acid levels. IUBMB Life. 2011;63:701–706. doi: 10.1002/iub.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang C, Anderson A, Cole GJ. Analysis of crosstalk between retinoic acid and sonic hedgehog pathways following ethanol exposure in embryonic zebrafish. Birth Defects Res A Clin Mol Teratol. 2015;103:1046–1057. doi: 10.1002/bdra.23460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kashyap B, Frey RA, Stenkamp DL. Ethanol-induced microphthalmia is not mediated by changes in retinoic acid or sonic hedgehog signaling during retinal neurogenesis. Alcohol Clin Exp Res. 2011;35:1644–1661. doi: 10.1111/j.1530-0277.2011.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eberhart JK, Parnell SE. The Genetics of Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res. 2016 doi: 10.1111/acer.13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacobson SW, Carr LG, Croxford J, Sokol RJ, Li TK, Jacobson JL. Protective effects of the alcohol dehydrogenase-ADH1B allele in children exposed to alcohol during pregnancy. J Pediatr. 2006;148:30–37. doi: 10.1016/j.jpeds.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 79.Viljoen DL, Carr LG, Foroud TM, Brooke L, Ramsay M, Li TK. Alcohol dehydrogenase-2*2 allele is associated with decreased prevalence of fetal alcohol syndrome in the mixed-ancestry population of the Western Cape Province, South Africa. Alcohol Clin Exp Res. 2001;25:1719–1722. [PubMed] [Google Scholar]
- 80.Warren KR, Li TK. Genetic polymorphisms: impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res A Clin Mol Teratol. 2005;73:195–203. doi: 10.1002/bdra.20125. [DOI] [PubMed] [Google Scholar]
- 81.Das UG, Cronk CE, Martier SS, Simpson PM, McCarver DG. Alcohol dehydrogenase 2*3 affects alterations in offspring facial morphology associated with maternal ethanol intake in pregnancy. Alcohol Clin Exp Res. 2004;28:1598–1606. doi: 10.1097/01.alc.0000141816.14776.97. [DOI] [PubMed] [Google Scholar]
- 82.McCarver DG, Thomasson HR, Martier SS, Sokol RJ, Li T. Alcohol dehydrogenase-2*3 allele protects against alcohol-related birth defects among African Americans. J Pharmacol Exp Ther. 1997;283:1095–1101. [PubMed] [Google Scholar]
- 83.Boyles AL, DeRoo LA, Lie RT, Taylor JA, Jugessur A, Murray JC, Wilcox AJ. Maternal alcohol consumption, alcohol metabolism genes, and the risk of oral clefts: a population-based case-control study in Norway, 1996-2001. Am J Epidemiol. 2010;172:924–931. doi: 10.1093/aje/kwq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brocardo PS, Gil-Mohapel J, Christie BR. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res Rev. 2011;67:209–225. doi: 10.1016/j.brainresrev.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 85.Das SK, Vasudevan DM. Alcohol-induced oxidative stress. Life Sci. 2007;81:177–187. doi: 10.1016/j.lfs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 86.Hernandez JA, Lopez-Sanchez RC, Rendon-Ramirez A. Lipids and Oxidative Stress Associated with Ethanol-Induced Neurological Damage. Oxid Med Cell Longev. 2016;2016:1543809. doi: 10.1155/2016/1543809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wentzel P, Eriksson UJ. Ethanol-induced fetal dysmorphogenesis in the mouse is diminished by high antioxidative capacity of the mother. Toxicol Sci. 2006;92:416–422. doi: 10.1093/toxsci/kfl024. [DOI] [PubMed] [Google Scholar]
- 88.Chernoff GF. The fetal alcohol syndrome in mice: maternal variables. Teratology. 1980;22:71–75. doi: 10.1002/tera.1420220110. [DOI] [PubMed] [Google Scholar]
- 89.Gilliam DM, Irtenkauf KT. Maternal genetic effects on ethanol teratogenesis and dominance of relative embryonic resistance to malformations. Alcohol Clin Exp Res. 1990;14:539–545. doi: 10.1111/j.1530-0277.1990.tb01196.x. [DOI] [PubMed] [Google Scholar]
- 90.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 91.Bonthius DJ, Tzouras G, Karacay B, Mahoney J, Hutton A, McKim R, Pantazis NJ. Deficiency of neuronal nitric oxide synthase (nNOS) worsens alcohol-induced microencephaly and neuronal loss in developing mice. Brain Res Dev Brain Res. 2002;138:45–59. doi: 10.1016/s0165-3806(02)00458-3. [DOI] [PubMed] [Google Scholar]
- 92.Bonthius DJ, Jr., Winters Z, Karacay B, Bousquet SL, Bonthius DJ. Importance of genetics in fetal alcohol effects: null mutation of the nNOS gene worsens alcohol-induced cerebellar neuronal losses and behavioral deficits. Neurotoxicology. 2015;46:60–72. doi: 10.1016/j.neuro.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karacay B, Mahoney J, Plume J, Bonthius DJ. Genetic absence of nNOS worsens fetal alcohol effects in mice. II: microencephaly and neuronal losses. Alcohol Clin Exp Res. 2015;39:221–231. doi: 10.1111/acer.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCarthy N, Wetherill L, Lovely CB, Swartz ME, Foroud TM, Eberhart JK. Pdgfra protects against ethanol-induced craniofacial defects in a zebrafish model of FASD. Development. 2013;140:3254–3265. doi: 10.1242/dev.094938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet. 2008;40:290–298. doi: 10.1038/ng.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- 97.Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- 98.Rattanasopha S, Tongkobpetch S, Srichomthong C, Siriwan P, Suphapeetiporn K, Shotelersuk V. PDGFRa mutations in humans with isolated cleft palate. Eur J Hum Genet. 2012;20:1058–1062. doi: 10.1038/ejhg.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell. 2002;2:103–113. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- 100.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swartz ME, Wells MB, Griffin M, McCarthy N, Lovely CB, McGurk P, Rozacky J, Eberhart JK. A screen of zebrafish mutants identifies ethanol-sensitive genetic loci. Alcohol Clin Exp Res. 2014;38:694–703. doi: 10.1111/acer.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011;35:469–478. doi: 10.1002/gepi.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Costa LG, Guizzetti M, Burry M, Oberdoerster J. Developmental neurotoxicity: do similar phenotypes indicate a common mode of action? A comparison of fetal alcohol syndrome, toluene embryopathy and maternal phenylketonuria. Toxicol Lett. 2002;127:197–205. doi: 10.1016/s0378-4274(01)00501-x. [DOI] [PubMed] [Google Scholar]
- 104.Toutant C, Lippmann S. Fetal solvents syndrome. Lancet. 1979;1:1356. doi: 10.1016/s0140-6736(79)91997-4. [DOI] [PubMed] [Google Scholar]
- 105.Wilkins-Haug l. Teratogen update: toluene. Teratology. 1997;55:145–151. doi: 10.1002/(SICI)1096-9926(199702)55:2<145::AID-TERA5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 106.Pearson MA, Hoyme HE, Seaver LH, Rimsza ME. Toluene embryopathy: delineation of the phenotype and comparison with fetal alcohol syndrome. Pediatrics. 1994;93:211–215. [PubMed] [Google Scholar]
- 107.Darling KW, Ackerman SL, Hiatt RH, Lee SS, Shim JK. Enacting the molecular imperative: How gene-environment interaction research links bodies and environments in the post-genomic age. Soc Sci Med. 2016;155:51–60. doi: 10.1016/j.socscimed.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen L, Zhou S. “CRASH”ing with the worm: insights into L1CAM functions and mechanisms. Dev Dyn. 2010;239:1490–1501. doi: 10.1002/dvdy.22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arevalo E, Shanmugasundararaj S, Wilkemeyer MF, Dou X, Chen S, Charness ME, Miller KW. An alcohol binding site on the neural cell adhesion molecule L1. Proc Natl Acad Sci U S A. 2008;105:371–375. doi: 10.1073/pnas.0707815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dou X, Wilkemeyer MF, Menkari CE, Parnell SE, Sulik KK, Charness ME. Mitogen-activated protein kinase modulates ethanol inhibition of cell adhesion mediated by the L1 neural cell adhesion molecule. Proc Natl Acad Sci U S A. 2013;110:5683–5688. doi: 10.1073/pnas.1221386110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Balaraman S, Tingling JD, Tsai PC, Miranda RC. Dysregulation of microRNA expression and function contributes to the etiology of fetal alcohol spectrum disorders. Alcohol Res. 2013;35:18–24. [PMC free article] [PubMed] [Google Scholar]
- 112.Mead EA, Sarkar DK. Fetal alcohol spectrum disorders and their transmission through genetic and epigenetic mechanisms. Front Genet. 2014;5:154. doi: 10.3389/fgene.2014.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sen A, Cingolani P, Senut MC, Land S, Mercado-Garcia A, Tellez-Rojo MM, Baccarelli AA, Wright RO, Ruden DM. Lead exposure induces changes in 5-hydroxymethylcytosine clusters in CpG islands in human embryonic stem cells and umbilical cord blood. Epigenetics. 2015;10:607–621. doi: 10.1080/15592294.2015.1050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mora-Zamorano FX, Klingler R, Murphy CA, Basu N, Head J, Carvan MJ., 3rd Parental Whole Life Cycle Exposure to Dietary Methylmercury in Zebrafish (Danio rerio) Affects the Behavior of Offspring. Environ Sci Technol. 2016;50:4808–4816. doi: 10.1021/acs.est.6b00223. [DOI] [PubMed] [Google Scholar]
- 115.Xu X, Weber D, Martin A, Lone D. Trans-generational transmission of neurobehavioral impairments produced by developmental methylmercury exposure in zebrafish (Danio rerio). Neurotoxicol Teratol. 2016;53:19–23. doi: 10.1016/j.ntt.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 116.Baker TR, Peterson RE, Heideman W. Using zebrafish as a model system for studying the transgenerational effects of dioxin. Toxicol Sci. 2014;138:403–411. doi: 10.1093/toxsci/kfu006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lombo M, Fernandez-Diez C, Gonzalez-Rojo S, Navarro C, Robles V, Herraez MP. Transgenerational inheritance of heart disorders caused by paternal bisphenol A exposure. Environ Pollut. 2015;206:667–678. doi: 10.1016/j.envpol.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 118.Volkova K, Reyhanian Caspillo N, Porseryd T, Hallgren S, Dinnetz P, Olsen H, Porsch Hallstrom I. Transgenerational effects of 17alpha-ethinyl estradiol on anxiety behavior in the guppy, Poecilia reticulata. Gen Comp Endocrinol. 2015;223:66–72. doi: 10.1016/j.ygcen.2015.09.027. [DOI] [PubMed] [Google Scholar]
- 119.Vignet C, Joassard L, Lyphout L, Guionnet T, Goubeau M, Le Menach K, Brion F, Kah O, Chung BC, Budzinski H, et al. Exposures of zebrafish through diet to three environmentally relevant mixtures of PAHs produce behavioral disruptions in unexposed F1 and F2 descendant. Environ Sci Pollut Res Int. 2015;22:16371–16383. doi: 10.1007/s11356-015-4157-8. [DOI] [PubMed] [Google Scholar]