

Abstract
We previously developed a pH-responsive copper-doxorubicin (CuDox) cargo in lysolipid-based temperature-sensitive liposomes (LTSLs). The CuDox complex is released from the particle by elevated temperature; however, full release of doxorubicin from CuDox requires a reduced pH, such as that expected in lysosomes. The primary goal of this study is to evaluate the cellular uptake and intracellular trafficking of the drug-metal complex in comparison with intact liposomes and free drug. We found that the CuDox complex was efficiently internalized by mammary carcinoma cells after release from LTSLs. Intracellular doxorubicin and copper were 6-fold and 5-fold greater, respectively, after a 0.5 h incubation with the released CuDox complex, as compared to incubation with intact liposomes containing the complex. Total cellular doxorubicin fluorescence was similar following CuDox and free doxorubicin incubation. Imaging and mass spectrometry assays indicated that the CuDox complex was initially internalized intact but breaks down over time within cells, with intracellular copper decreasing more rapidly than intracellular doxorubicin. Doxorubicin fluorescence was reduced when complexed with copper, and nuclear fluorescence was reduced when cells were incubated with the CuDox complex as compared with free doxorubicin. Therapeutic efficacy, which typically results from intercalation of doxorubicin with DNA, was equivalent for the CuDox complex and free doxorubicin and was superior to that of liposomal doxorubicin formulations. Taken together, the results suggest that quenched CuDox reaches the nucleus and remains efficacious. In order to design protocols for the use of these temperature-sensitive particles in cancer treatment, the timing of hyperthermia relative to drug administration must be examined. When cells were heated to 42°C prior to the addition of free doxorubicin, nuclear drug accumulation increased by 1.8-fold in cancer cells after 5 hours, and cytotoxicity increased 1.4-fold in both cancer and endothelial cells. Endothelial cytotoxicity was similarly augmented with mild hyperthermia applied prior to treatment with released CuDox. In summary, we find that the drug-metal complex formed in temperature-sensitive particles can be internalized by cancer and endothelial cells resulting in therapeutic efficacy that is similar to free doxorubicin, and this efficacy can be enhanced by elevated temperature.
Keywords: Doxorubicin, Liposome, Intracellular trafficking, Mild hyperthermia
Graphical abstract
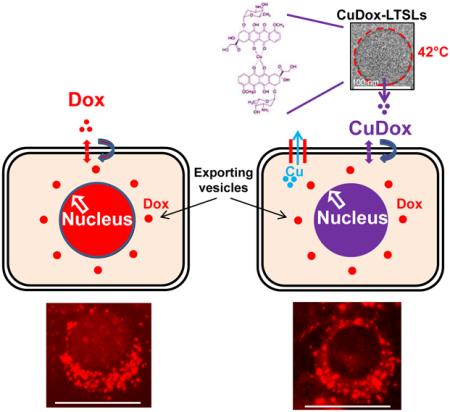
1. Introduction
Triggered release of chemotherapy from activatable drug carriers can provide an effective cancer treatment [1-8]. We have demonstrated that combining temperature-sensitive liposomes (TSLs) with ultrasound hyperthermia can produce a complete response in local cancer, with drug released in the tumor vasculature and interstitium impacting both endothelial and tumor cells [9, 10]. TSLs are typically formulated with bilayer-forming phospholipids and acyl moieties that melt between 40 and 42°C to facilitate cargo release under mild hyperthermia [11-14]. The permeable lipid shell limits circulation of the loaded particle to a few hours, even after coating with PEG lipids to extend systemic circulation [15, 16].
Stable circulation of particles is important to minimize systemic toxicity [17]. Active loading methods that result in drug precipitation or the formation of transition metal complexation can augment drug loading and further stabilize liposomal cargo [18]. Understanding the structure of such complexes within and outside of the liposomes and the functional implications of such structures remains an area of active investigation [19]. We previously demonstrated that complexation of doxorubicin (Dox) with copper in long-circulating liposomes composed of HSPC:DSPE-PEG2k:cholesterol (56:5:39, percent mole ratio) extended drug retention in blood, maintained the therapeutic effect and, more importantly, reduced drug accumulation in sensitive organs such as the heart [20]. Such a drug-metal complex is stable at physiological pH and releases the drug at the acidic pH associated with lysosomes. A further rationale for creation of a copper-doxorubicin (CuDox) complex inside liposomes prior to administration is to reduce the formation of free radicals resulting from the interaction of drug with metal ions in vivo, a primary mechanism proposed for the cardiac toxicity of Dox [21]. We leveraged the metal-drug complexation as a means to stabilize amphipathic Dox in a permeable lysolipid-containing temperature-activatable lipid formulation composed of DPPC:DSPE-PEG2k:MPPC (86:4:10, percent mole ratio) and capable of rapid release of drug [9, 10]. Overall, we found that the resulting lysolipid-containing TSLs encapsulating CuDox could create a durable and complete response and can be augmented with immunotherapy to treat multi-focal cancers [9, 10, 20].
Here, to further clarify the cellular trafficking mechanisms underlying the observed therapeutic efficacy of the drug-metal complex when released from TSLs, we examine whether the intact CuDox complex crosses the cell membrane and efficiently traffics within the cell. The inherent fluorescence of Dox allows us to visualize the cellular uptake and internalization process by both confocal and fluorescence microscopy. Additionally, Dox fluorescence is reduced when complexed with copper, facilitating studies of the free drug and the CuDox complex. Taking into account that interactions with cellular components alter the Dox fluorescence signal and can compromise accurate quantification, we measure Dox accumulation in whole cells and isolated subcellular fractions after restoration of Dox fluorescence.
Given that our goal is to create a temperature-sensitive particle and that hyperthermia will be employed to release the cargo, we also investigate the effect of the local temperature on efficacy. Free and liposomal Dox preparations, with or without copper complexation, are heated to release the drug, and the cellular uptake and intracellular trafficking are compared with those of non-heated drug treatments. Since mild hyperthermia alone may alter drug properties, or enhance drug internalization and cancer cell drug sensitivity [22, 23], we investigate the application of hyperthermic temperatures to both drug and cells.
2. Materials and methods
Further description of experimental procedures is found in the Supplementary Information.
2.1 Preparation of liposomal Dox
Lysolipid-containing temperature-sensitive liposomes (LTSLs) were formulated from DPPC:DSPE-PEG2k:MPPC (86:4:10, molar ratio). Liposomes were prepared as described previously [9, 10, 20]. Dox was added to ammonium sulfate-loaded (AS-LTSLs) or copper-loaded liposomes (Cu-LTSLs) to achieve a drug-to-lipid ratio of 0.2:1 (wt/wt) at 37°C overnight. Unencapsulated Dox was then separated from Dox-encapsulated liposomes using Sephadex G-75 spin columns with the appropriate buffer as described in the Supplementary Information. The purified liposomal Dox preparations from the AS (ASDox-LTSLs) and Cu-gluconate (CuDox-LTSLs) loading methods were concentrated using a Centrisart I centrifugal filtration device (100,000 Da cut-off, Sartorius AG., Gottingen, Germany). Copper liposomes prepared with 100 mM copper in triethanolamine (TEA) concentrations of 270, 540, and 810 mM created CuDox-LTSLs with intraliposomal pH of 7.4 (CuDox-LTSLs7.4), 8.4 (CuDox-LTSLs), and 8.8 (CuDox-LTSLs8.8), respectively. Loaded drug was released from liposomes after incubation of liposomes in the presence of 0.25% Triton X-100. Copper and Dox were dissociated using either 30 mM ethylenediaminetetraacetic acid (EDTA) at 55°C for 1.5 h or 20 mM citrate-buffered saline, pH 4.0 at 55°C, for 30 min. Dox concentration was quantified by measuring fluorescence intensity using a Tecan Infinite® M1000 Microplate Reader at excitation and emission wavelengths of 485 nm and 590 nm, respectively. Under the above-mentioned conditions, complete loading was achieved with a total of 0.2 mg Dox in 1 mg lipid when using 100 mM Cu-gluconate.
2.2 Copper transchelation and stability of copper-Dox complex in vitro
The CuDox complex was formed outside of liposomes by the addition of 15 μl of 100 mM copper gluconate to 75 μl of 2 mg/ml Dox solution in saline and incubation at 37°C. To this complex, we then added 1.5 ml of aqueous solutions of 0.5 mM bovine serum albumin (BSA, Sigma, St. Louis, MO) at pH values of 3-8, preincubated at 37°C to make the final concentrations of 0.172 mM Dox and 1 mM copper. Fluorescence intensity of free Dox was monitored over time at Ex: 485 nm and Em: 590 nm at 37°C and compared to that of free Dox solution in saline. The measured fluorescence intensities were corrected for variations in Dox fluorescence intensity with pH.
2.3 Cellular uptake and intracellular trafficking
The neu deletion (NDL) [24-26] and MET-1 [27] murine metastatic mammary carcinoma cell lines were obtained from the Borowsky Laboratory (UC Davis). Human malignant melanoma (H1 Melanoma) cells were obtained from the Thorsen laboratory (University of Bergen, Norway) [28]. Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD).
Cells were plated at 7 × 105 cells in 35-mm tissue culture dishes and used 48 h later at a confluency of 85%. For all cell experiments, drug treatments, washes and further incubations at 37°C were performed in phenol red-free media. Cells were treated with 20 μg of either free Dox or liposomal Dox in 1 ml of media (~34 μM). Briefly, cells and the drug treatments were separately incubated on ice for 30 min.
The culture media was removed and cold drug treatments in media or cold media alone (NT Control) were added to the cells and incubated on ice for another 30 min. Cold incubation was used to suppress the rapid internalization of drug, which normally occurs at physiological temperature, and to maximize adsorption and accumulation of free Dox, released CuDox or liposomal Dox onto the plasma membrane. The drug treatments and media were then removed and all treated and control cells were then washed two times with cold media replenished with fresh media and incubated at 37°C in a humidified 5% CO2 incubator to resume normal metabolism. Internalization of the adsorbed drug into the cytoplasm and subcellular distribution of the drug was then observed over time. In a subset of experiments, drug trafficking was also studied under reduced pH conditions by adjusting the pH of the media from 7.3 to 6.3 via addition of hydrochloric acid. For these experiments, all drug incubations, rinses and subsequent 37°C incubations were performed in pH 6.3 media.
The cellular uptake and intracellular trafficking of Dox were followed with a Zeiss LSM5 PASCAL confocal microscopy (40x objective; Carl Zeiss, Thornwood, NY). The cells were visualized live at the excitation wavelength of 488 nm and emission wavelength of 543 nm. Z-stacks of 8 images with a 0.5 μm thickness were recorded. The images were processed using Zeiss AIM Viewer, and relative accumulation of Dox in the nucleus or other intracellular compartments was quantified by identifying cellular regions-of-interest (ROIs) and measuring Dox fluorescence signal intensity using Image J software. At the indicated time points, NDL cells were labeled with LysoTracker® Blue DND-22 (Invitrogen, Inc., Carlsbad, CA) to stain lysosomes and with 4',6-diamidino-2-phenylindole (DAPI) to stain nuclei. For staining low-pH cellular compartments, such as lysosomes, media was removed and a solution of LysoTracker® Blue (5 μM final concentration in media) was added to live cells followed by incubation at 37°C for 30 min. The cells were washed two times in phosphate buffered saline (PBS) and fixed with a solution of 3% paraformaldehyde in PBS for 10 min at room temperature (20°C) and imaged at Ex: 373 nm and Em: 422 nm. To confirm nuclear drug accumulation and identify nuclear ROIs for Dox fluorescence quantification from images, cells were fixed as above and stained with DAPI at a final concentration of 600 nM at room temperature for 10 min. DAPI-stained nuclei were visualized at Ex: 358 nm and Em: 461 nm.
To eliminate the interaction of Dox with certain cellular components, such as DNA, proteins and lipids, which can have quenching or enhancing effects on Dox fluorescence, we also quantified the absolute fraction of free or liposomal Dox taken up by NDL cells after various incubation times using an acidified isopropanol protocol [22]. At the designated time points, cells were rinsed two times with cold media and detached by TrypLE™ Express (Invitrogen, Inc., Carlsbad, CA). A portion of the detached cells (~106 cells) in TrypLE™ Express were incubated with 25 μl of Triton X-100 (10% w/v) at room temperature for 15 min to permeabilize cellular membranes. To this cell suspension, 375 μl of acidified isopropanol solution (0.75 N HCl) was added to restore the fraction of quenched Dox fluorescence. The mixture was vortexed and incubated at -20°C overnight. A set of known serial dilutions of Dox was prepared in TrypLE™ Express and treated similarly to convert Dox fluorescence intensity into Dox concentration. After thawing, solutions were vortexed 5 min and Dox fluorescence intensity measured as described before and calculated as Dox concentration. A small portion of the detached cells for each treatment in TrypLE™ Express was also used to measure cell number using a hemocytometer. In some experiments, the detached cells in TrypLE™ Express were analyzed on a FACScan flow cytometer/Cell Quest software system (Beckon Dickson, San Jose, CA) to determine the cellular fluorescence of Dox.
2.4 Cell fractionation and isolation of nuclei
For isolation of nuclei, the protocol described by Nabbi and Riabowol [29] was followed with modifications. The entire procedure was performed on ice and ice-cold PBS (without Ca2+ and Mg2+) was used throughout. Cells (7-8 × 105) in 35-mm tissue culture dishes were rinsed once with ice-cold PBS and scraped off of the plate in 1 ml of PBS. The collected detached cells were pelleted at 10,000 rpm for 10 sec. The supernatant was removed and the cell pellet (~ 100 μl) was resuspended and lysed in 900 μl of PBS containing 0.2% Triton X-100. A fraction of 300 μl of the cell lysate was removed as the “Whole cell lysate” fraction. The remaining ~ 700 μl cell lysate was centrifuged at 10,000 rpm for 10 sec and a fraction of 300 μl of the supernatant was removed as the “Cytoplasmic” fraction. The remaining supernatant was discarded and 1 ml of PBS containing 0.1% Triton X-100 was added to the pellet. The suspension was again centrifuged at 10,000 rpm for 10 sec to pellet nuclei. The pellet was resuspended in 300 μl of PBS as the “Nuclei” fraction. The whole cell lysate and nuclei fractions were counted by hemocytometer and imaged at Ex: 488 nm and Em: 543 nm. To quantify Dox concentration in the fractions isolated from the cells treated with either free or liposomal Dox, 100 μl of each collected fraction were digested with a combination of 0.5% Triton X-100 and acidified isopropanol solution (0.75 N HCl) as described in section 2.3.
2.5 Cell viability measurements
Cells were plated at 4000 cells in 100 μl media per well in 96-well tissue culture plates 24 hours before the drug was added to the plates. Immediately prior to the drug treatment, plates were incubated for 30 min on ice. Media was removed and drug treatments added to each well in 100 μl cold media (n=10 wells/treatment). Plates were incubated for an additional 30 min on ice, rinsed twice with cold media and incubated for 24 h at 37°C in 5% CO2. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Invitrogen, Carlsbad, CA) was added to media at a concentration of 0.5 mg/ml, and cells were incubated 2 h at 37°C in a 5% CO2 incubator. Media was removed, and formazan crystals dissolved in 100 μl/well of DMSO (Sigma Aldrich, St. Louis, MO). Absorbance was measured using a Tecan (San Jose, CA) Infinite® M1000 microplate reader.
For drug IC-50 measurements, cells were plated at 2000 cells in 100 μl media per well in 96-well tissue culture plates. Plates were continuously incubated with free Dox in media at concentrations ranging between 0.0001 and 10 μM for 72 hours at 37°C in 5% CO2. Following a 72-h continuous drug incubation, cell viability was assessed by the MTT assay as described above. Best-fit curves and IC-50 values were calculated for concentration-response curves using GraphPad Prism software (GraphPad Inc., La Jolla, CA). Average IC-50 values were calculated from a minimum of three replicate experiments for each preparation.
2.6 Heat-activated release of CuDox complex and hyperthermia treatments involving Dox
In order to release the ASDox crystal and CuDox complex, liposomal Dox preparations, adjusted to pH 7.4, were heated at 42°C for 10 min either directly in media at 20 μg Dox/ml (~34 μM) or in buffer (saline or PBS) at 1 mg Dox/ml and then allowed to cool to 20°C. The released ASDox (Released ASDox), and released CuDox (Released CuDox) were then incubated separately on ice for 30 min. All treatments were applied at a final concentration of 20 μg Dox/ml (~34 μM) in cold media to cells that had been incubated on ice for 30 min prior to the addition of the drug. After the treatments were added to the cells, the combination was incubated on ice for an additional 30 min. Cells were then washed twice with cold media and transferred to a 37°C, 5% CO2 incubator.
2.7 Cell hyperthermia
In a separate experiment, NDL cells were heated to 42°C (hyperthermia, HT) before addition of free Dox. Cells were heated in media at 42°C for 0 (Free Dox) or 5 (Free Dox+Pre HT) or 10 and 15 min, cooled to 20°C for 5 min, and incubated on ice for 30 min. Cells were then incubated with cold free Dox in media for 30 min on ice, rinsed twice with cold media and transferred to a 37°C, 5% CO2 incubator.
After determination of optimal cellular pre-heating protocols in NDL cells, cell hyperthermia prior to addition of drug was also evaluated in HUVECs. For this experiment, HUVECs incubated in media, free Dox, CuDox-LTSLs or released CuDox were exposed to body temperature for 20 min (37°C), 42°C for 20 min (42°C), or 42°C for 5 min prior to and 20 min after drug exposure (42°C+Pre HT). All cells were then transferred to a 37°C, 5% CO2 incubator for 1 h, rinsed twice with media and incubated in media at 37°C for 24 h. Cell viability was assessed via MTT assay.
2.8 Inductively Coupled Plasma (quadrupole) Mass Spectrometry (ICP-MS)
Copper internalized by cells after incubation with liposomal copper, liposomal CuDox in LTSLs, released copper or CuDox was detected using Inductively Coupled Plasma (quadrupole) Mass Spectrometry (ICP-MS, Agilent Technologies, Santa Clara, CA) performed at the University of California, Davis/Interdisciplinary Center for Plasma Mass Spectrometry. For cell digestion, cells collected in 400 μl TrypLE™ Express were frozen in liquid nitrogen and lyophilized overnight. 100 μl of concentrated nitric acid (trace-metal-grade, 70%; Fisher Scientific, St. Louis, MO) was added to the dried samples and the mixture incubated for 2.5 h at 60°C. 100 μl of 30% hydrogen peroxide (Optima trace-metal-grade; Fisher Scientific, St. Louis, MO) was then added and the mixture incubated for 2 h at 55°C. To bring the samples to a final volume of 1 ml for analysis, 800 μl of distilled de-ionized water was then added.
2.9 Statistical analysis
Data points represent the average of triplicate measurements and the error bars are the standard deviations of the triplicate measurements. Statistical analysis between group-pairs and among multiple groups was performed using the two-tailed Student’s t-test assuming unequal variances, and one-way ANOVA followed by Tukey Post Hoc test, respectively. Statistical significance was set at p<0.05. The statistical differences are represented as * p<0.05, ** p<0.01, and *** p<0.001.
3. Results
3.1 CuDox complex is released by low pH and is stabilized by a greater intraliposomal pH
We first evaluated the stability of the released CuDox complex in a BSA solution designed to mimic the albumin concentration in blood. Using the described loading methods, copper formed a stable complex with Dox within liposomes resulting in quenched Dox fluorescence (Figure 1a). Once released and exposed to a reduced pH, the fluorescence of Dox increased, indicating a dissociation of Dox from copper (Figure 1a). Dox fluorescence was fully restored at pH values below 4, suggesting complete liberation of Dox. The released CuDox complex was stable for more than 24 h in the BSA solution.
Figure 1. Stability of CuDox complex at higher pH during loading and disruption by reduced pH after release.

CuDox liposomes were prepared with an intraliposomal pH of 7.4, 8.4, or 8.8 (CuDox-LTSLs7.4, CuDox-LTSLs, and CuDox-LTSLs8.8, respectively) and were compared with ASDox-LTSLs and free Dox. a) Dox fluorescence after disassociation of CuDox complex following exposure to BSA solutions with varied pH at 37°C. In a), CuDox complex was created by adding Dox to Cu/TEA solution (pH 7.4) in the absence of liposomes. Fluorescence intensity is presented as percent of the maximum fluorescence intensity of free Dox obtained at pH 3. b) Dox fluorescence after 30 min incubation of free drug or liposomal drugs with: HEPES/sodium chloride buffer at pH 7.4 and 20°C (RT), Triton X-100 in HEPES/ sodium chloride buffer at pH 7.4 and 42°C (TX, pH 7.4) or Triton X-100 in citrate/sodium chloride buffer at pH 4.0 and 42°C (TX, pH 4.0). c) Dox fluorescence of ASDox-LTSLs, and CuDox-LTSLs after 10 min incubation at 42°C at pH 4.0 in the absence of Triton X-100 as a percentage of the fluorescence measured with 0.25% Triton X-100 at pH 4.0. Release by heat was equivalent to release by Triton-X-100. d) CuDox-LTSLs were incubated in citric acid/NaCl buffer, pH 4.0, at 42°C to dissociate the released CuDox to free Dox. Drug release is presented as a percentage of the Dox fluorescence of free Dox at the same concentration under the same conditions. Statistical analyses were performed using one-way ANOVA followed by a Tukey Post Hoc test in (a) and (b) and using a Student’s t-test in (d). **p < 0.01, ***p < 0.001.
The stability of the CuDox complex formed within LTSLs was then assessed as a function of intraliposomal pH. Dox fluorescence was reduced for all intact liposomal Dox formulations as compared to free drug (Figure 1b). Upon lysing liposomes with Triton X-100 at pH 7.4, Dox fluorescence was fully restored for ASDox-LTSLs, but only partially restored for CuDox-LTSLs. Lysing CuDox liposomes at a pH of 4.0 fully released the drug and restored the fluorescence. When released at pH 7.4, CuDox dissociation at the time of release was greater for liposomes prepared at pH 7.4 than for those prepared at pH 8.4 and 8.8 (Figure 1b). Regardless of differences in intraliposomal condition, release by hyperthermia was equivalent to release by Triton X-100. All Dox-LTSLs released more than 90% of the encapsulated Dox upon heating at 42°C for 10 min, as assessed by fluorescence at a reduced buffer pH (Figure 1c, d). Although not shown, we tested stability of CuDox-LTSLs in vivo and found that the higher intraliposomal pH (8.4) was required to enhance drug retention in LTSLs in circulation, and therefore this pH was used to prepare liposomes for most of the studies reported here.
3.2. Liposomes traffic through lysosomes; free Dox traffics rapidly to the nucleus and is internalized more efficiently
We compared the cellular uptake and intracellular distribution of Dox for intact cells following a 30-min incubation on ice. Upon incubation at 37°C, free Dox fluorescence was detected in the nucleus, as demonstrated with DAPI nuclear staining at 24 h, and increased over time with a maximum after 24 h, (Supplementary Figure S1a, Figures 2a, b). Quantification of Dox fluorescence in the nucleus confirmed the suppression of membrane and intracellular trafficking of Dox at 4°C (Supplementary Figure S1b). Over time, a population of increasingly fluorescent punctate vesicle-like structures of similar shape and size were observed to spread into the cytoplasm away from the nucleus (Figure 2a (row 2)). Increasing the initial drug concentration also increased the nuclear accumulation of Dox (Supplementary Figure S1c-i) and significantly reduced cell viability as assessed by an MTT assay (Supplementary Figure S1c-ii). Nuclear Dox fluorescence was found to be highly correlated with viable cell number with a correlation coefficient of 0.99 (Supplementary Figure S1c-iii). Trafficking of free Dox in NDL cells was then evaluated in two other cancer cell lines: the mouse mammary carcinoma (MET-1) and human malignant melanoma (H1 Melanoma) cell lines. The IC-50 values of Dox obtained for NDL, MET-1, and H1 Melanoma cells were 0.011 ± 0.001, 0.068 ± 0.03, and 0.076 ± 0.008 μM, respectively. The increase in the nuclear accumulation of the drug over time was similar in all three cell lines with the highest values occurring after 24 h (data not shown). The appearance of fluorescent vesicles correlated with increased accumulation of drug in the nucleus of NDL and MET-1 cells, but was less pronounced in H1 Melanoma cells (data not shown).
Figure 2. Uptake and intracellular fate of free and liposomal Dox in NDL cells.

a) Confocal fluorescence images of NDL cells incubated with free Dox or liposomal drug (ASDox-LTSLs or CuDox-LTSLs). Cells were exposed to drug for 30 min at 4°C, washed and incubated with media for 15 min, 5 h and 24 h at 37°C (rows 1, 3-4). Fluorescence images of NDL cells treated with free Dox and co-stained with LysoTracker® Blue (red and green colors, respectively, row 2). Dox co-localized with LysoTracker® Blue appears yellow-orange. b) Time course of nuclear accumulation of drug for free Dox compared to liposomal Dox, presented as percent maximum nuclear fluorescence intensity of free Dox at 24 h. c) The total Dox associated with NDL cells after 30 min incubation with free or liposomal Dox at 4°C. Statistical analyses were performed using one-way ANOVA followed by a Tukey Post Hoc test. ***p < 0.001.
The uptake and intracellular distribution pattern of liposomal CuDox differed from that of free Dox and was difficult to visualize at early time points due to the quenched fluorescence of Dox (Figure 2a). In contrast to free Dox, nuclear accumulation was not observed at 15 min after treatment. Instead, Dox fluorescence was found in fine structures tightly packed in the perinuclear region, which co-localized with LysoTracker®. Nuclear accumulation was detected between 3 and 5 h after treatment, increased over 24 h (Figure 2b), and began to decline after 24 h (data not shown). For liposomal Dox, the maximum nuclear Dox accumulation was 10-20% of that quantified for free Dox (Figure 2b). Similar to free Dox treatment, following increased nuclear accumulation of drug, an increase in fluorescent punctate vesicle-like structures was observed. The outgoing fluorescent punctate vesicular structures were brighter, larger and more uniform in size compared to the incoming fine fluorescent structures observed in the perinuclear region immediately after addition of liposomal Dox (Figure 2a).
Further, 0.5 μg of the total 20 μg of free Dox applied to cells was associated with the cells after incubation on ice (Figure 2c), which is ~5-fold greater than the amount of Dox delivered by exposure to Dox-matched liposomal drug. Increasing the initial concentration of liposomes by 4-fold proportionally enhanced the amount of Dox associated with cells after ice incubation; however, the mass of Dox within cells exposed to 80 μg of liposomal Dox remained below that of cells exposed to 20 μg of free Dox (Figure 2c).
3.3. Released CuDox complex is internalized in a highly-efficient manner
We mimicked the in vivo thermal release of drug from activatable liposomes triggered by mild hyperthermia by heating CuDox-LTSLs at 42°C for 10 min and cooling to 4°C prior to incubation with cells. Cellular fluorescence resulting from incubation with the released CuDox complex was greater than that observed following incubation with the encapsulated CuDox complex (Figure 3a). As compared with free Dox, imaging demonstrated reduced nuclear fluorescence for cells incubated with the released CuDox complex; however, the fluorescence intensity was similar in punctate vesicle–like structures in the cytosol (Figures 3a). At later time points, cellular Dox fluorescence was similar for cells treated with free Dox and the released CuDox complex, as quantified by region-of-interest (ROI) analysis of each cell at 24 h after treatment (Figure 3b), and flow cytometry at the 24 h time point (Figure 3c). Further quantification of nuclear Dox fluorescence after 5 h via ROI image analysis revealed lower nuclear fluorescence for released CuDox-treated cells as compared to free-Dox treated cells (Figure 3d, p < 0.001).
Figure 3. Released CuDox accumulates efficiently in cancer cells.

a) Dox fluorescence images of adherent NDL cells in culture and isolated nuclei. Cells were treated with released CuDox, free Dox, or liposomal CuDox (CuDox-LTSL) and images were acquired 15 min, 5 h or 24 h after incubation at 37°C. b-c) NDL cellular fluorescence intensity in arbitrary units (AU) after 24 h incubation in media (NT Control) or treatments as above, and as quantified by (b) Image J or (c) flow cytometry. d) Nuclear Dox fluorescence across various treatments at 5 h via image-based fluorescence measurements of intact cells. e) Dox accumulation in whole cell lysate, isolated cytoplasm and isolated nuclei from cells treated with free Dox or released CuDox at 24 h. Dox quantification was performed in the presence of Triton X-100 and HCl-isopropanol. Statistical analyses were performed using one-way ANOVA followed by a Tukey Post Hoc test. ***p < 0.001, n.s. = not significant.
In isolated nuclei, quantification of Dox after restoration of Dox fluorescence confirmed the nucleus as the major subcellular destination of free Dox (Figure 3e). Following the separation of cellular components, we confirmed that Dox was lower in the nucleus and greater in the cytoplasm for CuDox-treated cells, each as compared with cells treated with free Dox (Figure 3e). Despite differences in the apparent subcellular drug distribution, total cellular Dox for free Dox and released CuDox treatments was again similar (Figure 3e).
3.4. Released CuDox is taken up by cells as an intact complex and dissociates within the cells
We then tracked the cellular uptake and fate of both copper and Dox within the same experiment in order to assess their disassociation. In this experiment, a combination of Triton X-100 and acidified isopropanol was used to dissociate Dox from cellular components in order to avoid any impact on Dox fluorescence. Given that the stability of the CuDox complex is reduced in an acidic environment, comparative studies were performed at pH 7.3 and 6.3, which was selected to mimic the acidic cancer tumor microenvironment (Figure 4).
Figure 4. Intracellular trafficking of Dox and copper in NDL cancer cells.

Cellular concentration of Dox and copper after treatment of NDL cells for 30 min on ice with: released CuDox, free Dox, or CuDox-LTSLs. Concentration of Dox (a, b) and copper (c, d) in cells incubated with various treatments and 0.5, 4 and 24 h incubation at 37°C in pH 7.3 media (a, c) or pH 6.3 media (b, d). Relative cellular Dox and copper concentrations from cells incubated with released CuDox (e, f) at 0.5, 4, 24 h after incubation at 37°C in pH 7.3 (e) and pH 6.3 (f) media. Statistical analyses were performed using one-way ANOVA followed by a Tukey Post Hoc test in a-d and Student’s t-test in e-f. *p < 0.05, **p < 0.01, ***p < 0.001.
At the early time point of 0.5 h, Dox fluorescence was greater for cells treated with released CuDox and free Dox as compared to intact CuDox-LTSLs; this difference was ~6- and 4-fold higher in neutral media and ~11- and 8-fold higher in pH 6.3 media, respectively (Figures 4a, b, p < 0.001). Similar trends for the relative Dox accumulation resulting from each treatment were observed with the two pH values although the accumulation of Dox was greater with the incubation at lower pH in all cases (Figure 4a, b). Although cellular Dox fluorescence was slightly higher for released CuDox than free Dox initially, the intensity was similar for equivalent treatments at later time points (Figures 4a, b), and the intensity gradually diminished over time for all treatments.
For copper, as measured by ICP-MS, intracellular copper was detected above background only in cells incubated with released CuDox (0.5, 4, 24 h) and with intact CuDox-LTSLs at 0.5 h; however, incubation with the released CuDox resulted in a much higher level of copper (~5- and 10-fold higher in pH 7.3 and 6.3 media, respectively) (Figures 4c, d, p < 0.001). Enhanced copper was not observed when cells were treated with the contents released from Cu-LTSLs (liposomal copper in LTSLs in the absence of Dox) (data not shown). Additionally, copper was not detected above background levels in NDL cells treated with free Dox (Figures 4c, d).
Copper was depleted from the cells at a faster rate than Dox (Figure 4e-f). Both the uptake and efflux of copper were greater in pH 6.3 media than neutral media, similar to the trend observed with Dox (Figures 4a-d). The experimental values obtained for depletion of cellular copper and Dox over time were fit with first-order kinetic and depletion constants were calculated as −0.028 s−1 and −0.057 s−1 in neutral pH and −0.043 s−1 and −0.11 s−1 in pH 6.3 media for Dox and copper, respectively (Figures 4e, f). Comparing the depletion constant rates indicates a two-fold faster depletion for copper than Dox. At the 0.5 h time point, the calculated intracellular molar ratio of copper to Dox was 1:13 in neutral media and 1:5 in lower-pH media, each of which is lower than the ratio at which the cells were initially treated (1:2).
3.5. Efficacy of free Dox and released CuDox
In order to predict the therapeutic protocol details that are likely to be most effective for temperature-sensitive delivery of Dox in vivo, we evaluated the effect of mild hyperthermia on the cells prior to drug incubation. We first evaluated trafficking of free drug in order to visualize nuclear fluorescence (which is initially quenched in the CuDox complex). Heating NDL cells to 42°C before adding free Dox at 4°C increased the nuclear Dox fluorescence by up to 1.8 fold (5 h after treatment) as compared to non-heated cells (p < 0.01, Supplementary Figures S2a-b and Figure 5a). Further, cytotoxicity was 1.4-fold higher in this pre-heated cohort as compared to non-heated control cells (p < 0.05, Figure 5b).
Figure 5. Timing and sequence of heat and free Dox exposure in NDL cells.

Effect of sequence of 42°C temperature (hyperthermia, HT) and free Dox exposure on (a) Dox trafficking into the nucleus as a function of time and (b) cytotoxicity after 24 h as quantified by MTT assay in NDL cells. Cells heated at 42°C and cooled before Dox incubation (Free Dox+Pre HT) were compared to cells incubated with Dox, but not exposed to elevated temperature (Free Dox). In each group, cells and free Dox were incubated on ice, rinsed 2x with cold media, and finally incubated at 37°C for 0, 3, 5 and 24 h. Statistical analyses were performed using one-way ANOVA followed by a Tukey Post Hoc test. *p < 0.05, **p < 0.01.
We next compared the efficacy of free Dox and released CuDox in human vascular endothelial cells (HUVECs) and NDL cells. Released CuDox exhibited similar cytotoxicity to that of free drug after 24 h in NDL cells (Figure 6a). Efficacy of the released CuDox was higher than that of encapsulated CuDox regardless of the loading pH (Figures 6a).
Figure 6. Cytotoxicity of Dox and released CuDox in NDL and vascular endothelial cells.

Cell viability following treatment with free Dox, liposomal CuDox loaded at intraliposomal pH 8.4 (CuDox-LTSLs) or loaded at varied intraliposomal pH (CuDox-LTSLs7.4 and CuDox-LTSLs8.8) and compared to released CuDox from the corresponding liposomal preparations described above. a) Viability of NDL cells at 24 h following treatment. Cell viability following all drug treatments was significantly lower than in the media-alone control (NT control). b) Viability of HUVECs 24 h after treatment. HUVECs were incubated in media (Media Only), free Dox, CuDox-LTSLs or released CuDox, exposed to body temperature for 20 min (37°C), 42°C for 20 min (42°C), or 42°C for 5 and 20 min prior to and after drug exposure, respectively (42°C+Pre HT). Cell viability was lower in all drug treated groups receiving the same hyperthermia protocol than in “Media Only” (p < 0.001). In (b), cell viability in all drug-treated groups was normalized to the corresponding “Media Only” control group. *p < 0.05, **p < 0.01, ***p < 0.001.
The efficacy of free Dox and released CuDox was also similar in HUVECs at 24 h after drug treatment (Figure 6b). Given that the CuDox complex is initially released within the vasculature as a result of hyperthermia, we also evaluated the viability of endothelial cells exposed to released CuDox following pre-treatment with hyperthermia. Across different drug concentrations, the viability of HUVECs was significantly reduced by applying mild hyperthermia prior to treatment with free Dox or released CuDox as compared to cells not exposed to hyperthermia (Figure 6b). The enhanced efficacy of free Dox after exposure to hyperthermia for 5 min prior to treatment is similar for NDL and endothelial cells (Figures 5b, 6b).
4. Discussion
We previously demonstrated the formation of an intraliposomal transition metal complex resulting from active loading of Dox in the presence of copper gluconate [20]. We chose to exploit this complex in both long circulating and lysolipid-containing temperature-sensitive liposomes (LTSLs) to extend the blood circulation of the drug, reduce its systemic toxicity, and facilitate tumor toxicity. Our previous results indicated that coupling CuDox thermally-sensitive particles with ultrasound hyperthermia produces a complete response in localized cancers [9]; however, the intracellular kinetics of the particles and released CuDox complex have not been established. Therefore, the cellular uptake and trafficking of CuDox encapsulated in or released from LTSLs was studied in NDL mammary carcinoma cells and endothelial cells. Most importantly, we found that the released CuDox complex is efficiently internalized by cancer and endothelial cells and retains efficacy.
While the central focus of this paper is to characterize a metal-drug complex (CuDox) that has been released from temperature-sensitive liposomes, comparisons with both free drug (Dox) and liposomal Dox trafficking required additional studies to quantify the differences in trafficking and efficacy of these alternative therapeutics. We emphasize that temperature-sensitive nanoparticles have the potential to stably carry a therapeutic to the region of interest and to rapidly release the cargo at the site, and therefore to avoid the limitations associated with the enhanced permeability and retention effect. Assessment of the cellular internalization of the released cargo, as compared with the nanoparticle or the native drug, is of high significance.
Cellular Internalization
Dox fluorescence is reduced when complexed with copper and we used this effect to study the differences in fluorescence over time for the various treatments. At later time points, total cellular fluorescence was equivalent for cells treated with released CuDox and free Dox. However, the nuclear fluorescence was consistently reduced for CuDox formulations as compared with free Dox. There are two potential reasons for this difference. CuDox may remain quenched in the nucleus or nuclear trafficking of CuDox may be reduced. Given the equivalent efficacy we have observed with released CuDox in vitro and CuDox-LTSLs in vivo, we hypothesize that CuDox remains quenched in the nucleus.
Measurements of intracellular copper were a second aspect of the study which confirmed that released CuDox is indeed internalized intact. Quantification of intracellular copper confirmed the internalization of copper at a significant level only in those cells treated with released CuDox. Mass spectrometry assays indicated that intracellular copper decreases more rapidly than intracellular Dox. The negligible amount of copper accumulated in cells treated with liposomal copper suggests that copper levels are tightly regulated in the cells [30] and that cellular internalization of copper is facilitated by Dox, perhaps via the flip-flop mechanism proposed for free Dox [31].
For the three cancer cell lines assessed here, free Dox fluorescence was first detected in the nucleus, as also reported in [32]; nuclear Dox fluorescence intensity has been correlated with drug cytotoxicity [33]. Free Dox has been reported to internalize via a flip-flop mechanism across the plasma membrane, rather than simple diffusion, and to undergo carrier-mediated translocation, such as via proteasomes, from the cytoplasm into the nucleus [31, 34, 35]. Both of these mechanisms are known to slow at low temperatures [36, 37]. In agreement with this proposed mechanism of transport, we found that free Dox trafficking across the plasma membrane and through the cytoplasm to the nucleus was dramatically suppressed by reducing membrane fluidity and cell metabolic activity during cold incubation.
Following incubation with liposomal Dox, fluorescence was first observed as a large population of small fluorescent importing lysosomes in the perinuclear region of the cell at 15 min after incubation, with nuclear fluorescence observed at the 5 h time point and reaching a plateau after 24 h. Thus, a distinct internalization pathway was observed for the liposomal Dox. A similar pattern of cellular trafficking and nuclear accumulation was observed for CuDox liposomes and Dox liposomes loaded via the ammonium sulfate method. Internalization of free Dox was more than 4-fold higher as compared to the liposomal drug, and the accumulation in the nucleus was much faster than liposomal Dox formulations (p < 0.001). Also as compared with treatment with liposomal CuDox preparations, internalization of released CuDox was more efficient. Intracellular Dox and copper were 6-fold and 5-fold greater, respectively, after a 0.5 h incubation with the released CuDox complex, as compared to incubation with intact liposomes containing the complex. However, this difference does not appear to be due to lysosomal entrapment of Dox, but rather differential cellular uptake of the liposomal drug compared to the free drug.
Later trafficking
At 24 h after treatment with all Dox formulations examined here, Dox fluorescence was associated with an increasing number of punctate vesicles, which appear to export the drug from the nucleus. These vesicles are larger than the drug-importing lysosomes observed at earlier time points. LysoTracker staining of lysosomes demonstrated the co-localization of Dox with a fraction of these lysosomes, which tended to spread into the cytoplasm and away from the nucleus over time. According to this and previously reported evidence, drug accumulation in the nucleus may trigger a cellular defense mechanism by which drug is effluxed from the nucleus and eventually from the cell [38-41].
Stability of the CuDox complex
We examined CuDox stability across various pH values in a BSA solution designed to mimic plasma. CuDox was stable above pH 7 for many hours, but rapidly dissociated to free Dox as the pH decreased below 7, with a complete liberation of free drug at pH 3, in agreement with literature reporting on the interaction of copper ions with Dox [42]. Therefore, the CuDox complex is anticipated to be stable in biological fluids and intracellular compartments with physiological pH and to liberate free drug in diseased tissues, tumors, and acidic intracellular compartments.
Efficacy
Although nuclear fluorescence was reduced when cells were incubated with the CuDox complex, as compared with free Dox, the released CuDox complex was efficacious. Given that the typical Dox therapeutic mechanism involves DNA intercalation, the results suggest that the CuDox complex traffics intact to the nucleus with fluorescence quenched by the accompanying copper ion. In contrast, the increasing number of punctate fluorescent vesicles in the cytoplasm over time supports the hypothesis that the nuclear CuDox complex is dissociated in drug-exporting lysosomes and that full fluorescence of Dox is observed only after the complex is exported from the nucleus to the low-pH vesicles. The mechanism underlying tumor cell death by released CuDox is not fully clear. A study using resonance Ramen spectroscopy has shown that copper (II)-Adriamycin can form a ternary complex with DNA [43]. Intercalation of Dox into DNA, as confirmed by X-ray studies [44, 45], followed by inhibition of the DNA synthesis or poisoning of topoisomerase II (TOP2A) [9, 10], is a major mechanism of Dox-induced cell death. Other mechanisms of cytotoxicity, including generation of free radicals leading to DNA and cell membrane damage, may also play a role [20, 46-48]. Therefore, efficacy resulting from a CuDox-DNA complex requires further investigation.
Development of an effective hyperthermia protocol
Inducing hyperthermia before treatment with free Dox enhanced nuclear Dox fluorescence by 1.8-fold and 1.4-fold after 5 h and 24 h, respectively, compared to cells maintained at 37°C (p < 0.01). The efficacy of free Dox and released CuDox were similarly enhanced in endothelial cells by the pre-application of mild hyperthermia. Kusumoto et al. reported a similar combinatorial heating-chemotherapeutic result with 2.75- and 1.64-fold greater cytotoxic effect of carboplatin and cisplatin against HeLa cells when cells were exposed to heat prior to drug administration compared to 0.67- and 0.92-fold change in cell viability for cells treated with drug before heat, respectively [23].
Our previous in vivo studies [9] indicated that elevating the tumor temperature prior to the addition of the activatable drug complex increased the treatment efficacy and resulted in enhanced extravasation of red blood cells within the tumor. Here, we demonstrate that one mechanism for the enhanced efficacy is the increased cellular internalization of the released CuDox complex. Other mechanisms for enhanced tumor efficacy in vivo include increased tumor blood flow and vascular permeability. We previously employed this strategy in vivo to treat aggressive syngeneic breast cancer in a murine model using ultrasound hyperthermia and achieved elimination of directly-treated tumor lesions with minimal systemic toxicity [9, 10].
5. Conclusions
Our overall goal is to create activatable liposomes with high serum stability and efficacy. In order to increase serum stability, a metal-drug complex (CuDox) was created. When CuDox was liberated from temperature-sensitive liposomes, the complex was efficiently internalized by cancer cells, with the metal and drug dissociating over time within the cells. The CuDox complex exerted similar cytotoxicity to that of free Dox. Moreover, heating human endothelial and murine NDL mammary cancer cells prior to addition of either Dox or released CuDox resulted in enhanced cellular sensitization to drug and thus overall cytotoxicity.
Supplementary Material
Acknowledgements
Funding was provided by NIHR01CA134659 and NIHCA199658. We appreciate the technical assistance of Austin M. Cole and Gry Hoffmann Barfod, University of California, Davis, Interdisciplinary Center for Plasma Mass Spectrometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kong G, Anyarambhatla G, Petros WP, Braun RD, Colvin OM, Needham D, Dewhirst MW. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Research. 2000;60:6950–6957. [PubMed] [Google Scholar]
- [2].Ponce AM, Viglianti BL, Yu D, Yarmolenko PS, Michelich CR, Woo J, Bally MB, Dewhirst MW. Magnetic resonance imaging of temperature-sensitive liposome release: Drug dose painting and antitumor effects. Journal of the National Cancer Institute. 2007;99:53–63. doi: 10.1093/jnci/djk005. [DOI] [PubMed] [Google Scholar]
- [3].Poon RTP, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin. Pharmacother. 2009;10:333–343. doi: 10.1517/14656560802677874. [DOI] [PubMed] [Google Scholar]
- [4].Poon RTP, Borys N. Lyso-thermosensitive liposomal doxorubicin: an adjuvant to increase the cure rate of radiofrequency ablation in liver cancer. Future Oncology. 2011;7:937–945. doi: 10.2217/fon.11.73. [DOI] [PubMed] [Google Scholar]
- [5].Staruch R, Chopra R, Hynynen K. In: Matsumoto Y, Crum LA, TerHaar GR, editors. Investigations into Thermally Mediated Drug Delivery Using a Preclinical System for MRI-Guided Focused Ultrasound; 10th International Symposium on Therapeutic Ultrasound.2011. pp. 163–167. [Google Scholar]
- [6].Staruch R, Chopra R, Hynynen K. Localised drug release using MRI-controlled focused ultrasound hyperthermia. International Journal of Hyperthermia. 2011;27:156–171. doi: 10.3109/02656736.2010.518198. [DOI] [PubMed] [Google Scholar]
- [7].Wood BJ, Poon RT, Locklin JK, Dreher MR, Ng KK, Eugeni M, Seidel G, Dromi S, Neennan Z, Kolf M, Black CDV, Prabhakar R, Libutti SK. Phase I Study of Heat-Deployed Liposomal Doxorubicin during Radiofrequency Ablation for Hepatic Malignancies. Journal of Vascular and Interventional Radiology. 2012;23:248–255. doi: 10.1016/j.jvir.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ponce AM, Vujaskovic Z, Yuan F, Needham D, Dewhirst MW. Hyperthermia mediated liposomal drug delivery. International Journal of Hyperthermia. 2006;22:205–213. doi: 10.1080/02656730600582956. [DOI] [PubMed] [Google Scholar]
- [9].Kheirolomoom A, Lai CY, Tam SM, Mahakian LM, Ingham ES, Watson KD, Ferrara KW. Complete regression of local cancer using temperature-sensitive liposomes combined with ultrasound-mediated hyperthermia. J. Control. Release. 2013;172:266–273. doi: 10.1016/j.jconrel.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kheirolomoom A, Ingham ES, Mahakian LM, Tam SM, Silvestrini MT, Tumbale SK, Foiret J, Hubbard NE, Borowsky AD, Murphy WJ, Ferrara KW. CpG expedites regression of local and systemic tumors when combined with activatable nanodelivery. J. Control. Release. 2015;220:253–264. doi: 10.1016/j.jconrel.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anyarambhatla GR, Needham D. Enhancement of the phase transition permeability of DPPC liposomes by incorporation of MPPC: A new temperature-sensitive liposome for use with mild hyperthermia. J. Liposome Res. 1999;9:491–506. [Google Scholar]
- [12].Ickenstein LM, Arfvidsson MC, Needham D, Mayer LD, Edwards K. Disc formation in cholesterol-free lippsornes during phase transition. Biochimica Et Biophysica Acta-Biomembranes. 2003;1614:135–138. doi: 10.1016/s0005-2736(03)00196-2. [DOI] [PubMed] [Google Scholar]
- [13].Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Research. 2000;60:1197–1201. [PubMed] [Google Scholar]
- [14].Sandstrom MC, Ickenstein LM, Mayer LD, Edwards K. Effects of lipid segregation and lysolipid dissociation on drug release from thermosensitive liposomes. J. Control. Release. 2005;107:131–142. doi: 10.1016/j.jconrel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- [15].de Smet M, Heijman E, Langereis S, Hijnen NM, Grull H. Magnetic resonance imaging of high intensity focused ultrasound mediated drug delivery from temperature-sensitive liposomes: An in vivo proof-of-concept study. J. Control. Release. 2011;150:102–110. doi: 10.1016/j.jconrel.2010.10.036. [DOI] [PubMed] [Google Scholar]
- [16].Paoli EE, Kruse DE, Seo JW, Zhang H, Kheirolomoom A, Watson KD, Chiu P, Stahlberg H, Ferrara KW. An optical and microPET assessment of thermally-sensitive liposome biodistribution in the Met-1 tumor model: Importance of formulation. J. Control. Release. 2010;143:13–22. doi: 10.1016/j.jconrel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Waterhouse DN, Tardi PG, Mayer LD, Bally MB. A comparison of liposomal formulations of doxorubicin with drug administered in free form: Changing toxicity profiles. Drug Safety. 2001;24:903–920. doi: 10.2165/00002018-200124120-00004. [DOI] [PubMed] [Google Scholar]
- [18].Gubernator J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opinion on Drug Delivery. 2011;8:565–580. doi: 10.1517/17425247.2011.566552. [DOI] [PubMed] [Google Scholar]
- [19].Schilt Y, Berman T, Wei X, Barenholz Y, Raviv U. Using solution X-ray scattering to determine the high-resolution structure and morphology of PEGylated liposomal doxorubicin nanodrugs. Biochimica et Biophysica Acta (BBA) - General Subjects. 2016;1860:108–119. doi: 10.1016/j.bbagen.2015.09.012. [DOI] [PubMed] [Google Scholar]
- [20].Kheirolomoom A, Mahakian LM, Lai CY, Lindfors HA, Seo JW, Paoli EE, Watson KD, Haynam EM, Ingham ES, Xing L, Cheng RH, Borowsky AD, Cardiff RD, Ferrara KW. Copper-Doxorubicin as a Nanoparticle Cargo Retains Efficacy with Minimal Toxicity. Mol. Pharm. 2010;7:1948–1958. doi: 10.1021/mp100245u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, Sawyer DB. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. 2008;26:3777–3784. doi: 10.1200/JCO.2007.14.9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Laginha KM, Verwoert S, Charrois GJR, Allen TM. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin. Cancer Res. 2005;11:6944–6949. doi: 10.1158/1078-0432.CCR-05-0343. [DOI] [PubMed] [Google Scholar]
- [23].Kusumoto T, Maehara Y, Baba H, Takahashi I, Kusumoto H, Ohno S, Sugimachi K. Sequence dependence of the hyperthermic potentiation of carboplatin-induced cytotoxicity and intracellular platinum accumulation in Hela-cells. British Journal of Cancer. 1993;68:259–263. doi: 10.1038/bjc.1993.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Miller JK, Shattuck DL, Ingalla EQ, Yen LL, Borowsky AD, Young LJT, Cardiff RD, Carraway KL, Sweeney C. Suppression of the Negative Regulator LRIG1 Contributes to ErbB2 Overexpression in Breast Cancer. Cancer Research. 2008;68:8286–8294. doi: 10.1158/0008-5472.CAN-07-6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. Embo Journal. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cardiff RD, Hubbard NE, Engelberg JA, Munn RJ, Miller CH, Walls JE, Chen JQ, Velasquez-Garcia HA, Galvez JJ, Bell KJ, Beckett LA, Li YJ, Borowsky AD. Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers. Lab. Invest. 2013;93:480–497. doi: 10.1038/labinvest.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Borowsky AD, Namba R, Young LJT, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff R, Gregg JP. Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior. Clinical & Experimental Metastasis. 2005;22:47–58. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- [28].Wang J, Daphu I, Pedersen PH, Miletic H, Hovland R, Mork S, Bjerkvig R, Tiron C, McCormack E, Micklem D, Lorens JB, Immervoll H, Thorsen F. A novel brain metastases model developed in immunodeficient rats closely mimics the growth of metastatic brain tumours in patients. Neuropathology and Applied Neurobiology. 2011;37:189–205. doi: 10.1111/j.1365-2990.2010.01119.x. [DOI] [PubMed] [Google Scholar]
- [29].Nabbi A, Riabowol K. Rapid isolation of nuclei from cells in vitro. Cold Spring Harbor Protocols. 2015 doi: 10.1101/pdb.prot083733. doi:10.1101/pdb.prot083733. [DOI] [PubMed] [Google Scholar]
- [30].Vulpe CD, Packman S. McCormick DB, editor. Cellular copper transport. Annual Review of Nutrition. 1995:293–322. doi: 10.1146/annurev.nu.15.070195.001453. [DOI] [PubMed] [Google Scholar]
- [31].Regev R, Eytan GD. Flip-flop of doxorubicin across erythrocyte and lipid membranes. Biochem. Pharmacol. 1997;54:1151–1158. doi: 10.1016/s0006-2952(97)00326-2. [DOI] [PubMed] [Google Scholar]
- [32].Coley HM, Amos WB, Twentyman PR, Workman P. Examination by laser-scanning confocal fluorescence imaging microscopy of the subcellular localization of anthracyclines in parent and multidrug-resistant cell-lines. British Journal of Cancer. 1993;67:1316–1323. doi: 10.1038/bjc.1993.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lopes de Menezes DE, Kirchmeier MJ, Gagne J-F, Pilarski LM, Allen TM. Cellular trafficking and cytotoxicity of anti-CD19-targeted liposomal doxorubicin in B lymphoma cells. J. Liposome Res. 1999;9:199–228. [Google Scholar]
- [34].Kiyomiya K, Matsuo S, Kurebe M. Proteasome is a carrier to translocate doxorubicin from cytoplasm into nucleus. Life Sciences. 1998;62:1853–1860. doi: 10.1016/s0024-3205(98)00151-9. [DOI] [PubMed] [Google Scholar]
- [35].Liu J, Zheng H, Tang M, Ryu Y-C, Wang X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. American Journal of Physiology-Heart and Circulatory Physiology. 2008;295:H2541–H2550. doi: 10.1152/ajpheart.01052.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bemporad D, Luttmann C, Essex JW. Computer simulation of small molecule permeation across a lipid bilayer: Dependence on bilayer properties and solute volume, size, and cross-sectional area. Biophys. J. 2004;87:1–13. doi: 10.1529/biophysj.103.030601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kampf JP, Cupp D, Kleinfeld AM. Different mechanisms of free fatty acid flip-flop and dissociation revealed by temperature and molecular species dependence of transport across lipid vesicles. Journal of Biological Chemistry. 2006;281:21566–21574. doi: 10.1074/jbc.M602067200. [DOI] [PubMed] [Google Scholar]
- [38].Gerasimenko JV, Gerasimenko OV, Petersen OH. Membrane repair: Ca2+-ellicited lysosomal exocytosis. Current Biology. 2001;11:R971–R974. doi: 10.1016/s0960-9822(01)00577-2. [DOI] [PubMed] [Google Scholar]
- [39].Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. Journal of Immunology. 2006;177:7849–7857. doi: 10.4049/jimmunol.177.11.7849. [DOI] [PubMed] [Google Scholar]
- [40].Minko T. HPMA copolymers for modulating cellular signaling and overcoming multidrug resistance. Adv. Drug Deliv. Rev. 2010;62:192–202. doi: 10.1016/j.addr.2009.12.002. [DOI] [PubMed] [Google Scholar]
- [41].Offenhaeuser C, Lei N, Roy S, Collins BM, Stow JL, Murray RZ. Syntaxin 11 Binds Vti1b and Regulates Late Endosome to Lysosome Fusion in Macrophages. Traffic. 2011;12:762–773. doi: 10.1111/j.1600-0854.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- [42].Greenaway FT, Dabrowiak JC. The Binding of Copper Ions to Daunomycin and Adriamycin. J. Inorg. Biochem. 1982;16:91–107. [Google Scholar]
- [43].Dutta PK, Hutt JA. Resonance raman-spectroscopic studies of adriamycin and copper(ii) adriamycin and copper(ii) adriamycin DNA complexes. Biochemistry. 1986;25:691–695. doi: 10.1021/bi00351a028. [DOI] [PubMed] [Google Scholar]
- [44].Frederick CA, Williams LD, Ughetto G, Vandermarel GA, Vanboom JH, Rich A, Wang AHJ. Structural comparison of anticancer drug DNA complexes-adriamycin and daunomycin Biochemistry. 1990;29:2538–2549. [PubMed] [Google Scholar]
- [45].Yang XL, Wang AHJ. Structural studies of atom-specific anticancer drugs acting on DNA. Pharmacology & Therapeutics. 1999;83:181–215. doi: 10.1016/s0163-7258(99)00020-0. [DOI] [PubMed] [Google Scholar]
- [46].Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007;23:15–25. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- [47].Dreher MR, Raucher D, Balu N, Colvin OM, Ludeman SM, Chilkoti A. Evaluation of an elastin-like polypeptide-doxorubicin conjugate for cancer therapy. J. Control. Release. 2003;91:31–43. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- [48].Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.