

Abstract
The liver maintains an immunologically tolerant environment as a result of continuous exposure to food and bacterial constituents from the digestive tract. Hepatotropic pathogens can take advantage of this niche and establish lifelong chronic infections causing hepatic fibrosis and hepatocellular carcinoma. Macrophages (Mϕ) play a critical role in regulation of immune responses to hepatic infection and regeneration of tissue. However, the factors crucial for Mϕ in limiting hepatic inflammation or resolving liver damage have not been fully understood. In this report, we demonstrate that expression of C‐type lectin receptor scavenger receptor‐AI (SR‐AI) is crucial for promoting M2‐like Mϕ activation and polarization during hepatic inflammation. Liver Mϕ uniquely up‐regulated SR‐AI during hepatotropic viral infection and displayed increased expression of alternative Mϕ activation markers, such as YM‐1, arginase‐1, and interleukin‐10 by activation of mer receptor tyrosine kinase associated with inhibition of mammalian target of rapamycin. Expression of these molecules was reduced on Mϕ obtained from livers of infected mice deficient for the gene encoding SR‐AI (msr1). Furthermore, in vitro studies using an SR‐AI‐deficient Mϕ cell line revealed impeded M2 polarization and decreased phagocytic capacity. Direct stimulation with virus was sufficient to activate M2 gene expression in the wild‐type (WT) cell line, but not in the knockdown cell line. Importantly, tissue damage and fibrosis were exacerbated in SR‐AI–/– mice following hepatic infection and adoptive transfer of WT bone‐marrow–derived Mϕ conferred protection against fibrosis in these mice. Conclusion: SR‐AI expression on liver Mϕ promotes recovery from infection‐induced tissue damage by mediating a switch to a proresolving Mϕ polarization state. (Hepatology 2017;65:32‐43).
Abbreviations
- AdOVA
adenovirus expressing ovalbumin
- arg1
arginase 1
- BMDM
bone‐marrow–derived macrophage
- CMV
cytomegalovirus
- DMEM
Dulbecco's modified Eagle's medium
- ECM
extracellular matrix
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- H&E
hematoxylin‐eosin
- HRP
horseradish peroxidase
- IL
interleukin
- IMDM
Iscove's modified Dulbecco's medium
- iNos
inducible nitric oxide synthase
- KC
Kupffer cell
- LDL
low‐density lipoprotein
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- Mϕ
macrophage(s)
- Mertk
mer receptor tyrosine kinase
- MIP
macrophage inflammatory protein
- MOI
multiplicity of infection
- mTOR
mammalian target of rapamycin
- pAb
polyclonal antibody
- PE
phycoerythrin
- Pen Strep
penicillin/streptomycin
- PFU
plaque‐forming units
- qPCR
quantitative polymerase chain reaction
- rAd5‐OVA
recombinant adenovirus type 5 expressing ovalbumin
- shRNA
short hairpin RNA
- SR‐AI
scavenger receptor AI
- srebf1
sterol regulatory element binding transcription factor 1
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild type
Because of constant exposure to typically inflammatory stimuli from the gut by the portal vein (i.e., debris from commensal bacteria), the liver has evolved out of necessity to maintain a tolerogenic environment.1 Subsequently, pathogens such as hepatitis C virus (HCV) have come to fill this niche and can establish lifelong chronic infections.2, 3 Although the liver is known to have remarkable regenerative capability, such persistent infections are characterized by liver fibrosis and cirrhosis, potentially leading to development of hepatocellular carcinoma (HCC).4 The phenomenon of liver tolerance has been variously ascribed to dysfunctional activation of several immune cell compartments, including macrophage(s) (Mϕ).5 The specific role of Mϕ activation and the factors that control Mϕ activation, however, have not yet been well defined. In addition to monocytes and Mϕ circulating throughout the bloodstream, the liver contains a specialized tissue‐resident Mϕ, the Kupffer cell (KC), which is physically integrated into the structure of the sinusoid.6 Liver Mϕ are responsible for maintenance of healthy tissue through phagocytic clearance of apoptotic cells and foreign materials and through tissue repair and remodeling during wound healing.7, 8 Critically, Mϕ are also major regulators of the inflammatory response to disease and infection, monitoring the microenvironment through an array of surface receptors and secreting appropriate cytokines and chemokines.9
Depending on the inflammatory insults they encounter, Mϕ populations can be directed to distinct phenotypical programs in a process known as Mϕ polarization.10 Classical activation is stimulated by microbial products and proinflammatory cytokines (interferon‐gamma and/or lipopolysaccharide [LPS] or tumor necrosis factor), and the resulting M1 Mϕ are characterized by high antigen presentation, high production of interleukin (IL)‐12 and IL‐23, and high production of nitric oxide and reactive oxygen intermediates.11 By contrast, alternative/M2 activation is mediated by IL‐4, IL‐10, and IL‐13 and is characterized by little‐to‐no secretion of proinflammatory cytokines, increased secretion of anti‐inflammatory cytokines, enhanced scavenging of cellular debris, and promotion of tissue remodeling and repair.12, 13 M2 Mϕ also up‐regulate several endocytic surface receptors, including scavenger receptor A (SR‐AI/CD204).11, 14
Scavenger receptors are phagocytic pattern recognition receptors that mediate clearance of both endogenous (modified host molecules, apoptotic cells) and exogenous (microbes, foreign particles) material.15 Scavenger receptor A exists in two isoforms (I and II) that are coexpressed mainly on Mϕ and have no functional differences; they are typically referred to collectively as SR‐AI.16, 17 Through its collagenous extracellular domain, SR‐AI is capable of binding natural ligands (lipoteichoic acid and LPS) as well as nonphysiological ligands (acetylated or oxidized low‐density lipoprotein [LDL] and maleylated bovine serum antigen).16 Indeed, scavenger receptor expression and function have been linked to a number of diseases, including atherosclerosis, nonalcoholic steatohepatitis, HCC, and transplant rejection.10, 18, 19
Many of these diseases may be linked to scavenger receptors through their involvement in Mϕ polarization: SR‐AI acts as both a marker and an activator of M2 activation.20 Notably, scavenger receptor function is also linked to HCV infection, in that both SR‐AI and SR‐B1 can recognize and endocytose HCV proteins.21, 22 The exact function of SR‐AI+ Mϕ in hepatic viral infection, however, remains unknown. In the present study, we investigate the role of SR‐AI expression by Mϕ in controlling tissue inflammation and repair during hepatic viral infection. This report shows that SR‐AI expression on liver Mϕ protects against infection‐induced tissue damage and fibrosis, possibly through mammalian target of rapamycin (mTOR)‐mediated modulation of M2 Mϕ polarization. These results provide insight into new targets for the design of therapeutic agents for chronic liver diseases caused by hepatic viral infections such as HCV.
Materials and Methods
MICE
Six‐ to 8‐week‐old female C57BL/6 and SR‐AI–/– (B6.Cg‐Msr1tm1Csk/J) mice were purchased from Taconic Farms (Hudson, NY) and The Jackson Laboratory (Sacramento, CA), respectively. Mice were housed in a pathogen‐free facility and routinely tested for mouse hepatitis virus and other pathogens. Animals were handled according to protocols approved by the University of Virginia (Charlottesville, VA) Institutional Animal Care and Use Committee.
VIRAL INFECTION
Replication‐defective recombinant adenovirus type 5 expressing ovalbumin under the human cytomegalovirus (CMV) promoter and lacking E1 and E3 genes were purchased from the Iowa Gene Transfer Vector Core (Iowa City, IA). Mice were injected intravenously with 5 × 107 IU of adenovirus expressing ovalbumin (AdOVA).
HEPATIC AND SPLENIC MONONUCLEAR CELL ISOLATION
Mononuclear cells were isolated for further experimentation as described.23 Briefly, livers were passed through a metal spleen screen and digested with 0.05% collagenase IV (Sigma‐Aldrich, St. Louis, MO), and intrahepatic mononuclear cells were purified by Histodenz density gradient centrifugation (Sigma‐Aldrich). Spleens were passed through a mesh spleen screen, and mononuclear cells were purified by Ficoll gradient.
FLOW CYTOMETRY AND LUMINEX ASSAY
Antibodies against major histocompatibility complex class II, Thy 1.2, F4/80, CD11b (eBioscience, San Diego, CA), and SR‐AI (R&D Systems, Minneapolis, MN) were used for surface staining. Cells (1.5 × 106) were blocked with anti‐CD16/CD32 (2.4G2; University of Virginia, Charlottesville, VA) and incubated with the appropriate antibodies for 30 minutes at 4°C in Iscove's modified Dulbecco's medium (IMDM) supplemented with 2% fetal bovine serum (FBS) and 0.1% NaN3. Cells were then washed and fixed in Cytofix/Cytoperm (BD Biosciences, Franklin Lakes, NJ), according to the manufacturer's instructions preceding flow cytometry analysis. All samples were run on a BD FACS Canto II (BD Immunocytometry Systems, San Jose, CA) and analyzed using FlowJo software (version 8.8.6; Tree Star Inc., Ashland, OR). For Luminex analysis, mononuclear cells were stained as above, but in the absence of NaN3 and without fixation. Cells were resuspended in Hank's buffered salt solution containing 1% fetal calf serum and 1 mM of ethylenediaminetetraacetic acid and sorted into F4/80hiCD11blo KCs or F4/80loCD11bhi Mϕ using a BD Influx Cell Sorter at the Flow Cytometry Core Facility (University of Virginia). Sorted cell populations were cultured overnight in IMDM supplemented with 100 U/mL of Pen Strep (penicillin/streptomycin), 10% Hyclone FBS, 2 mM of l‐glutamine, and 10 μM of β‐mercaptoethanol. The resulting supernatants were submitted to the Flow Cytometry Core Facility for analysis using the Luminex MAGPIX assay system (Luminex, Austin, TX).
FLUORESCENT MICROSCOPY
Samples were prepared for fluorescent microscopy as described.24 In brief, mouse livers were fixed with periodate‐lysine‐paraformaldehyde fixative and mounted in optimal cutting temperature medium before being sectioned at 5 μm, blocked in 2.4G2 solution, and stained with antibodies from BioLegend, eBioscience, and R&D Systems. Images were obtained with a Zeiss LSM‐700 confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany).
CELL CULTURE
RAW 264.7 cells were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 100 U/mL of Pen Strep at 37°C and 5% CO2. RAW cells were polarized by culturing 300,000 cells/well overnight in 24‐well plates before replacing the medium with complete culture medium supplemented with either LPS (300 ng/mL), IL‐4/IL‐13 (20 and 10 ng/mL), or dexamethasone (100 nM) and incubating for 2 hours at 37°C. For select experiments, RAW cells were cocultured overnight with AdOva in complete culture medium at a multiplicity of infection (MOI) of 0.5 or 5.0 plaque‐forming units (PFU) of AdOVA.
GENERATION OF msr1 KNOCKDOWN CELL LINE
A panel of four 29‐mer short hairpin RNA (shRNA) plasmids targeted against the murine msr1 gene was generated by and obtained from OriGene (Rockville, MD). Each of the four anti‐msr1 shRNA plasmids and the scramble control plasmid were packaged into lentiviral particles and transduced into RAW 264.7 cells following the manufacturer's instructions. The packaging plasmids, ENV pCMV‐VSVG, pRSV‐REV, and Gag/Pol pMDLg/pRRE, were kindly provided by Dr. Tim Bender (University of Virginia), amplified using OneShot TOP10 chemically competent Escherichia coli (Invitrogen, Carlsbad, CA), and isolated using an Endotoxin‐Free Plasmid Maxi Kit (Qiagen). Transduced RAW cells were isolated by puromycin selection and diluted to a single‐cell suspension before subculturing.
WESTERN BLOTTINGS
Cultured cells were lysed in buffer containing NaF, Na4P2O7, Na3VO4, and protease inhibitor cocktail V (EMD Millipore, Temecula, CA) to preserve protein phosphorylation. Proteins were resolved on Mini‐PROTEAN TGX precast gradient gels (BioRad, Berkeley, CA), transferred to polyvinylidene fluoride membranes, and incubated with rabbit anti‐phospho‐mTOR (monoclonal antibody [mAb]; Cell Signaling Technology, Danvers, MA), anti‐mTOR (mAb; Cell Signaling Technology), anti‐phospho‐MERTK (mer receptor tyrosine kinase; polyclonal antibody [pAb]; FabGennix, Frisco, TX), or anti‐MERTK (pAb; FabGennix). The blottings were then incubated with horseradish peroxidase (HRP)‐linked anti‐rabbit immunoglobulin G (pAb; Cell Signaling Technology) and HRP‐linked goat anti‐Actin (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized with an 80/20 mix of SuperSignal West Pico Chemiluminescent Substrate and Femto Chemiluminescent Substrate (Thermo Scientific, Rochester, NY). Densitometry was performed using ImageJ analysis software (National Institutes of Health, Bethesda, MD).
IN VITRO BEAD PHAGOCYTOSIS ASSAY
RAW and MSRC2 cells were polarized as described above for 2 hours and washed with serum‐free medium. A prewarmed suspension of phycoerythrin (PE)‐conjugated FluoroSphere carboxylated beads (Molecular Probes, Eugene, OR) was added to the cells (500 μL of serum‐free medium and 1.5 μL of beads per well), which were incubated for 2 hours at 37°C. Cells were washed three times with phosphate‐buffered saline and analyzed by flow cytometry. Quantification of internalized beads was determined by gating the PE histogram past the first peak to exclude beads stuck to the cell surface.
ADOPTIVE TRANSFER OF BONE‐MARROW–DERIVED MACROPHAGES
Bone marrow was extracted from femurs of wild‐type (WT) C57BL/6 mice and cultured for 2 hours at 37°C in DMEM supplemented with 10% FBS, 100 U/mL of Pen Strep, and 2 mM of l‐glutamine to adhere resident bone marrow Mϕ. Adherent cells were resuspended in culture medium containing 10% L‐292 medium and allowed to differentiate for 7 days. Medium was replaced with fresh differentiation medium every 2 days. Recipient SR‐AI–/– mice were treated with 100 μL of clodronate liposome suspension (Encapsula NanoSciences, Brentwood, TN) by tail vein injection 1 day preceding transfer of 2 × 106 differentiated bone‐marrow–derived macrophages (BMDMs), also by tail vein injection. Approximately 8 hours following BMDM transfer, recipients were infected with 5 × 107 PFU of AdOVA. Mice were sacrificed after 14 days for further experimentation.
STATISTICAL ANALYSIS
Statistical significance was determined using either the two‐tailed Student t test or one‐way analysis of variance, where appropriate. Analysis was performed using Prism software (GraphPad Software Inc., La Jolla, CA). Values of P < 0.05 were regarded as statistically significant. Asterisks (*, **, and ***) denote P < 0.05, P < 0.01, and P < 0.001, respectively.
Results
SR‐AI IS UP‐REGULATED ON Mϕ FOLLOWING HEPATOTROPIC VIRAL INFECTION
Expression of SR‐AI has been reported to modulate activation and polarization of Mϕ.20, 25 To assess the impact of SR‐AI expression on regulation of hepatic immune responses to infection and development of tissue damage, we injected 5e7 PFU of AdOVA into the tail vein of 6‐ to 8‐week‐old C57BL/6 mice. The virus travels along the tail vein to the liver where the large majority is taken up by Mϕ and hepatocytes, establishing a hepatotropic infection. In order to fully examine the phenotype of SR‐AI+ liver Mϕ, we first determined the tempo and specificity of SR‐AI expression in infection. Whole livers were harvested from AdOVA‐infected mice, and, following homogenization and density gradient centrifugation, liver Mϕ were separately identified as liver‐resident KCs (F4/80hiCD11blo) or nonresident circulating Mϕ (F4/80loCD11bhi) by flow cytometry (Fig. 1A). Increased expression of SR‐AI was observed on both KC and nonresident liver Mϕ 7 days after AdOVA infection, a time point coinciding with viral clearance and the beginning of tissue repair (Fig. 1B,C). In contrast, there was no up‐regulation of SR‐AI on splenic Mϕ following infection (Fig. 1B,C), suggesting that the specific up‐regulation of SR‐AI on liver Mϕ was possibly a result of viral recognition at the site of infection. Importantly, fluorescent microscopy of histochemically stained liver sections confirmed up‐regulation of SR‐AI detected by flow cytometry and also revealed that SR‐AI+ liver Mϕ coexpressed the M2 surface marker, YM‐1 (Fig. 1D). Taken together, these results suggest that SR‐AI may trigger signaling involved in alternative activation in liver Mϕ during hepatotropic viral infection.
Figure 1.
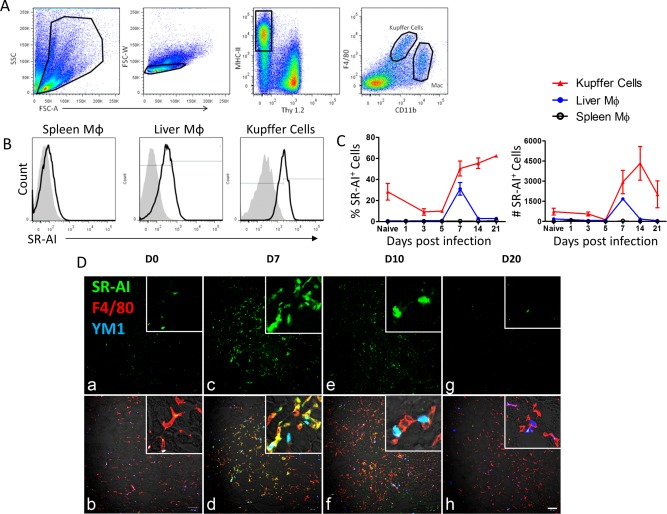
SR‐AI is up‐regulated on Mφ following hepatic viral infection. (A) Flow cytometry gating strategy for liver macrophages. Mononuclear cells were separated from whole‐liver homogenate by density gradient centrifugation, and live singlets were gated on Thy1.2–MHC‐II+. F4/80hiCD11bmid cells were identified as liver‐resident Kupffer cells and F4/80midCD11bhi cells were identified as nonresident macrophages. (B) SR‐AI surface expression (black trace) versus isotype control (gray histogram) in spleen Mφ, liver Mφ, and KCs at day 7 postinfection. (C) Time course of frequency and number of SR‐AI+ cells (determined by gating on isotype control) during AdOVA infection. Data points are mean ± SEM of n=3 mice. (D) Immunofluorescence microscopy of sections from AdLacZ infected mouse liver at 0, 7, 10, and 20 days postinfection (100× magnification and scale bar = 100 μm; insert, ×200 magnification). Panels (A), (C), (E), and (G) show SR‐AI single‐surface staining in green; panels (B), (D), (F), and (H) show merged staining of SR‐AI (green), F4/80 (red), and YM1 (blue).
SR‐AI MODULATES Mϕ ACTIVATION UPON VIRAL INSULT
We next investigated whether the increase in SR‐AI expression following infection indeed correlated with a shift in Mϕ activation. Luminex analysis of fluorescence‐activated cell sorting (FACS)‐sorted KCs and circulating Mϕ showed that production of the M2 cytokine, IL‐10, by both populations was increased on day 7 postinfection (Fig. 2A). KCs tended to produce higher levels of IL‐10 compared to nonresident Mϕ. Several proinflammatory mediators typical of M1 activation (IL‐6, macrophage inflammatory protein [MIP]‐1α, and MIP‐1β) were also secreted by these cells in both naïve and infected conditions, suggesting that liver Mϕ may assume an “M2‐like” intermediate phenotype with some M1 characteristics (Fig. 2A).
Figure 2.
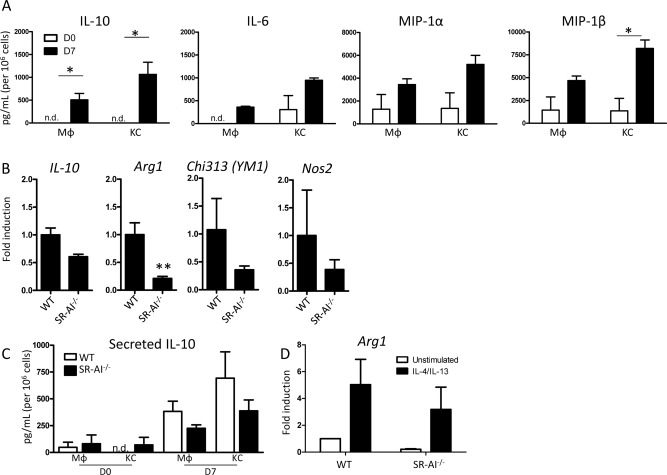
SR‐AI modulates Mφ activation upon viral insult. (A) Luminex quantification of cytokines and chemokines in supernatants collected from FACS‐sorted liver KCs and Mφ from infected WT mice following overnight culture. Data are mean ± SEM of n = 3 mice. (B) qPCR analysis for expression of M2‐related genes in Mφ‐enriched mononuclear cell fractions from WT and SR‐AI–/– livers on day 7 postinfection. Expression levels were calculated by the delta‐delta threshold cycle method and normalized to hypoxanthine guanine phosphoribosyl transferase expression; data are mean ± SEM for n = 3 mice. (C) Luminex data of IL‐10 levels in supernatants from sorted Mφ or KC from WT and SR‐AI–/– mice at day 7 postinfection. Data are mean ± SEM for n=3 mice. (D) Arginase 1 qPCR analysis of Mφ from infected WT or SR‐AI–/– livers cultured for 2 hours in either plain media or media with IL‐4 and IL‐13 to induce M2 polarization. Expression levels were calculated as in (A); data are mean ± SEM for n = 3 mice.
Based on the finding that SR‐AI+ Mϕ in the liver exhibited M2‐like characteristics, we examined the status of Mϕ activation in WT and SR‐AI–/– animals to determine whether SR‐AI expression contributes to M2 polarization. Indeed, when Mϕ‐enriched mononuclear liver cell fractions from infected animals were analyzed by quantitative polymerase chain reaction (qPCR), liver Mϕ in SR‐AI–/– mice were impaired in their expression of the M2 genes, arginase 1 (arg1), chi313 (YM‐1), and il‐10 (Fig. 2B). Interestingly, expression of the M1 gene, nos2, was also reduced in cells obtained from SR‐AI–/– mice compared to WT controls. When sorted SR‐AI–/– Mϕ and KCs were analyzed by Luminex, both populations up‐regulated IL‐10 secretion following infection, but to a lesser degree than their WT counterparts (Fig. 2C). We next asked whether SR‐AI–/– Mϕ had lost the ability to become alternatively activated by attempting to force M2 polarization by 2‐hour stimulation with IL‐4 and IL‐13. Both the WT and SR‐AI–/– Mϕ were capable of up‐regulating arg1 expression in the presence of these strong M2 stimuli, but the level of Arg‐1 expression by SR‐AI–/– cells was still lower than WT cells (Fig. 2D).
MICE DEFICIENT IN SR‐AI DEVELOP EXACERBATED INFECTION‐INDUCED LIVER TISSUE DAMAGE AND FIBROSIS
Given the timing of the appearance of SR‐AI+ Mϕ in the course of infection and their potential role in wound repair and tissue remodeling following injury and infection, we performed histological examinations on liver sections from WT and SR‐AI–/– mice to interrogate possible protective qualities of SR‐AI expression. Postinfection liver tissue damage appeared dramatically more severe in the absence of SR‐AI. Hematoxylin‐eosin (H&E) staining of liver tissue sections showed increased inflammatory infiltrates in SR‐AI–/– mice compared to WT at 7 and 14 days postinfection (Fig. 3A). Infection‐induced hepatocyte DNA damage was also increased in the absence of SR‐AI as measured by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Fig. 3B). Collagen staining by Trichrome C revealed deposition of extracellular matrix (ECM) between cells (in blue) in the knockout liver 14 days postinjection (Fig. 3C). No positive staining was observed in the WT liver at day 14 or in either mouse at day 7 postinfection. Together, these data suggest that expression of SR‐AI on liver Mϕ plays a protective role in viral infection. SR‐AI+ liver Mϕ may thus comprise an alternatively activated subset involved in wound repair and tissue remodeling.
Figure 3.
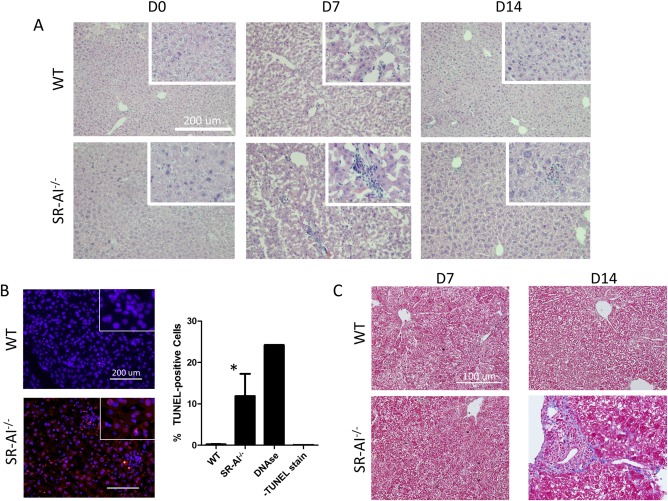
Infection‐induced tissue damage is more severe in the absence of SR‐AI. (A) H&E staining of liver sections obtained from WT and SR‐AI–/– mice 0, 7, or 14 days after tail vein injection of 5e7 PFU of AdOVA (100× magnification and scale bars = 200 μm; insert, ×200 magnification). Images are representative of three independent experiments. (B) Visualization of cell damage by TUNEL staining of WT and SR‐AI–/– liver sections 7 days postinfection (100× magnification and scale bars = 200 μm; insert, ×200 magnification). Staining was quantified by dividing the number of positive red stained cells by the total number of blue counterstained cells. Data are mean ± SEM; * P < 0.05 versus WT mice. (C) Trichrome staining for collagen in liver sections from WT and SR‐AI–/– mice 7 and 14 days postinfection (100× magnification and scale bars = 200 μm). Images are representative of three independent experiments.
SR‐AI EXPRESSION IS REQUIRED FOR MODULATION OF Mϕ ACTIVATION BY mTOR AND MAINTAINING PHAGOCYTIC ABILITY
In order to further investigate a direct effect of SR‐AI on Mϕ activation and function, we established a cell line that recapitulated SR‐AI–/– mice. To accomplish this, plasmids containing shRNA sequences targeted against the msr1 gene were loaded into lentivirus particles by a lentiviral packaging cell line. These lentiviruses were then administered to the RAW 264.7 murine Mϕ cell line, after which individually infected cells were clonally expanded and tested for knockdown efficiency by flow cytometry of SR‐AI and qPCR of msr1. Subclones MSRC1 and MSRC2 showed significant knockdown of msr1 message, but MSRC2 showed more dramatic reduction in SR‐AI surface expression than that in MSRC1 (Fig. 4A,B). Stable SR‐AI knockdown persisted through at least 20 passages in culture, as well as through freeze‐thaw cycles (data not shown). The RAW 264.7 cell line (hereafter referred to as RAW cells) can be reliably polarized in vitro by stimulation with LPS (M1) or IL‐4/IL‐13 (M2). We confirmed Mϕ polarization using real‐time PCR for the prototypical M1 and M2 genes, arginase (arg1) and inducible nitric oxide synthase (iNos [nos2]; Supporting Fig. S1A). Notably, msr1 gene expression appeared to track with M2 polarization (Supporting Fig. S1B). When exposed to M2 stimuli, RAW cells exhibit robust up‐regulation of arg1 and chi313 (encoding YM1) whereas MSRC2 cells show minimal up‐regulation, in agreement with the results obtained from SR‐AI–/– mice (Fig. 4C). The lipid metabolism gene, sterol regulatory element binding transcription factor 1 (srebf1), was included as an irrelevant control and remained unchanged in the knockdown cell line under all stimulation conditions (Fig. 4D).
Figure 4.
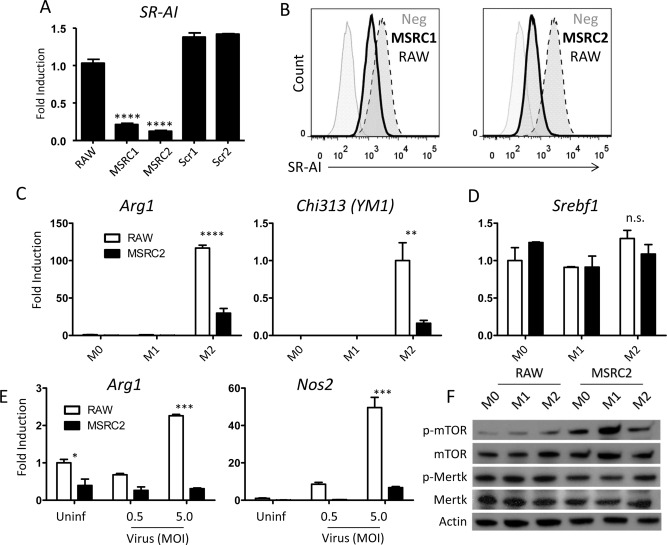
Generation of a stable MSR knockdown cell line. (A) qPCR analysis of relative Msr1 expression in two subcultures of small interfering RNA (siRNA)‐transfected cells and two subcultures transfected with scrambled control plasmids. Expression levels were calculated by the delta‐delta threshold cycle method and normalized to hypoxanthine guanine phosphoribosyl transferase expression, then normalized to expression of untransfected RAW cells. Data are mean ± SEM for n = 3; **** P < 0.00005. (B) Flow analysis of SR‐AI expression after transfection with lentivirally packaged anti‐Msr1 siRNA. The bold trace represents transfected RAW cell subclones whereas the dotted trace represents WT RAW cells. (C,D) qPCR analysis comparing arg1, chi313, and srebf1 expression by stable knockdown MSRC2 cells to that of untransfected RAW cells. Expression levels were calculated as in (A). Data are mean ± SEM for n = 3; ** P < 0.005; **** P < 0.00005. (E) Arg1 and Nos2 gene expression of MSRC2 and untransfected RAW cells following overnight coculture with 0, 0.5 MOI, or 5.0 MOI of AdOVA in complete media. Expression levels were calculated as in (A); data are mean ± SEM for n = 3. (F) Western blotting of phosphorylated and total mTOR and Mertk from whole cell lysates of RAW or MSRC2 cells incubated for 2 hours in either plain media (M0), LPS (M1), or IL‐4 and IL‐13 (M2). Images are representative of three independent experiments. Abbreviation: p, phosphorylated.
Although stimulation with LPS or IL‐4/IL‐13 are reliable ways to induce specific aspects of Mϕ polarization, we speculated that viral infection represented a unique mode of stimulation by SR‐AI‐mediated recognition, and therefore we attempted to stimulate RAW and MSRC2 cells by directly adding AdOVA in vitro. Overnight incubation with virus was enough to stimulate up‐regulation of both arg1 and nos2, and the nos2 effect was dose dependent with viral MOI (arg1 expression actually decreased slightly at 0.5 MOI when compared to uninfected). Strikingly, Arg1 expression by MSRC2 cells remained unchanged upon addition of virus, and nos2 up‐regulation was only observed at 5.0 MOI and to a very small degree when compared to RAW cell expression (Fig. 4E). Interestingly, RAW cells exhibited decreased phagocytosis with 0.5 MOI of virus compared to uninfected cells, but this deficit was absent with 5.0 MOI of virus. However, at a higher dose of virus, bead uptake by MSRC2 cells progressively decreased and MSRC2 cells treated with 5.0 MOI of virus were able to internalize fewer beads than their RAW counterparts (Supporting Fig. S2A,B).
We next sought to investigate the mechanism by which SR‐AI expression alters Mϕ gene expression. Although many studies implicate SR‐AI signaling in biological processes, the exact mechanism remains unclear. Previous reports that activation of mTOR regulates scavenger receptor expression led us to investigate whether mTOR phosphorylation could, in turn, be regulated by expression of SR‐AI.26, 27 When analyzed by western blotting, SR‐AI knockdown cells exhibited enhanced mTOR phosphorylation compared to SR‐AI sufficient cells under all stimulation conditions (Fig. 4F and Supporting Fig. S3A). Although SR‐AI possesses no recognized intracellular signaling motif, it has been suggested that SR‐AI associates with Mertk in order to transduce signals.28 Indeed, western blotting analysis revealed Mertk phosphorylation to be impaired in SR‐AI knockdown cells stimulated with M1 or M2 cocktails as compared to their WT counterparts (Fig. 4F and Supporting Fig. S3B). Taken together, these results suggest that activation of Mertk may lead to inhibition of the mTOR pathway and be involved in SR‐AI‐mediated alteration of Mϕ activation.
TRANSFER OF SR‐AI+ Mϕ INTO SR‐AI‐DEFICIENT MICE PROTECTS AGAINST INFECTION‐INDUCED TISSUE DAMAGE AND FIBROSIS
We next investigated whether SR‐AI+ Mϕ are able to protect from development of fibrosis in SR‐AI–/– mice. First, SR‐AI–/– mice were treated with clodronate to deplete the endogenous liver Mϕ population before receiving an adoptive transfer of WT BMDMs intravenously (Fig. 5A). Transfer of SR‐AI+ Mϕ derived from WT mice was verified by the presence of SR‐AI+ Mϕ in SR‐AI–/– recipient mice (Fig. 5C). Animals were then infected with AdOVA for 14 days, and the degree of liver fibrosis was assessed by Trichrome C staining. Remarkably, whereas SR‐AI–/– mice accumulated a significant degree of fibrosis, knockout mice that received WT BMDMs before infection did not develop fibrosis similar to that of WT mice (Fig. 5B). These results suggest that manipulation of SR‐AI signaling and adoptive transfer of SR‐AI+ Mϕ may represent potential therapeutic targets for preventing development of hepatic fibrosis.
Figure 5.
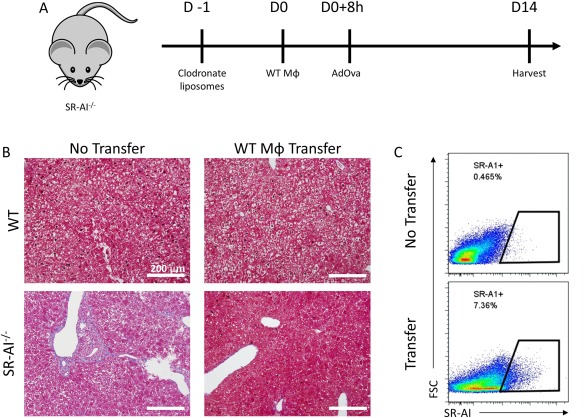
Transfer of SR‐AI+ Mϕ protects against infection‐induced tissue damage. (A) Experimental design for adoptive BMDM transfer and infection. (B) Trichrome staining for collagen in liver sections from WT and SR‐AI–/– mice 14 days postinfection (100× magnification and scale bars = 200 μm). Images representative of 3 mice. (C) Confirmation of successful transfer of SR‐AI+ BMDMs into SR‐AI–/– mice by flow cytometry.
Discussion
The liver is a highly regenerative organ with a unique immune repertoire that houses over 80% of the body's macrophages, including liver‐resident KCs and noncirculating Mϕ. These Mϕ play a unique role in maintaining homeostatic immune tolerance in the liver and, as described in this report, orchestrate inflammatory responses and restore tissue homeostasis following injury. Here, we describe the functional characteristics of SR‐AI+ Mϕ and their protective role in fibrosis development during hepatotropic viral infection. SR‐AI expression steadily increases at later time points in infection, which parallels the ability of SR‐AI+ Mϕ to produce anti‐inflammatory mediators, express M2‐like markers, perform efficient phagocytosis, and limit deposition of fibrotic tissue. Collectively, these observations identify SR‐AI as a key regulator and potential target in preventing liver fibrosis.
As a scavenger receptor, SR‐AI can bind a variety of polyanionic ligands, including LDL, bacterial products, polysaccharides, and nucleic acids. Interestingly, M2‐like features were reduced in SR‐AI–/– mice (both in vivo and under M2‐polarizing conditions ex vivo) and in vitro in cultured Mϕ with deficient SR‐AI expression. Given that each of these conditions is likely to have a different composition of SR‐AI ligands, the similarities in Mϕ phenotypes indicate that the identity of the ligands might not be crucial for determining the polarizing function of SR‐AI. Rather, SR‐AI signaling upon engagement with specific ligands may play a pivotal role in influencing M2‐like Mϕ polarization. However, expression of iNOS (typically associated with M1 polarization) was decreased in Mϕ from the SR‐AI–/– animals, tracking with the M2‐like phenotype exhibited by SR‐AI+ Mϕ. Differences in the quality and quantity of SR‐AI ligands may underlie this mosaic polarization phenotype. Furthermore, redundancy in scavenger receptor repertoires and other polarizing factors may be driving these intermediate polarization states. Indeed, the loss of M2‐like features was more pronounced in cell lines lacking expression of SR‐AI when compared to SR‐AI–/– mice, underscoring that Mϕ polarization in vivo includes a spectrum of activation states rather than absolute M1 or M2 phenotypes. Recent reports have also shown that production of proinflammatory mediators by M2 Mϕ subsets is not uncommon.31, 32, 33 It is therefore likely that the polarization state of SR‐AI+ liver Mϕ following viral infection represents a crossover M2‐like state with some M1 features. SR‐AI modulates this entire phenotype, so when arginase induction is inhibited in SR‐AI‐deficient animals and cell lines, iNOS expression tracks with this phenotype. Moreover, the inhibition of M2 polarization in SR‐AI‐deficient Mϕ RAW cells occurred in the complete absence of potential physiological stimuli, such as apoptotic cells or viral particles. This finding indicates that in addition to its role during infection, SR‐AI contributes to the M2 polarization signaling pathway even under homeostatic conditions.
Intriguingly, SR‐AI was differentially expressed in liver Mϕ and KCs: SR‐AI expression peaked in liver Mϕ at day 7 postinfection and returned to basal levels by day 14. In contrast, KC expression of SR‐AI was maintained at high levels from day 7 to 21 postinfection. These differences prompt interesting questions regarding distinct functions of these two Mϕ populations. A likely explanation of these differences is that KCs are the predominant M2‐like cells mediating tissue recovery in hepatotropic viral infection given that they continue to express high levels of the M2‐promoting marker, SR‐AI. KCs are thought to arise from liver‐resident yolk‐sac–derived precursors; meanwhile, nonresident liver Mϕ are derived from monocyte precursors from the bone marrow. Although there is limited information regarding expression of SR‐AI during Mϕ development, it is possible that expression of SR‐AI and other M2 effectors are programmed into liver‐resident Mϕ precursors. However, deposition of fibrotic tissue was minimized upon transfer of SR‐AI+ BMDMs into SR‐AI–/– mice, suggesting that the local tolerogenic environment may also induce SR‐AI and other M2‐like properties in Mϕ originating from extrahepatic sources. Furthermore, the balance of M1 and M2 Mϕ at the site of infection is directly related to the development and progression of tissue injury. Whereas proresolving M2 Mϕ subsets are responsible for the resolution of inflammation and clearance and remodeling of scar tissue, M1 Mϕ (or even other M2 Mϕ subsets) can actively contribute to production of ECM and further tissue damage. Our studies report that SR‐AI+ liver Mϕ are necessary for optimal recovery from infection, as measured by the presence of inflammatory infiltrates, fibrotic lesions, and hepatocellular DNA damage (Fig. 3A‐C).
The molecular and cellular cues that program Mϕ activation thus require further investigation and may have significant implications for development and function of these cells. Our findings implicate SR‐AI expression as a possible driver of alternative Mϕ activation in hepatotropic viral infection. The balance between M1 and M2 Mϕ activation (and additional subtypes of M2 Mϕ) is a key determinant of recovery from tissue injury and progression to fibrosis. Consequently, the idea that the scavenger receptor, SR‐AI, acts as a switch to turn on resolution of inflammation and repair of tissue injury is a promising avenue to understand Mϕ polarization in the pathogenesis of chronic liver diseases.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28873/suppinfo.
Supporting Information
Potential conflict of interest: Nothing to report.
Supported by National Institute of Health grants R01DK063222 and U19AI083024.
REFERENCES
- 1. Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev 2000;174:21–34. [DOI] [PubMed] [Google Scholar]
- 2. Dustin LB, Cashman SB, Laidlaw SM. Immune control and failure in HCV infection—tipping the balance. J Leukoc Biol 2014;96:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoofnagle JH. Course and outcome of hepatitis C. Hepatology 2002;36(5 Suppl 1):S21–S29. [DOI] [PubMed] [Google Scholar]
- 4. Manickam C, Reeves RK. Modeling HCV disease in animals: virology, immunology and pathogenesis of HCV and GBV‐B infections. Front Microbiol 2014;5:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crispe IN. Immune tolerance in liver disease. Hepatology 2014;60:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strauss O, Dunbar PR, Bartlett A, Phillips A. The immunophenotype of antigen presenting cells of the mononuclear phagocyte system in normal human liver‐‐a systematic review. J Hepatol 2015;62:458–468. [DOI] [PubMed] [Google Scholar]
- 7. Chen Q, Xue Y, Sun J. Kupffer cell‐mediated hepatic injury induced by silica nanoparticles in vitro and in vivo. Int J Nanomedicine 2013;8:1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You Q, Holt M, Yin H, Li G, Hu CJ, Ju C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem Pharmacol 2013;86:836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boltjes A, Movita D, Boonstra A, Woltman AM. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J Hepatol 2014;61:660–671. [DOI] [PubMed] [Google Scholar]
- 10. Labonte AC, Tosello‐Trampont AC, Hahn YS. The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells 2014;37:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004;25:677–686. [DOI] [PubMed] [Google Scholar]
- 12. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009;27:451–483. [DOI] [PubMed] [Google Scholar]
- 13. Alfano M, Graziano F, Genovese L, Poli G. Macrophage polarization at the crossroad between HIV‐1 infection and cancer development. Arterioscler Thromb Vasc Biol 2013;33:1145–1152. [DOI] [PubMed] [Google Scholar]
- 14. Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003;3:23–35. [DOI] [PubMed] [Google Scholar]
- 15. Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev 2016;269:44–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bowdish DM, Gordon S. Conserved domains of the class A scavenger receptors: evolution and function. Immunol Rev 2009;227:19–31. [DOI] [PubMed] [Google Scholar]
- 17. Martínez VG, Moestrup SK, Holmskov U, Mollenhauer J, Lozano F. The conserved scavenger receptor cysteine‐rich superfamily in therapy and diagnosis. Pharmacol Rev 2011;63:967–1000. [DOI] [PubMed] [Google Scholar]
- 18. Yu X, Guo C, Fisher PB, Subjeck JR, Wang XY. Scavenger receptors: emerging roles in cancer biology and immunology. Adv Cancer Res 2015;128:309–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012;217:492–502. [DOI] [PubMed] [Google Scholar]
- 20. Hu Y, Zhang H, Lu Y, Bai H, Xu Y, Zhu X. Class A scavenger receptor attenuates myocardial infarction‐induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res Cardiol 2011;1311–1328. [DOI] [PubMed] [Google Scholar]
- 21. Beauvillain C, Meloni F, Sirard JC, Blanchard S, Jarry U, Scotet M, et al. The scavenger receptors SRA‐1 and SREC‐I cooperate with TLR2 in the recognition of the hepatitis C virus non‐structural protein 3 by dendritic cells. J Hepatol 2010;52:644–651. [DOI] [PubMed] [Google Scholar]
- 22. Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J Hepatol 2011;54:48–55. [DOI] [PubMed] [Google Scholar]
- 23. Dolina JS, Braciale TJ, Hahn YS. Liver‐primed CD8+ T cells suppress antiviral adaptive immunity through galectin‐9‐independent T‐cell immunoglobulin and mucin 3 engagement of high‐mobility group box 1 in mice. Hepatology 2014;59:1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krueger PD, Kim TS, Sung SS, Braciale TJ, Hahn YS. Liver‐resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J Immunol 2015;194:3213–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X, Zong G, Zhu L, Jiang Y, Ma K, Zhang H, et al. Deletion of class A scavenger receptor deteriorates obesity‐induced insulin resistance in adipose tissue. Diabetes 2014;63:562–577. [DOI] [PubMed] [Google Scholar]
- 26. Fruhwürth S, Krieger S, Winter K, Rosner M, Mikula M, Weichhart T, et al. Inhibition of mTOR down‐regulates scavenger receptor, class B, type I (SR‐BI) expression, reduces endothelial cell migration and impairs nitric oxide production. Biochim Biophys Acta 2014;1841:944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mwaikambo BR, Yang C, Chemtob S, Hardy P. Hypoxia up‐regulates CD36 expression and function via hypoxia‐inducible factor‐1‐ and phosphatidylinositol 3‐kinase‐dependent mechanisms. J Biol Chem 2009;284:26695–26707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Todt JC, Hu B, Curtis JL. The scavenger receptor SR‐A I/II (CD204) signals via the receptor tyrosine kinase Mertk during apoptotic cell uptake by murine macrophages. J Leukoc Biol 2008;84:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gensel JC, Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res 2015;1619:1–11. [DOI] [PubMed] [Google Scholar]
- 30. Keeler GD, Durdik JM, Stenken JA. Localized delivery of dexamethasone‐21‐phosphate via microdialysis implants in rat induces M(GC) macrophage polarization and alters CCL2 concentrations. Acta Biomater 2015;12:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohtsuki T, Kimura K, Tokunaga Y, Tsukiyama‐Kohara K, Tateno C, Hayashi Y, et al. M2 macrophages play critical roles in progression of inflammatory liver disease in hepatitis C virus transgenic mice. J Virol 2015;90:300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saha B, Kodys K, Szabo G. Hepatitis C virus‐induced monocyte differentiation into polarized M2 macrophages promotes stellate cell activation via TGF‐β. Cell Mol Gastroenterol Hepatol 2016;2:302–316.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poczobutt JM, De S, Yadav VK, Nguyen TT, Li H, Sippel TR, et al. Expression profiling of macrophages reveals multiple populations with distinct biological roles in an immunocompetent orthotopic model of lung cancer. J Immunol 2016;196:2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dansako H, Yamane D, Welsch C, McGivern DR, Hu F, Kato N, et al. Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll‐like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS Pathog 2013;9:e1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeWitte‐Orr SJ, Collins SE, Bauer CM, Bowdish DM, Mossman KL. An accessory to the ‘Trinity’: SR‐As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS Pathog. 2010;6: e1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28873/suppinfo.
Supporting Information