

Introduction
Cystic fibrosis is a life limiting disease caused by defective or deficient cystic fibrosis trans-membrane conductance regulator (CFTR) activity. The recent FDA approval of lumacaftor combined with ivacaftor (Orkambi) targets patients with the F508del-CFTR. The question remains, is this breakthrough combination therapy the ‘magic-bullet’ cure the vast majority of patients with CF? This review covers the contemporary clinical and scientific knowledge-base for Orkambi and highlights the emerging issues from recent conflicting literature reports.
Cystic fibrosis (CF)
CF is an autosomal recessive genetic disease with an incidence of 1:3,500 in Caucasian populations while less common in non-white populations (1-4). The disease affects the exocrine mucus glands of the lung, liver, pancreas, and intestines causing progressive multi-system failure, such as loss of lung function and pancreatic insufficiency (1, 4-6). Even though a slew of medications such as antibiotics, anti-inflammatories and mycolytics have increased the life expectancy of patients with CF, there is still a long way to go until people with CF reach appreciable levels of quality of life (7). With the inevitable progression of the disease patients often require more intensive therapy, which prior to the development of cystic fibrosis transmembrane conductance regulator (CFTR) drugs, only targeted the symptoms (8). In the clinical setting, the most common methods to measure CFTR function are the assessments of sweat chloride concentration and nasal potential difference (9). Sweat chloride is a promising biomarker as sweat glands do not seem to be subjected to the secondary damage from CF abnormalities which can occur at an early age (unlike i.e. lung and gastrointestinal tract tissue). Notably, sweat chloride concentration has been proposed as an index of CFTR function (10). This suggestion arises from the assumption that greater residual CFTR function leads to lower sweat chloride concentrations, as well as protection against severe lung disease. However, sweat chloride concentration alone does not necessarily predict a milder pulmonary course of the disease (10). Clinical manifestations of CF include chronic lung infection and inflammation with the loss of lung function that eventually results in respiratory failure (4). Thick mucus accumulation in the lung promotes bacterial infections (4, 7). Major pathogens in CF lungs commonly include Pseudomonas aeruginosa followed by Haemophilus influenzae, Staphylococcus aureus, Burkholderia pseudomallei, and Stenotrophomonas maltophilia (4, 7, 11). Thick mucus also obstructs the pancreatic duct eventually leading to complete closure by scar tissue which results in exocrine pancreatic insufficiency (4, 12, 13). Insufficient secretion of pancreatic enzymes leads to malabsorption of fats and protein that results in deficiencies of fat soluble vitamins (i.e. A, D, E, and K), leading to cachexia that predisposes patients to infection (4). In general, patients with CF with pancreatic sufficiency are associated with milder expression of the CF phenotype (1, 3, 12). One consequence of pancreatic insufficiency namely, distal intestinal obstructive disease is caused by partial or complete bowel obstruction caused by inspissated faeces (4). This condition necessitates surgery in 10% of newborns with CF (13). Gastro-intestinal reflux disease or rectal prolapses are also more common in patients with CF than in the general population (14). The second most common cause of death in patients with CF is liver disease (15). One in four patients with CF over the age of four years display liver function related issues and 6-8% suffer from potentially fatal liver disease (15). The spectrum of liver abnormalities is very broad ranging from neonatal cholestasis, benign increase in circulating liver enzymes, cholelithiasis, steatosis, focal biliary cirrhosis, or multilobular biliary cirrhosis with portal hypertension (15).
In the US and Europe CF affects ~30,000 and ~35,800 people with a prevalence of 0.797/10,000 (US) and 0.737/10,000 (EU), respectively (2, 16, 17). Ireland is the unfortunate exception with a prevalence of 2.98/10,000 (2). The mean annual health cost for patients with CF is highly dependent on the severity and progression of the disease (18). The base annual cost, not including the cost of Kalydeco or Orkambi, for patients with CF with mild, moderate, and severe disease are US $10,151, US $25,647, and US $33,691, respectively (18). In Europe the annual cost per patient varies depending on the country i.e. €21,144 in Bulgaria, €30,123 in Italy, or €53,256 in Germany (19). The majority of costs are accounted for by hospitalized inpatients (58%), followed by pharmaceuticals (29%), medical services (10%), complications (2%), and diagnostic testing (1%) (18).
At present, there is no cure for CF. Due to the complex disease manifestations and multi-organ involvement, patient management usually requires large number of medications for symptomatic treatment of respiratory infections, inflammation, the clearance of mucus, and nutritional maintainance (1, 4). In recent years, medical and technological improvements like early diagnosis, specialist care, advanced treatment methods, and lung transplants have resulted in an increase in median survival age to now ~37.5 years in total (in the US). This is a marked improvement from the 1980s when the median survival age was ~20 years (4). This shift furthermore impacts on socioeconomic aspects such as independent adults with this lethal disease entering the workforce or wishing to start their own families.
Notwithstanding, there is still a high unmet medical need for effective treatment that targets the underlying mechanism of the diseased state itself (20). The progressive nature of CF combined with the life-long healthcare costs are major factors in favour for the development of novel CF therapeutics that modulate CFTR function (18). Orkambi, the novel lumacaftor-ivacaftor combination is the first CF therapeutic that actually treats the disease itself as opposed to only managing the symptoms. Excitingly, this new therapeutic potentially represents the ‘magic bullet’ patients with CF and respiratory clinicians have been waiting for. Worryingly, a number of conflicting reports have emerged that overshadow the clinical efficacy of this purported wonder drug (21, 22). Can Orkambi live up to its promise of allowing patients with CF to finally ‘catch a breath’? This review surveys the current clinical and pre-clinical knowledge-base on Orkambi, and provides a critical point-of-view of the benefits and potential caveats of this revolutionary new CFTR corrector-modulator combination.
Mode of action
An understanding of the mode of action of Orkambi necessitates the background into the biology and mechanism of the CF disease state itself.
CF is caused by mutations in the gene that encodes the CFTR protein, which is expressed in epithelial cells of various tissues including the lung (3). The full length CFTR protein is a multi-domain membrane protein consisting of two membrane spanning domain (MSD1&2) and two nucleotide binding domains (NBD1&2) linked together by a phosphorylation regulated (R) domain (Figure 1A) (23). The MSD forms an anion-selective pore and the dimerized NBDs mediate the ATP-binding, -hydrolysis and ATP-dependent gating by the phosphorylation of the CFTR protein (23). CFTR function is regulated by the cyclic adenosine monophosphate (cAMP)-dependent protein kinase A. The CFTR functions to conduct hydrochloride across the cytoplasmic membrane (24, 25). A deficiency in bicarbonate secretion results in poor solubility of luminal mucus; leading to the production of abnormal mucus secretion which damages the lungs, liver, pancreas, and intestine resulting in multi-organ failure (4, 5, 13). Furthermore, due to a mutation in the CFTR the majority of men with CF suffer from associated congenital bilateral absence of the vas deferens which results in obstructive azoospermia (26).
Figure 1A.

Model of the proposed structure of the cystic fibrosis transmembrane conductance regulator (CFTR) channel. The CFTR is a member of the ABC transporter family that functions a Cl- channel. It is located in the apical membrane of epithelia. The CFTR is composed of five domains which include two membrane-spanning domains (MSD-1 and -2), two nucleotide-binding domains (NBD-1 and -2), and a regulatory (R) domain.
One in 22 people of European descent carry one gene for CF making it one of the most common autosomal recessive genetic diseases (3). More than 1900 CFTR protein mutations have been identified and are categorized into functional classes I, II, III, IV, V, and VI (Figure 1B) (1, 3). Broadly speaking, the class I, II, and III mutations lead to the classic CF phenotype with pancreatic insufficiency and a non-functional CFTR; while IV, V, and VI mutations produce a partially functional CFTR and are associated with a milder form of the disease (1, 3, 13). Class I mutations result in the expression of a defective CFTR with a complete loss of function (13). The most common mutation found in 70% of patients with CF (Caucasians of European descent) is the class II F508 deletion. Class III mutations (i.e. G551D) result in defective regulation of channel opening (13, 27). The G551D mutation produces a CFTR protein that is localized on the epithelial membrane but fails to open (28). The G551D-CFTR is missense mutation targeted by Kalydeco is seen in approximately 4% of patients with CF and is the third most common mutation found in class III (3). A clinical study conducted by Vertex convincingly demonstrated the clinical efficacy of ivacaftor in a cohort of 167 G551D-CFTR CF patients (16, 29). Patients received 150 mg ivacaftor or placebo every 12 h with fat containing food for 48 weeks. The ivacaftor treatment group displayed a 10.6% improvement in ppFEV1 in patients ≥12 years (P<0.0001) and 12.5% (P<0.0001) in patients aged 6-11 years compared to placebo which continued through a 48-week period.
Figure 1B.
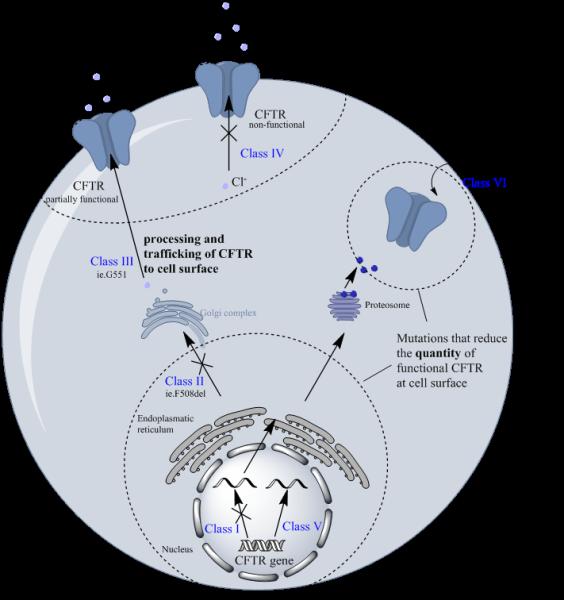
Schematic diagram showing the outcomes of CFTR mutation classes I, II, III, IV, V, and VI. Mutation that reduce the trafficking of functional CFTR to the cell surface are classed as III and IV. Mutations that reduce the total quantity of cellular CFTR are classed as I, II, V, VI.
The class II mutation causes aberrant processing of the CFTR protein, which leads to either its trafficking to the incorrect location or the presentation of non-functional mature glycoform of the CFTR (Figure 1B) (13). The F508del mutation is by far the most common mutation in the population with CF, with an overall prevalence of 70% and about 50% homozygous patients (16), positioning itself as a major target for CFTR corrector/modulator therapeutics (28). This mutation produces a severe defect in the processing and trafficking of CFTR resulting in little to no CFTR protein presentation on the cell surface (16, 25, 30-36), and additionally leads to abnormal opening of the channel in the limited amounts of CFTR that actually makes it to cell surface (28). Class IV mutations result in defective ion conductance by the CFTR (13). Class V mutations marginally decrease the amount of functional CFTR at cell surface and class VI mutations impact on the stability of the CFTR at the cell surface (37-40). Notwithstanding, patients with class V and VI mutations still retain some functional CFTR expression (Figure 1B).
Serendipitously, the concept for the development of CFTR modulators originated from background research on the class II F508del-CFTR mutation. The F508del mutation in NBD1 leads to a decrease in the thermostability of the CFTR (23, 41, 42). Moreover, the F508del mutation perturbs the native interactions between NBD1 and the coupling helix of MSD2 (23). The breakthrough finding was that mis-folding of the defective F508del-CFTR could be corrected by lowering the temperature (<30°C) or by employing chemical chaperones (e.g. glycerol) (43-48). For the first time, this indicated that defective CFTR function could be restored, this in turn led to extensive efforts by Vertex to identify small molecule CFTR modulators (43-48). Mechanistically, CFTR modulators fall into three classes: 1) suppressors that prevent premature termination of protein synthesis (i.e. ataluren), 2) correctors that partially correct folding and processing defects (i.e. lumacaftor), and 3) potentiators that increase channel gating and conductance (i.e. ivacaftor).
Eckford et al (49) were the first to report that ivacaftor binds directly to the CFTR protein causing CFTR channel opening via a non-conventional ATP-independent mechanism (49). In CHO (Chinese hamster ovary) and human CFPAC-1 (Human Caucasian pancreatic adenocarcinoma) cell culture models lumacaftor has been shown to increase of the amount of functional CFTR present at cell surface by ~34% (50, 51). Van Goor et al (32) showed that in cultured human bronchial epithelial cells lumacaftor improved F508del-CFTR trafficking to the cell surface and furthermore enhanced chloride secretion up to 14% (32). Furthermore, Ren et al (50) proved that lumacaftor stabilized the F508del-CFTR N-terminal domain (the MSD-1 region) and partially restored the function. Coincidently, Kopeikin et al (51) have shown that the main effect of ivacaftor on F508del-CFTR is to increase the ATP-dependent opening rate of the channel. The authors purport that this results from the destabilization of the closed-state, thereby shifting the balance towards the open state (Figure 2) (51). On a purely mechanistic level, there is substantial evidence in favour of combining the CFTR potentiator ivacaftor with the CFTR corrector lumacaftor. However, as will be outlined in the proceeding discussion, on a pharmacokinetic level, the combination of ivacaftor with lumacaftor may not be a marriage made in heaven.
Figure 2.
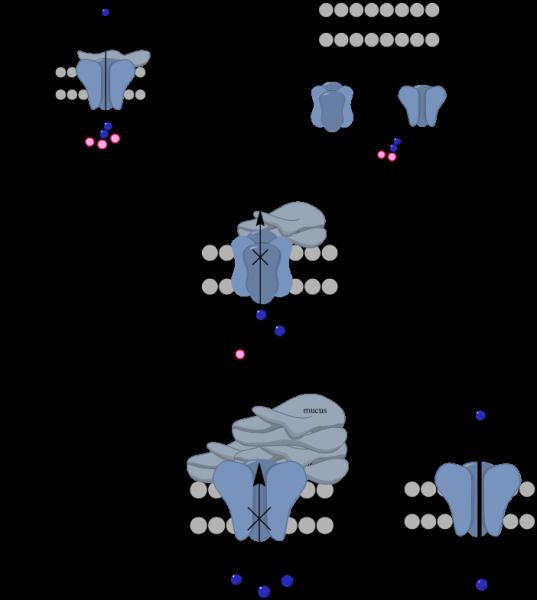
Mutant F508del CFTR channel after prolonged Orkambi treatment. Left, Orkambi therapy improves long-term CFTR function by acting to enhance the trafficking of CFTR to the cell surface and enhancing Cl− influx intracellularly. Right, alternatively, prolonged Orkambi therapy could causes destabilization and accelerated turnover of CFTR, resulting in less functional CFTR being presented on the cell surface.
Orkambi - ‘the promised land of clinical efficacy’
The three pillars of CF therapy are 1) maintaining lung function, 2) reducing the frequency of pulmonary exacerbations, and finally 3) improving nutritional status. (16, 52). Ivacaftor (VX-770) is the first FDA-approved CFTR potentiator (53-55). Ivacaftor functions as a G551D-CFTR potentiator producing an increased channel opening probability to enhance chloride influx (49, 56). Although Vertex's first-ofits-kind CFTR potentiator drug Kalydeco (ivacaftor) represents a major breakthrough in CF therapy, its target patient collective is limited to around 4% of patients with CF (G551D mutation) [glycine (G) in position 551 is replaced by aspartic acid (D)] (3, 21). Additionally Ivacaftor was later approved for other gating mutations such as G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, or S549R (57).
To solve this limitation, Vertex has developed the novel corrector-potentiator combination Orkambi (lumacaftor-ivacaftor) which targets the vast majority of CF patients carrying the homozygous F508del-CFTR. The F508del is an in frame deletion of the CFTR gene which results in the loss of phenylalanine (F) at position 508 (25, 30-36). Lumacaftor (VX-809) is a first of its kind CFTR corrector that facilitates processing and trafficking of the F508del-CFTR protein to increase the amount at the epithelial cell surface (Table 1) (16). This strategy combines a CFTR corrector which rescues F508del-CFTR to the cell surface with a potentiator that increases the channel gating open probability (50), effectively expands the treatment window to ~28% of the CF population (58). In the US alone, Orkambi now expands the patient collective from only 1,950 patients that were treatable with Kalydeco to 15,000 patients with homozygous F508del-CFTR mutation (16, 25, 30-32). In the Vertex clinical phase 3 studies 103 and 104 (TRAFFIC and TRANSPORT) (Table 2) involving 1,108 homozygous F508del-CFTR patients, Orkambi was shown to decrease sweat chloride secretion and increase ppFEV1 (percent predicted forced expiratory volume in 1 second) over placebo (16). According to Vertex's FDA briefing, Orkambi proved to be superior over placebo in all key secondary endpoints such as change in ppFEV1 (5.3% difference to placebo), change in Body Mass Index (0.36% difference to placebo), change in CF questionnaire revised respiratory domain score (2.9% difference to placebo), reduction in sweat chloride (−11% compared to placebo), and number of pulmonary exacerbations (0.7 % vs 1.2 % placebo) (16, 59). Notably, the ppFEV1 was sustained over a 48-week period (16, 50). Encouragingly, patients receiving Orkambi experienced less lung infections requiring hospitalization [−61% (P<0.0001)] or intravenous (IV) antibiotics [−56% (P<0.0001)] (16).
Table 1.
Overview of the major metabolites of ivacaftor and lumacaftor
| Chemical properties | ||
| Ivacaftor | ||
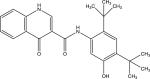
|
PubChem CID | 16220172 |
| Chemical names | Ivacaftor; VX-770; Kalydeco; 873054-44-5; VX770; N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide | |
| Molecular formula | C24H28N2O3 | |
| Molecular weight | 392.49 g/mol | |
| Metabolism | primarily metabolized by CYP3A4 into M1 and M6 | |
| Plasma protein binding | 99% plasma protein bound, primarily to HSA and AGP | |
| Hydroxymethyl-Ivacaftor (M1) | ||
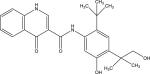
|
PubChem CID | Not found |
| Chemical names | N-(2-(tert-Butyl)-5-hydroxy-4-(1-hydroxy-2methylpropane-2-yl)phenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide | |
| Molecular formula | C24H28N2O4 | |
| Molecular weight | 408.49 g/mol | |
| Metabolism | Ivacaftor oxidation by CYP3A4 to M1 | |
| Plasma protein binding | Highly bound but lesser extent than ivacaftor | |
| Ivacaftor-Carboxylate (M6) | ||
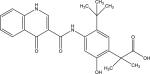
|
PubChem CID | 46917263 |
| Chemical names | UNII-38WUF8D79H; 38WUF8D79H; 1246213-24-0; 2-(5-(tert-Butyl)-2-hydroxy-4-(4-oxo-1,4-dihydroquinoline-3-carboxamido)phenyl)-2-methylpropanoic acid; Ivacaftor (m6); Ivacaftor carboxylate; | |
| Molecular formula | C24H26N2O5 | |
| Molecular weight | 422.47 g/mol | |
| Metabolism | Ivacaftor oxidation by CYP3A4 to M6 | |
| Plasma protein binding | Highly bound but lesser extent than ivacaftor | |
| Lumacaftor | ||
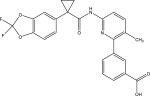
|
PubChem CID | 16678941 |
| Chemical names | 936727-05-8; Lumacaftor; VX-809; VX809; VX 809; 3-(6-(1-(2,2-Difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid; | |
| Molecular formula | C24H18F2N2O5 | |
| Molecular weight | 452.41 g/mol | |
| Metabolism | not extensively metabolized | |
| Plasma protein binding | 99% plasma protein bound, primarily to HSA | |
Table 2.
Chronology of the randomized, double-blind, placebo-controlled Orkambi phase 2 and 3 studies in patients bearing a F508del-mutation of the CFTR
| Type | Name | Objectives | Treatment | Patient Collective | Duration | Endpoints |
|---|---|---|---|---|---|---|
| Phase 2A | 101 | Safety and tolerability of LUMA | LUMA 25, 50, 100, 200 mg qd; orally | 89 patients with a homozygous F508del-CFTR mutation | 28 d |
Primary endpoints: • Change of CFTR function measured by sweat chloride concentration • Safety Secondary endpoints: • Absolute change in predicted FEV1 percentage • Change in sweat chloride concentration from baseline • Change in CFQ-R score |
| Phase 2 | 102 | Safety and tolerability of LUMA-IVA combination | • Cohort 1: LUMA 200 mg qd followed by LUMA 200 mg qd/ IVA 150 or 250 mg q12h • Cohort 2: LUMA 200, 400, 600 mg qd followed by LUMA/IVA 250 mg q12h • Cohort 3: LUMA 400 mg q12h followed by LUMA 400 mg q12h/ IVA 250 mg q12h • Cohort 4: LUMA 400 mg q/12h followed by LUMA 400 mg qd/ IVA 250 mg q12h |
312 patients with homozygous (cohort 1 &3) or heterozygous (cohort 4) F508del-CFTR mutation (cohort 2 mixed) | • Cohort 1: 14 d mono- followed by 7 d combination therapy • Cohort 2 & 3: 28 d mono-followed by 28 d combination therapy • Cohort 4: 56 d of combination therapy |
|
| TRAFFIC and TRANSPORT studies | ||||||
| Phase 3 | 103 | Efficacy and safety of LUMA-IVA combination | LUMA 600 or 400 mg qd/ IVA 250 mg q12h | 549 patients with a homozygous F508del-CFTR mutation | Up to 24 w + 5 d |
Primary endpoints: • Absolute change from baseline in predicted FEV1 percentage at week 24 Secondary endpoints: • Relative change from baseline in predicted FEV1 percentage (for week 16 & 24) • Absolute change from baseline in BMI percentage at week 24 • Absolute change from baseline in CFQ-R score at week 24 • Percentage of patients with a relative increase from baseline of ≥5% in the percentage predicted FEV1 • Number of pulmonary exacerbations through week 24 • Safety |
| Phase 3 | 104 | Efficacy and safety of LUMA-IVA combination | LUMA 600 or 400 mg qd/ IVA 250 mg q12h | 559 patients with a homozygous F508del-CFTR mutation | Up to 24 w + 5 d | |
| Phase 3 | 105 | Long term efficacy and safety of LUMA-IVA combination | • Part A: LUMA 600 or 400 mg qd/ IVA 250 mg q12h • Part B: LUMA 400 mg qd/IVA 250 mg q12h |
• Part A: 1031 patients with a homozygous F508del-CFTR mutation (including patients from study 103/104 • Part B: 119 patients with a heterozygous F508del-CFTR mutation |
~96 w | |
Orkambi phase 2 efficacy results
Lumacaftor by itself went into a phase 2 clinical study, where patients with CF with homozygous F508del-CFTR mutations were randomly assigned 25-200 mg/day lumacaftor for 4 weeks (Table 2) (43). The study findings revealed that lumacaftor produced a dose-dependent, rapid and sustained reduction in sweat chloride levels (P=0.0013). The reduction in sweat chloride values was rapid and sustained, with changes seen within 7 days of lumacaftor therapy. The mean change from baseline in sweat chloride concentration (mmol/l) were +2.2 in the placebo group, −0.5 (25 mg), −3.7 [50 mg (95% CI −7.1 to −0.28, P=0.03)], −2.3 (100 mg); and −6.6 [200 mg (95% CI −10.27 to −2.83, P=0.0008)] (20). However, the study did not show sufficient clinical benefit due to increased pulmonary exacerbation rates, and disappointing FEV1-scores [percentage changes relative from baseline FEV1: placebo +0.07, lumacaftor 25 mg (−2.46), 50 mg (−2.15), 100 mg (+0.32), and 200 mg (+0.47)] and CFQ-R scores [respiratory domain score after 28 days: placebo +4.5, lumacaftor 25 mg (−5.2), 50 mg (−6.3), 100 mg (−1.3), and 200 mg (+2.2)] (20, 43). Lumacaftor alone did not show sufficient clinical benefit, including no improvement in lung function or quality of life, over the course of the phase 2 study. Therefore, lumacaftor monotheraphy did not proceed into the phase 3 trials.
In further phase 2 trials, study 102 efficacy results studies reported by Boyle et al (37) (Table 2) showed that Orkambi therapy was able to achieve an improvement in lung function (measured as FEV1) for a collective of 312 CF patients suffering from homozygous F508del-CFTR mutation (initial lung function 40-90% FEV1) (37, 60). In cohort 1 (Table 2), 62 homozygous F508del-CFTR patients received 200 mg lumacaftor once daily, combined with 150 or 250 mg doses of ivacaftor (twice daily). Both of these treatment groups showed a reduction of sweat chloride [~9.1 mmol/L (p<0.001)] compared to placebo; however, no significant changes in FEV1 were observed (60, 61). In cohorts 2 and 3, mean sweat chloride concentrations did not decrease significantly between day 28 and day 56 [cohort 2: p=0.889 (Lumacaftor 200 mg once per day, ivacaftor 250 mg every 12 h); p=0.664 (Lumacaftor 400 mg once per day, ivacaftor 250 mg every 12 h); p=0.218 (Lumacaftor 600 mg once daily, ivacaftor 250 mg every 12 h) and cohort 3: p=0.544 (Lumacaftor 400 mg-Ivacaftor 250 mg every 12 h)]; compared to baseline. However a pre-planned secondary analysis, Boyle et al observed that the total change in sweat chloride over the entire study period of 56 days decreased significantly in the lumacaftor 400 mg and the lumacaftor 600 mg group for 28 days followed by the addition of ivacaftor 250 mg for a further 28 days: −9.1 mmol/L [95% CI −13.3 to −4.9; p<0.001;change from baseline)] and −8.9 mmol/L [(−13.1 to −4.7; p<0.001(change from baseline)), respectively. For the patients from cohort 3 the total change in sweat chloride over the entire study period was −10.3 mmol/L (−16.7 to −4.0 p=0.002). These reductions were significant compared with placebo [(−11.1, −18.5 to −3.7, p=0.00 ;(treatment vs. placebo)] (60). Most importantly, in cohort 2, patients treated with lumacaftor 600 mg once daily, displayed an improvement in FEV1 (change from baseline) of 5.6% (p=0.013) (60). In cohort 3, FEV1 only improved during the combination period by 7.7% (p=0.003). These dose ranging studies suggest that lumacaftor should be dosed at a higher ratio to ivacaftor, which leads us to the phase 3 studies.
Orkambi phase 3 efficacy results
The multi-centre phase 3 studies led by Wainwright, Elborn, Ramsay, and Boyle et al (58) corroborated the phase 2 findings (58). They conducted two phase 3, randomized, double-blind, placebo-controlled studies to investigate the effectiveness of lumacaftor combined with ivacaftor in 1,108 patients with CF 12 years or older that had the homozygous F508del-CFTR mutation and a FEV1 of 40-90% (58). In contrast to the phase 2 study, the patients were given a high dose of lumacaftor 600 mg once daily or 400 mg every 12 hours in combination with ivacaftor 250 mg every 12 hours for an extended duration of 24 weeks (58). The non-placebo patients displayed an increase in FEV1 within 15 days of the initiation of therapy that was sustained throughout 24 weeks. The reported findings in The New England Journal of Medicine showed a significant improvement of predicted FEV1 (change from baseline) in the lumacaftor-ivacaftor group over placebo (3.3% for the 600 mg once daily and 2.8% for the 400 mg twice daily, respectively [p<0.001 for all groups]); less pulmonary exacerbations (39% fewer exacerbations in the 400 mg twice daily lumacaftor group compared to placebo [p<0.001] and 30% fewer exacerbation in the 600 mg once daily lumacaftor group compared to placebo [p=0.001]). Similarly to the phase 2 studies, the phase 3 study patients displayed an overall lower rate of hospitalization and/or the use of IV antibiotics (58, 61).
Pharmacokinetics
Orkambi is prescribed as an oral dose twice daily every 12 h in the form of a tablet (200 mg lumacaftor and 125 mg ivacaftor) (16, 32-36). After administration, lumacaftor and ivacaftor are absorbed in the gastrointestinal tract and exhibit peak plasma concentrations (Tmax) in ~3-6 h and ~4 h, respectively (29). The systemic exposure of lumacaftor is approximately 2-fold higher in healthy individuals compared to patients with CF (Table 3) (59). The difference in AUC might be attributed to absorption difficulties in patients with CF due to deficiencies in pancreatic enzymes, impaired bowel movement or thick mucus accumulation. However, experimental data is needed to validate these postulates. The exposure of ivacaftor in healthy or patients with CF is similar (29). The exposure of lumacaftor and ivacaftor is increased when given with high-fat containing food (29). After a single dose with high-fat containing food, lumacaftor exposure was ~2-fold higher and ivacaftor exposure was ~3-fold higher compared to those administration on a fasting stomach, respectively (Table 3) (59). It is a common phenomenon that the oral absorption of lipophilic drugs is improved by dietary fats, which leads us to the postulate that the oral absorption (the oral bioavailability of Orkambi ranging from 30-100%) (29) of Orkambi could be optimized via a lipid-based formulation (16, 29, 53). Presently, there is very little information regarding the peak and steady-state plasma concentrations of Kalydeco or Orkambi. Schneider et al., have recently published a HPLC-LC/MS method for the monitoring of exposure-response relationships of ivacaftor and lumacaftor (62). Given the noted metabolism of ivacaftor and lumacaftor monitoring of exposure-response relationships is requisite to achieve optimizing dosage regimens for Kalydeco or Orkambi therapy. Hence, there remains the question of whether current dosage regimens are achieving optimal exposure levels and/or overdosing is an issue.
Table 3.
| ORKAMBI | |||
|---|---|---|---|
| Lumacaftor | Ivacaftor | ||
| Absorption | single oral dose | exposure increases by 2-fold if taken with fat-containing food | exposure increases by 3-fold if taken with fat-containing food |
| multiple oral dose in combination | exposure increase proportional to dose from 200 mg/24h to 400 mg/24h | exposure increase with dose from 150 mg/12h to 250 mg/12h | |
| Distribution | 99% plasma protein bound, primarily to HSA | 99% plasma protein bound, primarily to HSA and AGP | |
| Half-life | 26 h | 12 h (in combination with lumacaftor 9 h) | |
| Metabolism | not extensively metabolized | primarily metabolized by CYP3A4 into M1 and M6 | |
| Excretion | 51% unchanged in faeces; minimal urinary excretion | 87·8% excreted in faeces after metabolic conversion to mainly Ml and M6; minimal urinary excretion of ivacaftor and metabolites | |
| Drug-drug interactions | CYP3A4 | inducer | Substrate |
| CYP2C9 | inhibitor | potential inhibitor | |
| P-glycoprotein | inducer/ inhibitor | weak inhibitor | |
| Side effects (37, 73) | ORKAMBI (after 24 weeks of treatment) | ||
| 14% | dyspnea, nasopharyngitis, nausea | ||
| 8-12% | respiration abnormal, fatigue, upper respiratory tract infections, diarrhea | ||
| <7% | Influenza, rhinorrhea, flatulence, rash, blood creatine phosphokinase increase | ||
| Ivacaftor (after 48 weeks of treatment) | |||
| 24% | - | headache | |
| 22-15% | - | Oropharyngeal pain, upper respiratory tract infection, nasal congestion, abdominal pain | |
| 15-9% | - | Nasopharyngitis, diarrhoea, rash, nausea, dizziness | |
| Potential drug-drug interactions for Orkambi (37) | |||
| CF Drug Class | Interaction to be expected with Orkambi | Dose adjustment | Alternative treatment option (no drug-drug interactions to be expected due to different metabolism and/or route of elimination) |
| Antibiotics | Clarithromycin Erythromycin Telithromycin |
Preferentially: seek alternative treatment option | Ciprofloxacin Azithromycin Levofloxacin Aztreonam Ceftazidim Colistimethate Colistin Tobramycin Sulfamethoxazol/ Trimethoprim |
| Antifungals | Itraconazole Ketoconazole Posaconazole Voriconazole |
Preferentially: seek alternative treatment option | Fluconazole |
| Anti-allergies and systemic corticosteroids | Montelukast Prednisone Methylprednisone |
Dose adjustment for Montelukast is not recommended. Increase dose of Prednisone and Methylprednisone |
Budesonide Fluticasone Cetirizine |
| Anti-inflammatives | Ibuprofen | Increase dose of Ibuprofen | |
| Antidepressants | Citalopram Escitalopram Sertaline |
Increase dose of these antidepressants | |
| Hormonal contraceptives | Estrogen Progesterone |
Hormonal contraceptives should not be relied on. | |
| Proton pump inhibitors, H2-blockers, Antiacids | Omeprazole Esomeprazole Lansoprazole |
Increase dose of the -prazoles | |
Ivacaftor and lumacaftor are both very hydrophobic drugs and as such are ~99% bound to plasma proteins, which significantly limits the free (active) drug concentration (1). Ivacaftor is highly bound to human serum albumin (HSA) and α1-acid glycoprotein (AGP) (1). Similarly, lumacaftor preferentially binds to HSA (29, 59). Schneider et al have investigated the impact of co-administered CF drugs on the plasma protein binding of ivacaftor (1). Using surface plasmon resonance and fluorimetric binding assays that measure the displacement of site-selective probes, they have showed that interactions between ivacaftor are to be expected with ducosate, montelukast, ibuprofen, dicloxacillin, omeprazole and loratadine due to their ability to strongly compete for the ivacaftor binding sites on HSA and AGP. The half-life of lumacaftor administered as Orkambi is about 26 h in patients with CF. The half-life of ivacaftor administered as Kalydeco is 12 h and ~9 h when administered as Orkambi (29, 59). Due to high plasma protein binding there is a strong possibility that co-administered CF drugs could compete with ivacaftor or lumacaftor for the same plasma protein binding sites and impact on the free drug concentration. This in turn could lead to variable free drug plasma concentrations and thereby impact therapeutic outcomes (1). Similarly, the shorter half-life of ivacaftor when administered as Orkambi may be due to the displacement from its plasma protein binding sites by lumacaftor, which in turn leads to increased free drug and clearance. Our group has reported that such drug-drug interactions between ivacaftor and other CF drugs can be expected with docusate, montelukast, ibuprofen, dicloxacillin, omeprazole, and loratadine. Ideally, Kalydeco and Orkambi should be administered in a staggered dosage regimen with these CF drugs to maximize free drug concentrations and clinical efficacy (1).
Ivacaftor is heavily metabolized in humans, primarily by CYP3A4 into 11 metabolites found in bile, urine, plasma, and faeces (53, 59). Ivacaftor is primarily metabolized into active hydroxymethyl-ivacaftor (M1) and inactive ivacaftor-carboxylate (M6) by oxidation (Table 1) (53, 59). A disconcerting aspect of the metabolism of Orkambi are the potentially complex interactions between lumacaftor and ivacaftor themselves and with the supplementing medications to treat the symptoms of this multisystem disease (Table 3) (52). Notably, strong CYP3A4 inhibitors such as clarithromycin or -azole antifungals have been reported to increase exposure of ivacaftor (16). The product information inset states that in the first week of initiating Orkambi or Kalydeco therapy in conjunction with these drugs, half of the dose of ivacaftor/lumacaftor is recommended (Table 3) (59). Similarly, the concomitant use of strong CYP3A4 inducers such as rifampin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St John's Wort (hypericum perforatum) are not recommended as the exposure of ivacaftor is decreased by ~57% (59). Lumacaftor is not heavily metabolized with the majority being excreted unchanged in the faeces (16, 29). The approximately 10% of lumacaftor that is metabolized occurs via oxidation and glucuronidation (29). In the FDA and EMA reports for Orkambi, Vertex mentions lumacaftor itself is an inducer of the cytochrome P450 drug metabolizing enzymes (16, 29). Paradoxically ivacaftor is a substrate of cytochrome P450 3A4 (CYP3A4) which leads us to another potential antagonistic drug-drug interaction between the actual components of Orkambi themselves (i.e. lumacaftor versus ivacaftor) (29, 59). To this end we have found that the steady-state plasma concentration of ivacaftor in patients receiving Orkambi therapy is significantly lower than the levels we measured in patients receiving Kalydeco therapy (62).
Side effects
In general oral administration of Orkambi showed an acceptable tolerability profile when added to the standard therapy of patients with CF aged ≥ 12 years in the 24-week TRAFFIC and TRANSPORT studies (58). However, to date the 120-week extension of these trials, called PROGRESS study is still ongoing. Aside from the benefits of Orkambi, it is also essential to examine the other side of the coin, its potential caveats. Ironically, the major adverse effects reported for patients undergoing Orkambi therapy versus placebo were dyspnoea (14%:7.8%) and respiratory chest tightness (9.8%:5.9%) (16, 29, 59). Diarrhoea, nausea, and upper respiratory tract infections have also been reported as adverse effects (16, 59). Notwithstanding, Vertex suggests that most respiratory adverse effects resolve within the first few weeks of treatment (16, 59). Additionally, elevation of liver enzymes and hepatobiliary disorders were reported in some patients compared to placebo (0.9%) (16). This could restrict Orkambi treatment, as up to 35% of patients with CF suffer from liver disease, a known clinical manifestation as a result of CFTR dysfunction in their biliary tract cells (15, 16, 63-66). Coincidently, Vertex's pooled phase 2 studies 103 and 104 were discontinued due to the patients exhibiting elevated liver transaminases (0.5%) and respiratory events (0.7%) (16). If the benefits of therapy outweigh the risks, Orkambi could be used carefully at a lower dosage and importantly, with close monitoring in patients with advanced liver disease (59). Thus, liver function monitoring is recommended prior to and during the course of Orkambi therapy.
Due to their lipophilicity, ivacaftor and lumacaftor could potentially cross the blood brain barrier. Pharmacodynamic studies in rats have shown that ivacaftor functions as an inhibitor of the monoamine transporter and binds serotonin 5-HT2C receptors (16), suggesting that Kalydeco and Orkambi could possess neuropharmacological activity. To date, no adverse data for Orkambi in pregnant women has been reported; currently Orkambi is listed as a pregnancy category ‘B’ drug that requires additional human trials (61, 67). Kaminski et al (68) were the first to report a successful uncomplicated pregnancy during Kalydeco therapy for a single patient with CF (68). However, breastfeeding mothers should remain cautious, as both ivacaftor and lumacaftor have been detected in the milk of lactating rats (61, 67).
A beneficial side effect of ivacaftor is its antimicrobial activity (69-71). Schneider et al have shown synergistic antibacterial activity of ivacaftor in combination with polymyxin B against a panel of polymyxin-resistant Gram-negative pathogens that commonly colonize the lungs of patients with CF, including strains that were resistant to the quinolone antibiotic ciprofloxacin, which is structurally related to ivacaftor (69, 70). As polymyxins (in particular colistin) are commonly used in patients with CF for treatment of lung infections caused by multidrug-resistant P. aeruginosa, such a synergistic killing effect plays a key role in minimising any potential resistance to polymyxins (70).
Ivacaftor versus lumacaftor
Although the aforementioned TRANSPORT and TRAFFIC clinical studies have confirmed that Orkambi therapy improves F508del-CFTR function from day 1 to day 56 of therapy, the reasons for the discouraging long-term results (>24 weeks) were not obvious from the available clinical data (TRANSPORT and TRAFFIC studies for long term efficacy and safety are still ongoing). Recently, evidence has emerged from a number of independent laboratories suggesting that prolonged exposure to ivacaftor counteracts the corrector function of lumacaftor by destabilising the lumacaftor-rescued mature glycoform of F508del-CFTR (21, 22, 72). Such inhibitory interactions are especially concerning given that lumacaftor only partially restores (11-15%) F508del-CFTR surface expression (21, 22). The in vitro studies by Gentzsch et al (22, 43) suggested that chronic administration of ivacaftor caused a dose-dependent reversal of lumacaftor-mediated CFTR correction in homozygous F508del human primary bronchial epithelial cells. They reported that in cells chronically treated with 5 μM lumacaftor, acute exposure to ivacaftor (5 μM) resulted in an increase of CFTR mediated ion transport within minutes (22). Whereas, chronic treatment (24-48 h) of lumacaftor rescued cells with 5 μM ivacaftor resulted in a decrease in channel conductance (22). This result reflected the destabilization of corrected ΔF508 CFTR by ivacaftor, dramatically increasing its turnover rate. Chronic ivacaftor treatment furthermore reduced mature wild-type CFTR levels and function. These findings demonstrate that chronic treatment with CFTR potentiators and correctors may have unexpected effects that cannot be predicted from short-term studies. Furthermore, it was demonstrated that the CFTR-mediated short circuit current increases after addition of ivacaftor to corrector-rescued F508del-CFTR, albeit this effect was transient (22). These findings may be an indicator of a rapidly decreasing quantity of functional protein at the apical membrane (22). They also showed that the loss of corrected-rescued function of F508del-CFTR treated with lumacaftor in combination with ivacaftor was reflected in reduced chloride secretion responses (22). In their Western blot the amount of mature F508del-CFTR was actually diminished and instead the F508del-CFTR protein only appeared as an immature band B (22). Therefore, it is essential that we gain further knowledge of the interaction and interference between CFTR potentiators and CFTR correctors.
Another recent study by Veit et al (21) indicated that ivacaftor reduces the correction efficacy of lumacaftor. Their results indicated that ivacaftor destabilizes the native CFTR protein in immortalized and primary human respiratory epithelia (21). The authors purport that destabilization is caused by ivacaftor itself which could further play an essential role in the faulty assemblage of the CFTR protein. Furthermore, this implies that the lumacaftor rescued F508del-CFTR is still defective and not functionally equivalent to the native CFTR (73). These findings show that chronic treatment with Orkambi may have unexpected long-term effects that are not predictable from the clinical studies.
Cost versus benefit
Affordability is a key issue with Orkambi therapy (28). As the price is supposed to be representative of the compensation Vertex would expect to cover their development costs (due the small patient collective), we would assume the price of Kalydeco to have gone down with the introduction of Orkambi (28). However, pricing for both drugs remains extraordinary high, despite the low cost of goods (ivacaftor and lumacaftor are easily accessible synthetically and therefore the cost of development is presumably low). However, we must take into account the high development costs which would help justify the high price of these drugs. Orkambi treatment is estimated at $259,000 USD per year, which is lower than the established pricing of Kalydeco at $307,000 USD per year (61, 74). Given the price differential, it would be more economically prudent for patients with G551D-CFTR to simply adopt Orkambi therapy. However, Orkambi has yet to be approved for patients with G551D-CFTR. Moreover, given that patients with G551D-CFTR express CFTR on the epithelial cell surface, this may not prove successful due to the increase in CFTR turnover rates reported with Orkambi therapy (Figure 2) (21, 22, 45). As patients are forced to take Orkambi or Kalydeco as a lifelong therapy, the impact on the health care budget is staggering (40). In view of the modest clinical outcomes reported for Orkambi therapy, the cost versus benefit becomes questionable.
Perspective-Can CF patients finally catch a breath?
Orkambi represents a first-of-its-kind breakthrough CF treatment strategy. However, since its release a number of key questions remain unanswered. First and foremost, unlike the experience with Kalydeco, patients receiving Orkambi therapy displayed only modest improvements in lung function and pulmonary exacerbations (16). Secondly, antagonistic drug-drug interactions could potentially limit Orkambi's clinical efficacy (57). Despite these concerns, the long-term benefits of Orkambi require further assessment and we are all curiously awaiting the results of the ongoing-longterm TRANSPORT and TRAFFIC studies for these two revolutionary CF drugs.
Acknowledgments
This study is supported by a research grant to J.L and T.V. from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (R01AI111965). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J. L. is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow, and T. V. is an Australian NHMRC Industry Career Development Level 2 Research Fellow.
Footnotes
Authorship Contributions
Wrote or contributed to the writing of the manuscript:
Schneider, E.K., Reyes-Ortega, F., Li, J., Velkov, T.
References
- 1.Schneider EK, Huang JX, Carbone V, Baker M, Azad MA, Cooper MA, et al. Drug-drug plasma protein binding interactions of ivacaftor. Journal of molecular recognition : JMR. 2015;28(6):339–48. doi: 10.1002/jmr.2447. [DOI] [PubMed] [Google Scholar]
- 2.Farrell PM. The prevalence of cystic fibrosis in the European Union. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2008;7(5):450–3. doi: 10.1016/j.jcf.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Dean M, Santis G. Heterogeneity in the severity of cystic fibrosis and the role of CFTR gene mutations. Human genetics. 1994;93(4):364–8. doi: 10.1007/BF00201659. [DOI] [PubMed] [Google Scholar]
- 4.Solomon M. Leatte PN, editor. Cystic fibrosis-update on diagnosis and treatment from Cystic fibrosis etiology, diagnosisn and treatments. Treatments for Cystic fibrosis. 2009 [Google Scholar]
- 5.Tizzano EF, Buchwald M. CFTR expression and organ damage in cystic fibrosis. Annals of internal medicine. 1995;123(4):305–8. doi: 10.7326/0003-4819-123-4-199508150-00009. [DOI] [PubMed] [Google Scholar]
- 6.O'Sullivan BP, Flume P. The clinical approach to lung disease in patients with cystic fibrosis. Seminars in respiratory and critical care medicine. 2009;30(5):505–13. doi: 10.1055/s-0029-1238909. [DOI] [PubMed] [Google Scholar]
- 7.Ratjen F, Doring G. Cystic fibrosis. Lancet (London, England) 2003;361(9358):681–9. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 8.Conway SP, Pond MN, Hamnett T, Watson A. Compliance with treatment in adult patients with cystic fibrosis. Thorax. 1996;51(1):29–33. doi: 10.1136/thx.51.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Accurso FJ, Van Goor F, Zha J, Stone AJ, Dong Q, Ordonez CL, et al. Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2014;13(2):139–47. doi: 10.1016/j.jcf.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis PB, Schluchter MD, Konstan MW. Relation of sweat chloride concentration to severity of lung disease in cystic fibrosis. Pediatric pulmonology. 2004;38(3):204–9. doi: 10.1002/ppul.20054. [DOI] [PubMed] [Google Scholar]
- 11.Registry UC Registry UD, editor. US CFF Data Registry 2002. 2002 [Google Scholar]
- 12.McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet (London, England) 2003;361(9370):1671–6. doi: 10.1016/S0140-6736(03)13368-5. [DOI] [PubMed] [Google Scholar]
- 13.Goodman BE, Percy WH. CFTR in cystic fibrosis and cholera: from membrane transport to clinical practice. Advances in physiology education. 2005;29(2):75–82. doi: 10.1152/advan.00035.2004. [DOI] [PubMed] [Google Scholar]
- 14.Scott RB, O'Loughlin EV, Gall DG. Gastroesophageal reflux in patients with cystic fibrosis. The Journal of pediatrics. 1985;106(2):223–7. doi: 10.1016/s0022-3476(85)80291-2. [DOI] [PubMed] [Google Scholar]
- 15.Colombo C. Liver disease in cystic fibrosis. Current opinion in pulmonary medicine. 2007;13(6):529–36. doi: 10.1097/MCP.0b013e3282f10a16. [DOI] [PubMed] [Google Scholar]
- 16.FDA VERTEX Pharmaceuticals Incorporated, editor. Sponsor Briefing Document: ORKAMBI (Lumacaftor/Ivacaftor) for the Treatment of Cystic Fibrosis in Patients Age 12 Years and Older Who are Homozygous for the F508del Mutation in the CFTR Gene. commitee FACBMV FP-Ada. 2015:98. [Google Scholar]
- 17.Registry CFFP . In: Annual Data Report to the Center Directors. Foundation CF, editor. Bethesda, Maryland: 2013. [Google Scholar]
- 18.van Gool K, Norman R, Delatycki MB, Hall J, Massie J. Understanding the costs of care for cystic fibrosis: an analysis by age and health state. Value in health : the journal of the International Society for Pharmacoeconomics and Outcomes Research. 2013;16(2):345–55. doi: 10.1016/j.jval.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Chevreul K, Michel M, Brigham KB, Lopez-Bastida J, Linertova R, Oliva-Moreno J, et al. Social/economic costs and health-related quality of life in patients with cystic fibrosis in Europe. The European journal of health economics : HEPAC : health economics in prevention and care. 2016;17(Suppl 1):7–18. doi: 10.1007/s10198-016-0781-6. [DOI] [PubMed] [Google Scholar]
- 20.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–8. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Science translational medicine. 2014;6(246):246ra97. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cholon DM, Quinney NL, Fulcher ML, Esther CR, Jr., Das J, Dokholyan NV, et al. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Science translational medicine. 2014;6(246):246ra96. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckford PD, Ramjeesingh M, Molinski S, Pasyk S, Dekkers JF, Li C, et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chemistry & biology. 2014;21(5):666–78. doi: 10.1016/j.chembiol.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 24.Wine JJ, Brayden DJ, Hagiwara G, Krouse ME, Law TC, Muller UJ, et al. Cystic fibrosis, the CFTR, and rectifying Cl-channels. Advances in experimental medicine and biology. 1991;290:253–69. doi: 10.1007/978-1-4684-5934-0_25. discussion 69-72. [DOI] [PubMed] [Google Scholar]
- 25.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–4. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 26.Sokol RZ. Infertility in men with cystic fibrosis. Current opinion in pulmonary medicine. 2001;7(6):421–6. doi: 10.1097/00063198-200111000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiological reviews. 1999;79(1 Suppl):S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 28.Jones AM, Barry PJ. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm? Thorax. 2015;70(7):615–6. doi: 10.1136/thoraxjnl-2015-207369. [DOI] [PubMed] [Google Scholar]
- 29.EMA Assessment report ORKAMBI (ivacaftor/lumacaftor) European medicines agency EMEA/H/C/003954/0000. 2015 [Google Scholar]
- 30.Denning GM, Ostedgaard LS, Cheng SH, Smith AE, Welsh MJ. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. The Journal of clinical investigation. 1992;89(1):339–49. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. The Journal of biological chemistry. 1994;269(41):25710–8. [PubMed] [Google Scholar]
- 32.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(46):18843–8. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H, et al. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). The New England journal of medicine. 1990;323(22):1517–22. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 34.Kerem E, Kerem B. Genotype-phenotype correlations in cystic fibrosis. Pediatric pulmonology. 1996;22(6):387–95. doi: 10.1002/(SICI)1099-0496(199612)22:6<387::AID-PPUL7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 35.McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest. 2006;130(5):1441–7. doi: 10.1378/chest.130.5.1441. [DOI] [PubMed] [Google Scholar]
- 36.MacKenzie T, Gifford AH, Sabadosa KA, Quinton HB, Knapp EA, Goss CH, et al. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry. Annals of internal medicine. 2014;161(4):233–41. doi: 10.7326/M13-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. The Lancet Respiratory medicine. 2013;1(2):158–63. doi: 10.1016/S2213-2600(12)70057-7. [DOI] [PubMed] [Google Scholar]
- 38.Castellani C, Cuppens H, Macek M, Jr., Cassiman JJ, Kerem E, Durie P, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2008;7(3):179–96. doi: 10.1016/j.jcf.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–65. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 40.Bosch B, De Boeck K. Searching for a cure for cystic fibrosis. A 25-year quest in a nutshell. European journal of pediatrics. 2015 doi: 10.1007/s00431-015-2664-8. [DOI] [PubMed] [Google Scholar]
- 41.Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S, et al. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein science : a publication of the Protein Society. 2010;19(10):1917–31. doi: 10.1002/pro.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, et al. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein science : a publication of the Protein Society. 2010;19(10):1932–47. doi: 10.1002/pro.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Ma T. F508del-cystic fibrosis transmembrane regulator correctors for treatment of cystic fibrosis: a patent review. Expert opinion on therapeutic patents. 2015;25(9):991–1002. doi: 10.1517/13543776.2015.1045878. [DOI] [PubMed] [Google Scholar]
- 44.Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P & T : a peer-reviewed journal for formulary management. 2014;39(7):500–11. [PMC free article] [PubMed] [Google Scholar]
- 45.Merk D, Schubert-Zsilavecz M. Repairing mutated proteins--development of small molecules targeting defects in the cystic fibrosis transmembrane conductance regulator. Expert opinion on drug discovery. 2013;8(6):691–708. doi: 10.1517/17460441.2013.788495. [DOI] [PubMed] [Google Scholar]
- 46.Deeks ED. Ivacaftor: a review of its use in patients with cystic fibrosis. Drugs. 2013;73(14):1595–604. doi: 10.1007/s40265-013-0115-2. [DOI] [PubMed] [Google Scholar]
- 47.Pasyk S, Molinski S, Yu W, Eckford PD, Bear CE. Identification and validation of hits from high throughput screens for CFTR modulators. Current pharmaceutical design. 2012;18(5):628–41. doi: 10.2174/138161212799315957. [DOI] [PubMed] [Google Scholar]
- 48.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nature reviews Drug discovery. 2009;8(2):153–71. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. The Journal of biological chemistry. 2012;287(44):36639–49. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren HY, Grove DE, De La Rosa O, Houck SA, Sopha P, Van Goor F, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Molecular biology of the cell. 2013;24(19):3016–24. doi: 10.1091/mbc.E13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kopeikin Z, Yuksek Z, Yang HY, Bompadre SG. Combined effects of VX-770 and VX-809 on several functional abnormalities of F508del-CFTR channels. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2014;13(5):508–14. doi: 10.1016/j.jcf.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Sawicki GS, Sellers DE, Robinson WM. High treatment burden in adults with cystic fibrosis: challenges to disease self-management. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2009;8(2):91–6. doi: 10.1016/j.jcf.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.EMA Assessment report KALYDECO ivacaftor European Medicines Agency EMA/473279/2012 EMEA/H/C/002494//0000.
- 54.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eckford PD, Bear CE. Targeting the regulation of CFTR channels. The Biochemical journal. 2011;435(2):e1–4. doi: 10.1042/BJ20110461. [DOI] [PubMed] [Google Scholar]
- 56.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18825–30. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cholon DM, Esther Charles R, Gentzsch Martina. Efficacy of lumacaftor ivacaftor for the treatment of Cystic Fibrosis patients homozygous for the F508del-CFTR mutation. Expert Review of Precision Medicine and Drug Development. 2016 doi: 10.1080/23808993.2016.1175299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. The New England journal of medicine. 2015;373(3):220–31. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.VERTEX Highlights of prescribing information ORKAMBI tablets for oral use. Vertex prescribing infomation. 2015 [Google Scholar]
- 60.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. The Lancet Respiratory medicine. 2014;2(7):527–38. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 61.Brewington JJ, McPhail GL, Clancy JP. Lumacaftor alone and combined with ivacaftor: preclinical and clinical trial experience of F508del CFTR correction. Expert review of respiratory medicine. 2016;10(1):5–17. doi: 10.1586/17476348.2016.1122527. [DOI] [PubMed] [Google Scholar]
- 62.Schneider EK, Reyes-Ortega Felisa, Wilson John W., Kotsimbos Tom, Li Jian, Velkov Tony. Development of HPLC and LC-MS/MS methods for the analysis of ivacaftor, its major metabolites and lumacaftor in plasma and sputum of cystic fibrosis patients treated with ORKAMBI or KALYDECO. J Chrom B. 2016 doi: 10.1016/j.jchromb.2016.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology (Baltimore, Md) 1999;30(5):1151–8. doi: 10.1002/hep.510300527. [DOI] [PubMed] [Google Scholar]
- 64.Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. Journal of hepatology. 2004;41(6):920–5. doi: 10.1016/j.jhep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Feranchak AP. Hepatobiliary complications of cystic fibrosis. Current gastroenterology reports. 2004;6(3):231–9. doi: 10.1007/s11894-004-0013-6. [DOI] [PubMed] [Google Scholar]
- 66.Moyer K, Balistreri W. Hepatobiliary disease in patients with cystic fibrosis. Current opinion in gastroenterology. 2009;25(3):272–8. doi: 10.1097/MOG.0b013e3283298865. [DOI] [PubMed] [Google Scholar]
- 67.Goss CH, VanDevanter DR. CFTR modulators and pregnancy: Our work has only just begun. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2016;15(1):6–7. doi: 10.1016/j.jcf.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 68.Kaminski R, Nazareth D. A successful uncomplicated CF pregnancy while remaining on Ivacaftor. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2016;15(1):133–4. doi: 10.1016/j.jcf.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 69.Reznikov LR, Abou Alaiwa MH, Dohrn CL, Gansemer ND, Diekema DJ, Stoltz DA, et al. Antibacterial properties of the CFTR potentiator ivacaftor. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2014;13(5):515–9. doi: 10.1016/j.jcf.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider EK, Azad Mohammad, A. K., Han Mei-Ling, Zhou Qi (Tony), Wang Jiping, Huang Johnny X, Cooper Matthew A., Doi Yohei, Baker Mark A., Bergen Phillip J., Muller Mark, Li Jian, Velkov Tony. An “Unlikely” Pair: The Antimicrobial Synergy of Polymyxin B in Combination with the Cystic Fibrosis Transmembrane Conductance Regulator Drugs KALYDECO and ORKAMBI. ACS Infectious Disease. 2016 doi: 10.1021/acsinfecdis.6b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bernarde C, Keravec M, Mounier J, Gouriou S, Rault G, Ferec C, et al. Impact of the CFTR-potentiator ivacaftor on airway microbiota in cystic fibrosis patients carrying a G551D mutation. PloS one. 2015;10(4):e0124124. doi: 10.1371/journal.pone.0124124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matthes E, Goepp J, Carlile GW, Luo Y, Dejgaard K, Billet A, et al. Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). British journal of pharmacology. 2016;173(3):459–70. doi: 10.1111/bph.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baroni D, Zegarra-Moran O, Moran O. Functional and pharmacological induced structural changes of the cystic fibrosis transmembrane conductance regulator in the membrane solved using SAXS. Cellular and molecular life sciences : CMLS. 2015;72(7):1363–75. doi: 10.1007/s00018-014-1747-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health technology assessment (Winchester, England) 2014;18(18):1–106. doi: 10.3310/hta18180. [DOI] [PMC free article] [PubMed] [Google Scholar]