

ABSTRACT
EYA1 is known to be overexpressed in human breast cancer, in which the Myc protein is also accumulated in association with decreased phospho-T58 (pT58) levels. We have recently reported that EYA1 functions as a unique protein phosphatase to dephosphorylate Myc at pT58 to regulate Myc levels. However, it remains unclear whether EYA1-mediated Myc dephosphorylation on T58 is a critical function in regulating Myc protein stability in breast cancer. Furthermore, EYA1's substrate specificity has remained elusive. In this study, we have investigated these questions, and here, we report that depletion of EYA1 using short hairpin RNA (shRNA) in breast cancer cells destabilizes the Myc protein and increases pT58 levels, leading to an increase in the doubling time and impairment of cell cycle progression. In correlation with EYA1-mediated stabilization of cMyc and reduced levels of pT58, EYA1 greatly reduced cMyc-FBW7 binding and cMyc ubiquitination, thus providing novel insight into how EYA1 acts to regulate the FBW7-mediated Myc degradation machinery. We found that the conserved C-terminal haloacid dehalogenase domain of EYA1, which has been reported to have only tyrosine phosphatase activity, has dual phosphatase activities, and both the N- and C-terminal domains interact with substrates to increase the catalytic activity of EYA1. Enzymatic assay and nuclear magnetic resonance (NMR) analysis demonstrated that EYA1 has a striking conformation preference for phospho-T58 of Myc. Together, our results not only provide novel structural evidence about the conformation specificity of EYA1 in dephosphorylating phosphothreonine in Myc but also reveal an important mechanism contributing to Myc deregulation in human breast cancer.
KEYWORDS: EYA1, threonine phosphatase, Myc, degradation, FBW7, cell proliferation, breast cancer, deregulation
INTRODUCTION
The transcription factor Eyes absent (EYA) represents the prototype of a novel class of eukaryotic aspartyl protein tyrosine phosphatases (1–3) that play important roles in development and disease (4–6). While the conserved C-terminal haloacid dehalogenase domain of EYA (ED), which contains the signature motif for the aspartate-based serine/threonine (Ser/Thr) phosphatase family (1–3, 7), has been reported to have only tyrosine phosphatase activity (8), a recent study reported that the N terminus of EYA (NT), which bears no sequence similarity to any known Ser/Thr phosphatase families, has Thr phosphatase activity (8). Therefore, it remains unclear which domain of EYA contains residues for substrate binding and catalytic activity and how EYA achieves substrate specificity. It is also unclear how EYA uses its phosphatase activity to regulate distinct cellular processes, and very little is known about its physiological substrates.
We recently reported that EYA1 regulates Myc levels through its Thr phosphatase activity (9). Phosphorylation of Myc at S62 and T58 is critical for controlling Myc turnover in diverse cellular processes, such as the cell cycle and tumorigenesis (10). The Ser/Thr residues that are followed by proline (S/T-P) in many proteins are phosphorylated by mitotic kinases at the G2-to-M phase transition of the cell cycle (11), which is known to trigger many of the structural modifications during mitosis. Phosphorylated S/T-P motifs in polypeptides exist in cis or trans configuration owing to the proline peptide bond. The cis/trans conversion of the phospho-S/T-P (pS/T-P) motif is mediated by PIN1, which is an essential mitotic phosphorylation-directed proline isomerase (12). PP2A regulates Myc turnover by targeting phospho-S62 (pS62)-P, and its activity is enhanced by PIN1 (13, 14). However, the conformation specificity of EYA1 on phospho-T58 (pT58)-Myc and the sequence responsible for catalytic activity are unknown. Furthermore, although EYA is known to be overexpressed in many cancer cells (4), its activity in regulating Myc stability has not been studied.
In this study, we have integrated molecular, structural, biochemical, and loss-of-function analyses to define EYA1's conformation specificity in targeting pT58-Myc and its function in the cell cycle. We demonstrate for the first time that the conserved C-terminal haloacid dehalogenase ED has dual-specificity phosphatase (DUSP) activity and that both the NT and ED interact with substrates to increase the catalytic activity of EYA1. Enzymatic assay and nuclear magnetic resonance (NMR) spectroscopy analysis indicated that EYA1 has striking conformation preference for phospho-T58 of Myc. Depleting EYA1 using short hairpin RNA (shRNA) in human breast cancer cells destabilizes Myc and increases pT58 levels, leading to an increase in the doubling time and impairment of cell cycle progression. We found that EYA1 affects FBW7-Myc binding to regulate the FBW7-mediated Myc degradation machinery. Thus, our results indicate a critical function of EYA1 in regulation of Myc protein stability and cell proliferation, providing an important mechanism contributing to Myc deregulation in human breast cancer.
RESULTS
EYA1's N- and C-terminal domains interact with substrates to increase catalytic activity.
To map the region in EYA1 required for substrate binding and dephosphorylation, we first performed mutational analysis of EYA1 (Fig. 1A) in targeting phospho-threonine/serine/tyrosine peptides (Fig. 1C). EYA1 wild-type or mutant proteins expressed in 293 cells were purified and confirmed on SDS-PAGE using Coomassie blue staining (Fig. 1B). Purified EYA1 showed activity on pT58-Myc peptide that was ∼2.5-fold lower than that on phosphotyrosine pY-H2AX peptide (a known substrate for EYA's tyrosine phosphatase activity) (5) (Fig. 1D). In contrast, EYA1 exhibited little or no activity on pS62-Myc peptide or phospho-S2-CTD peptide (YSPTSPSYpS2PTSPS) (the C-terminal domain [CTD] of RNA polymerase II) (Fig. 1D). This result is in agreement with a previous study in which the CTD peptides YSPTSPSYpS2PTpS5PS and YSPTSPSYSPpT4SPS were used to show that EYA is much weaker as a serine phosphatase, as its activity is ∼10-fold lower than its threonine phosphatase activity (8). The NT or ED of EYA1 alone had reduced activity toward pT58 (Fig. 1E), suggesting that either the NT or ED has threonine phosphatase activity. The C-terminal haloacid dehalogenase ED contains the signature motif DXDXT (amino acids [aa] 327 to 331) for phosphatase activity found in the aspartate-based Ser/Thr phosphatases (Fig. 1A) (2, 3, 7), and the D327N (Asp327-to-Asn) mutation was previously reported to be a tyrosine-phosphatase-dead mutation within the ED (1, 5). In agreement with this, the D327N mutation almost abolished EYA1's tyrosine phosphatase activity toward pY-H2AX (Fig. 1F). Interestingly, the D327N mutation also decreased EYA1's threonine phosphatase activity on pT58 (Fig. 1E), suggesting that the D327 in the signature motif DXDXT (aa 327 to 331) conserved in the aspartate-based Ser/Thr phosphatases is also important for EYA1's threonine phosphatase activity. Replacement of a set of four conserved tyrosine residues in the NT with alanine (Y4A) (Fig. 1A), which has been reported to disrupt EYA4's threonine phosphatase activity (8), greatly reduced not only EYA1's threonine phosphatase activity in dephosphorylating pT58 (Fig. 1E) but also its tyrosine phosphatase activity toward pY-H2AX, which became comparable to that of ED alone (Fig. 1F). While the ED is the catalytic domain for tyrosine phosphatase activity, the domain alone exhibited weaker activity than the full-length EYA1 in dephosphorylating pY-H2AX (Fig. 1F). Combination of the Y4A and D327N mutations further decreased its activity on pT58 (Fig. 1E). These data indicate that the conserved C-terminal haloacid dehalogenase ED that contains the signature motif for the aspartate-based Ser/Thr phosphatase family reveals the hallmarks of a dual-specificity phosphatase, but both domains appear to interact with substrates to regulate EYA1's phosphatase activity.
FIG 1.

The conserved C-terminal haloacid dehalogenase EYA domain has dual tyrosine and threonine phosphatase activities. (A) Diagrams of EYA1 and its mutants used for this study. Three short signature motifs for the phosphatase subgroup of the haloacid dehalogenase superfamily (1–3, 7) are marked: motif I (aa 327 to 331), DXDXT/S; motif II (aa 497 to 500), hhh(T/S), where “h” is a hydrophobic residue; and motif III (aa 533 to 559), K(Xn)-hhhhGDXXX(D/E), which is less conserved. Among the three short motifs, the first motif, DXDXT/S (red), is also highly conserved in aspartate-based Ser/Thr phosphatase family members (7). Another signature catalytic motif, CX5R (CXXXXXR; aa 532 to 559), conserved in DUSPs, is marked in blue (27). Asterisks indicate the location of the D327N mutation in the ED. (B) Coomassie staining of an SDS-PAGE gel showing the purified FLAG-EYA1 wild type and its mutant proteins and cMyc proteins from HEK293 cells using anti-FLAG M2 beads. (C) Nonphosphorylated and phospho-Myc peptides, as well as control peptides phospho-S2-RNA PII CTD and phospho-Y-γH2AX, used for enzymatic assays. Phosphorylation modification of amino acid residues (pS/T/Y; red) is shown. (D) Dephosphorylation of different peptides by EYA1. (E and F) Dephosphorylation of pT58-Myc (E) or pY-H2AX (F) peptide by EYA1 and its mutants. All experiments were performed in triplicate, and the error bars represent standard deviations (SD).
To further examine the importance of the Y4 and D327 residues for EYA1's threonine phosphatase activity, we introduced Y4A in the NT alone and D327N in the ED alone (Fig. 1A) and performed an in vitro phosphatase assay. While the Y4A mutation markedly reduced the NT's threonine phosphatase activity (to 15.9% ± 2.1% of that of the wild-type NT), the D327N mutation only decreased the ED's threonine phosphatase activity (Fig. 1E; see Fig. 3A). This result suggests that the ED contains an additional residue(s), other than D327 alone, that is important for its threonine phosphatase activity.
FIG 3.
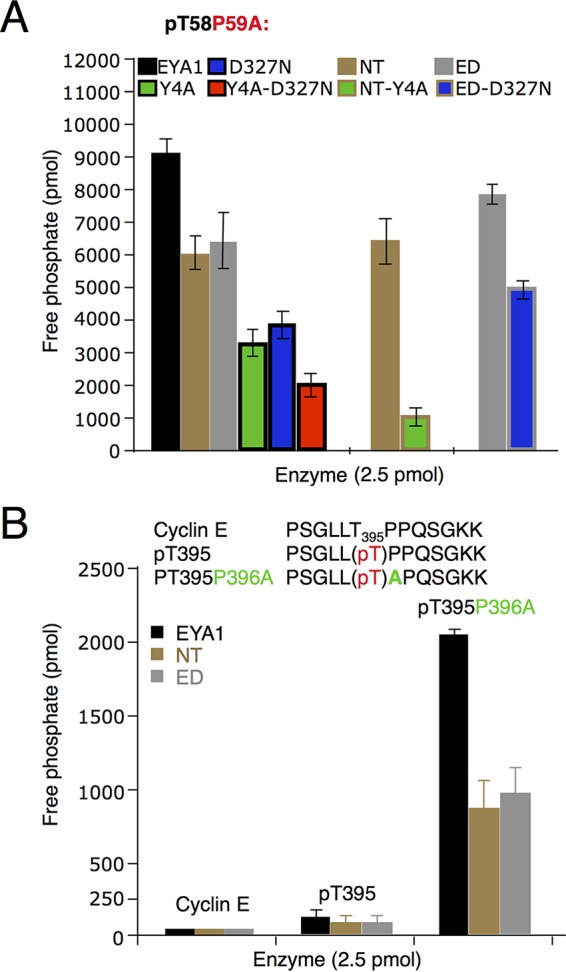
Dephosphorylation of different peptides by EYA1 and its mutants. Shown are Myc pT58P59A mutant (A) and cyclin E nonphosphorylated and pT395 wild-type and pT395A mutant (B) peptides. The error bars represent SD. P, ≤0.005.
EYA1 shows substrate preferences in catalyzing the phosphothreonine peptides.
To investigate EYA1's conformation specificity in targeting pT58-Myc, we synthesized a series of phosphoserine/threonine-containing peptide substrates with distinct mutations introduced flanking T58 or S62 (Fig. 2A) and assayed their dephosphorylation by EYA1. Mutation of pT58 to pS58 or pY58 significantly decreased the rate of dephosphorylation by EYA1 (Fig. 2B). However, mutation of the isomerization site P59 to alanine (pT58-P59A), which blocks conformational exchange (15), drastically increased the rate of dephosphorylation on T58 by EYA1 to ∼18-fold higher than that on pT58-Myc (Fig. 2B).
FIG 2.

Substrate preference of EYA1 and optimum pH for EYA1's phosphatase activity. (A) Peptides used for enzymatic assay. Phosphorylation modification of amino acid residues (pS/T/Y; red) and substitution of different residues (green) are shown. Replacement of Thr58 or Ser62 with glutamic acid (E) mimics the phosphorylated T58 or S62. (B) Structural effects of substrates on EYA1 phosphatase activity. The released phosphate was quantified by measuring the OD620. (C) Optimum pH of EYA1 phosphatase activity for dephosphorylating phospho-T58, phospho-T58A mutant, or phospho-Y-γH2AX peptide. (D) Comparison of kinetic constants of EYA1 phosphatase on pT58 and pT58P59A. The range of peptide concentrations was 0, 50, 100, 200, 400, 600, and 800 μM. All experiments were performed in triplicate, and the error bars represent SD. P values were calculated using StatView t tests.
We next measured EYA1's phosphatase activity under different pHs to define the optimum pH for its tyrosine- or threonine phosphatase activity. EYA1 showed the highest tyrosine phosphatase activity with pY-H2AX at pH ∼7.25, whereas its threonine phosphatase activity potentiated at higher pH, with an optimum pH of ∼8.0 (Fig. 2C). The Michaelis-Menten constant (Km) of EYA1 for pT58-P or pT58-P59A peptides was 1,582 ± 350 μM or 268 ± 33 μM, respectively, and a P59-to-A mutation resulted in up to a 23-fold increase in the kcat/Km ratio (Fig. 2D). Either NT or ED alone showed weaker activity on the pT58-P59A mutant peptide (Fig. 3A). While either the D327N or Y4A mutation alone decreased EYA1's activity, a combination of both mutations led to a further reduction in its activity (Fig. 3A). Similarly, the NT-Y4A mutation almost abolished the NT's threonine phosphatase activity, while the ED-D327N mutation only decreased the ED's threonine phosphatase activity. These data further indicate that, besides D327, there is an additional residue(s) in the ED that is important for the ED's threonine phosphatase activity and that both the ED and the NT domain interact with the phosphopeptide substrates to increase the catalytic activity of EYA1.
We next asked if EYA1 has similar conformation preferences on the phosphothreonine-proline in other proteins. Cyclin E also contains a conserved phosphorylation site consensus sequence, ϕXϕϕϕT395PPXS399, with φ representing a hydrophobic residue, and double phosphorylation at T395/S399 is known to be critical for controlling cyclin E stability (16). We synthesized nonphosphorylated (PSGLLTPPQSGKK) and phosphothreonine (PSGLLpT395PPQSGKK) cyclin E wild-type (WT) and P396-to-A mutant (PSGLLpT395APQSGKK) peptides and examined their dephosphorylation by EYA1 (Fig. 3B). Mutation of P396A (pT395-P396A) similarly increased EYA1's phosphatase activity (Fig. 3B), further suggesting that EYA1 has conformation specificity in targeting pT58-Myc.
Conformation specificity of EYA1 in dephosphorylating pT58-Myc.
The relatively weak activity on pT58-Myc but marked increase on pT58-P59A suggests that Pro59-to-Ala mutation may cause conformational change of the peptide, which makes the peptide more sensitive to dephosphorylation by EYA1. To test this hypothesis, we performed NMR spectroscopy analysis to examine the effects of T58 phosphorylation and P59-to-A mutation on the peptide backbone conformation. NMR analysis showed that, consistent with the observation that the trans conformation is far more energetically favorable at equilibrium than the cis (17), 88% of the pT58-P motif in the pT58-Myc peptide or 87% of the pT58-A motif in the pT58-P59A mutant peptide exists in the trans configuration (Fig. 4A). Phosphorylation of T58 caused major chemical shift changes for T58-Hα/Hβ proton resonances and, to a lesser extent, for T58-Hγ and P59-Hδ resonances (Fig. 4B). However, the P59A mutation induced dramatic chemical shift changes of pT58-Cα/Hα resonances (at 4.40 ppm/60.0 ppm to 4.10 ppm/62.0 ppm) and nuclear Overhauser effect (NOE) signals detected between A59Hα and P57Hδ1/Hδ2 (Fig. 4B), thus providing direct structural evidence that P59A mutation causes pronounced local backbone conformational changes at the pT58 site.
FIG 4.

NMR analysis. (A) The majority of the pT58-P motif in the pT58-Myc peptide or the pT58-A motif in the pT58-P59A mutant peptide is in the trans isomer. (B) Comparison of two-dimensional (2D) 1H-nuclear Overhauser effect spectroscopy (NOESY) NMR spectra of the wild-type T58 (left) and pT58P59 (middle) or pT58P59A (right) mutant peptides. Note that the intramolecular NOE peak annotations are indicated. The NOESY spectra were all collected with a mixing time of 300 ms on a 500-MHz Bruker NMR spectrometer at 288 K.
We next investigated the potential anti/syn conformation conversion at the pT58 site by defining a γ virtual backbone dihedral angle between two carbonyl groups of two consecutive peptide bonds (O=Ci—Ci + 1=O), using molecular dynamics (MD) simulation (Fig. 5A to C). A dihedral angle between ±90° and 180° represents an anti conformation (i.e., on opposite sides), while a dihedral angle between 0° and ±90° represents a syn conformation (i.e., on the same side). The calculated γ virtual dihedral angle of P57pT58 of the pT58 peptide showed an anti conformation characterized by two well-defined patterns that fluctuated between −168° ± 9° and 161° ± 13° and at about −122° ± 13°, respectively, for ∼97% of the MD simulation. A syn conformation that contributed less was shown by a third pattern at about 4° ± 20° for ∼3% of the MD simulation, which suggests an incipient turn backbone formation in the pT58 peptide at the pT58 site from anti to syn conformation (Fig. 5B). In the case of the pT58P59A peptide, the calculated γ virtual dihedral angle of P57pT58 is different from the corresponding angles of the wild-type and pT58 peptides, indicating an anti-to-syn conformation conversion (Fig. 5C). The syn conformation has an average γ virtual dihedral angle of 25° ± 19°, representing ∼73% of the MD simulation, and the anti conformation has an average γ virtual dihedral angle that fluctuates between −157° ± 19° and 162° ± 20°, representing ∼27% of the MD simulation (Fig. 5D). Taken together, the computational studies of the wild-type, pT58, and pT58-P59A Myc peptides have revealed that the pT58-P59A peptide results in a backbone turn conformation precisely at the pT58 site, causing an anti-to-syn conformation conversion, shown in Fig. 5. This makes the pT58-P59A peptide more sensitive to dephosphorylation by EYA1, due to the favorable syn conformation of its two carbonyl groups of two consecutive peptide bonds formed by P57 and pT58, and pT58 and P59A, respectively, which could explain the robust EYA1 phosphatase activity of 18-fold compared to the pT58-P59 peptide.
FIG 5.

MD simulation. (A to C) Histograms of a γ backbone dihedral angle between two carbonyl groups of two consecutive peptide bonds (O=Ci—Ci + 1=O) formed by Pro4 and Thr5 (Pro57 and Thr58). A γ backbone dihedral angle close to 180° represents an anti conformation, while a dihedral angle close to 0° represents a syn conformation. (A) WT peptide, with mainly anti conformation along the MD simulation. Although most of the time the virtual dihedral angles of P57T58 (P4T5) of the wild-type peptide fluctuated between −161° ± 10° and 162° ± 14° along the whole MD simulation (overall, ∼49% of the MD simulation), there were two other substrates that converged on average to −122° ± 14° (∼50% of the MD simulation) and to −10° ± 22° (∼1% of the MD simulation). (B and C) pT peptide, with mainly anti conformation along the MD simulation (B), and pTA peptide, with ∼73% of the MD simulation corresponding to the syn conformation and 27% to the anti conformation (C). (D) Representative structures of anti-to-syn conformations. MD simulation studies revealed that the pT58P59A peptide results in a backbone turn conformation precisely at the pT58 site, causing an anti (i.e., on opposite sides)-to-syn (i.e., on the same side) conformation conversion between two carbonyl groups of two consecutive peptide bonds. The pT58P59 peptide exists in anti carbonyl group conformation, whereas the pT58P59A peptide adopts a syn carbonyl group conformation. The structure of each peptide, pT58 and pT58P59A, is shown.
PIN1 appears to inhibit EYA1-mediated dephosphorylation of Myc.
The mitotic-specific prolyl isomerase PIN1 catalyzes isomerization of the cis/trans conversion of the pT/S-P motifs to facilitate dephosphorylation mediated by PP2A (15, 18). We thus examined whether PIN1 regulates EYA1-mediated dephosphorylation. Addition of PIN1 led to only ∼16 to 17% reduction of EYA1 activity in dephosphorylating pT58-P Myc peptide (Fig. 6A). As this reduction of EYA1 activity by PIN1 was reproducible in multiple independent experiments, PIN1 may reduce the cis substrate population to an even lower level to inhibit EYA1 activity. To further investigate this, we next used purified cMyc protein (Fig. 1B) for an in vitro phosphatase assay. PIN1 itself did not directly induce dephosphorylation of cMyc compared to the samples in the presence of bovine serum albumin (BSA) (Fig. 6B). In contrast to enhanced PP2A-mediated dephosphorylation of Myc by PIN1 (13), EYA1-dependent dephosphorylation at T58 appeared to be inhibited by the addition of PIN1 (Fig. 6B). When overexpressed in 293 cells, consistent with a previous observation that addition of PIN1 destabilizes Myc (13), coexpression of cMyc with PIN1 shortened Myc's half-life (Fig. 6C). In contrast, overexpression of EYA1 stabilized Myc in 293 cells (Fig. 6C) (9). However, when cMyc was overexpressed with both EYA1 and PIN1, EYA1-mediated stabilization of Myc was also inhibited (Fig. 6C).
FIG 6.

Effect of PIN1 on EYA1-mediated dephosphorylation of Myc. (A) (Top) In vitro phosphatase assay of pT58-Myc wild-type peptide in the absence or presence of PIN1. (Bottom) Quantification of EYA1's threonine phosphatase activity and comparison between EYA1 alone and EYA1 together with PIN1. Addition of PIN1 reduced EYA1 activity to 83.1% ± 1.84% of its activity without PIN1. The experiment was performed in triplicate and repeated three times, and the data were reproducible. The error bars represent SD; the P value (<0.001) was calculated using StatView t tests. (B) PIN1 decreases EYA1-mediated Myc dephosphorylation. (Top) EYA1 dephosphorylates Myc at T58. Either BSA or PIN1 was added in the absence or presence of EYA1. (Bottom) Relative dephosphorylation ratios of pT58 (left) and pS62 (right) according to the quantification of Western blot data. (C) Western blot analysis. His-cMyc alone or together with Flag-Pin1 was transfected into 293 cells or Flag-Eya1-transfected stable 293 cells, and 40 h posttransfection, the cells were treated with cycloheximide (CHX) for different times before harvest. t1/2, half-life of cMyc. (D) Western blot analysis. His-cMyc or the His-P59A mutant was transfected into 293 cells or Flag-Eya1-overexpressing stable 293 cells cells, and 40 h posttransfection, the cells were treated with CHX for different times before harvest. t1/2, half-life of cMyc. Anti-pT58 failed to detect the phosphorylated-T58-P59A mutant protein. The arrow indicates the cMyc products with higher molecular weights in the P59A-transfected cells.
As EYA1 had much higher threonine phosphatase activity on a pT58P59A mutant peptide and the P59A mutation blocks cis/trans conformational exchange, we sought to use the T58P59A mutant protein as a control to further examine the effect of PIN1 in EYA1-mediated stabilization of Myc in vitro and in vivo. However, the anti-pT58 antibody failed to detect phospho-T58-P59A (Fig. 6D), which is likely due to the structure change at the pT58 position caused by the P59A mutation, as demonstrated by our NMR analysis (Fig. 5). When the T58P59A mutant was transfected into 293 cells, we detected not only the cMyc protein with a molecular weight similar to that of the wild-type protein but also the cMyc products with higher molecular weights (Fig. 6D, arrow). These cMyc products also appeared to accumulate in the presence of EYA1 (Fig. 6D). This molecular weight change upon overexpression in vivo and infeasibility of using anti-pT58 antibody for detection made it impossible for a dephosphorylation assay using purified cMycP59A and assaying for the effect of PIN1 on EYA1-mediated dephosphorylation of the mutant protein to lead to a specific outcome.
We next asked if EYA1 and PIN1 physically interact. Coimmunoprecipitation (coIP) revealed no physical interaction between EYA1 and PIN1 (Fig. 7A). Both the NT and the ED of EYA1 showed binding to the Myc protein with affinities similar to that of PIN1 or full-length EYA1 (Fig. 7B). The T58A mutation, which blocks phosphorylation at T58 and decreases Myc binding with PIN1 (to ∼65.3% ± 2.08%) (13) (Fig. 5B), also reduced binding to either the NT (to ∼73.5% ± 3.34% that of wild-type protein) or ED (to ∼56.2% ± 3.9% that of wild-type protein) (Fig. 7B). Endogenously, pT58 showed higher-affinity binding to EYA1 (Fig. 7C). Although binding is not equivalent to activity, these results demonstrate that both domains of EYA1 bind to the pT58 region in the Myc protein. The inhibition of EYA1 activity by addition of PIN1 could be partially due to substrate-binding competition between EYA1 and PIN1, which in turn may reduce EYA1 activity in dephosphorylating pT58-Myc. Nonetheless, our results suggest that EYA1 belongs to a class of threonine phosphatases that is different from PP2A, whose activity is enhanced by PIN1.
FIG 7.

EYA1 does not interact with PIN1 but interacts with cMyc, and the T58A mutation affects Myc interaction with EYA1. (A) CoIP analysis of cell lysates from HEK293 cells transfected with the indicated plasmids. GFP, green fluorescent protein. (B) (Left) CoIP analysis of T58 phosphorylation on EYA1-Myc binding. PIN1 was used as a control. (Right) Ratios of immunoprecipitated/input Myc (left y axis) and T58A (right axis) were quantified from three independent experiments. The error bars represent SD, and P values were calculated using StatView t tests. (C) CoIP analysis. Lysates from cells transfected with Flag-Pin1 and His-cMyc or His-T58A, Flag-Eya1ED/His-cMyc or His-T58A, Flag-Eya1NT/His-cMyc or His-T58A, or Flag-Eya1/His-cMyc or His-T58A were precipitated with anti-Flag and detected using anti-His for His-cMyc or -T58A and anti-Flag for Flag-PIN1, -EYA1, -ED, or NT on Western blots. Inputs, 5% of the amount used for IP.
Depletion of EYA1 causes Myc destabilization and impairs cell cycle progression in human breast cancer.
EYA has cancer-promoting activity, and EYA1's phosphatase function is essential for breast cancer cell proliferation by maintaining cyclin D1 (19). Myc is known to be accumulated in ∼70% of human cancers, and a recent study reported that Myc protein stability is also increased in breast cancer, which is associated with decreased pT58 levels (20). However, how pT58 levels and Myc stability are regulated in breast cancer remains largely unclear. We therefore investigated EYA1's phosphatase activity on Myc stability and pT58 levels in the human breast cancer cell line MDA-MB-231, in which both EYA1 and Myc are overexpressed (19, 20). We used two distinct sets of EYA1 shRNAs (shEYA1), each of which encoded a mix of four shEYA1s, to transfect the MDA-MB-231 cells and established stable cell lines (shEYA1-MDA-MB-231 cells) through puromycin drug selection (see Materials and Methods). Control scramble shRNA-MDA-MB-231 stable cell lines were similarly established. Western and quantification analyses confirmed that EYA1 in the shEYA1-KD MDA-MB-231 cells was depleted to 21% ± 5.6% of those in control shRNA-MDA-MB-231 cells (Fig. 8A). Depletion of EYA1 decreased Myc (40.3% ± 2.4%) and cyclin D1 (40.8% ± 1.1%) levels but increased pT58 levels (194.7% ± 6.0%) compared to those in the control shRNA-MDA-MB-231 cells (Fig. 8A). In contrast, knockdown (KD) of EYA1 also slightly reduced pS62 levels (78.1% ± 9.7%). The shEya1-KD MDA-MB-231 cells showed an increase in the doubling time (Fig. 8B) and a reduced number of cycling cells and half-life of cMyc (Fig. 8B and C). Similar results were observed in mouse embryonic fibroblasts (MEFs) isolated from Eya1-null embryos (Fig. 9). The shEYA1-KD MDA-MB-231 cells released from mitotic arrest with nocodazole into medium progressed from G1 to S but did so somewhat incompletely or more slowly than control cells, and a certain population of the cells appeared to be arrested in G2/M phase (Fig. 8D). Collectively, these data identify an important mode of EYA1 in regulating Myc stability and cell growth in human breast cancer.
FIG 8.

EYA1 regulates Myc stability in breast cancer. (A) Western blot of cell lysates from passage 3 scramble shRNA- or shEya1-transfected MDA-MB-231 (left) and quantification (right). Protein levels in MDA-MB-231 cells transfected with scramble shRNA were set to 100%. P values were calculated using StatView t tests. (B) (Left) Graphic representation of cell numbers determined on the indicated days. The numbers were consistent in triplicate, and averages are shown. (Right) FACS analysis of cells at day 4. (C) Western blot of cells treated with CHX for different times. t1/2, half-life of cMyc. (D) Flow cytometry of scramble shRNA- or shEYA1-MDA-MB-231 cells released into medium from G2/M arrest by nocodazole and Western blot of cell lysates with anti-cyclin A and β-actin as a loading control. The error bars indicate SD.
FIG 9.

EYA1 regulates Myc stability in MEFs. (A) Western blot of cell lysates from wild-type (+/+) and Eya1−/− MEFs at passage 2. (B) Quantification of Western blot in panel A. The error bars represent SD, and the P values were calculated using StatView t tests. (C) (Left) Graphic representation of wild-type and Eya1−/− MEFs from passage 3 on the indicated days. Eya1−/− MEFs stopping growing at passage 3; 2 × 105 cells were plated in a 60-mm plate (in triplicate) and were counted after each day of growth for 4 days. The numbers were consistent in triplicate, and averages are shown. (Right) Flow cytometry analysis of wild-type and Eya1−/− MEFs at passages 1, 2, and 3. (D) Western blot analysis of cell lysates from passage 2 wild-type or Eya1−/− MEFs collected at the indicated time points after treatment with CHX. The cells were starved in low serum for 3 days before treatment with CHX. t(1/2), half-life of cMyc.
Inhibition of cMyc ubiquitination and interaction with FBW7 in the presence of EYA1.
There is substantial evidence that T58 phosphorylation-dependent degradation of cMyc is mediated by the F-box protein FBW7, a component of the SCFFBW7 ubiquitin ligase and a tumor suppressor (13, 21, 22). To test the role of EYA1 in FBW7-mediated cMyc degradation, we investigated whether EYA1 affects cMyc-FBW7 interaction and cMyc ubiquitination. HA-Ub alone or together with His-cMyc was transfected into 293 cells or Eya1-overexpressing stable 293 cells (23), and cell lysates were immunoprecipitated (IP) using anti-cMyc beads. Consistent with our previous observation that EYA1 physically interacts with and stabilizes cMyc through dephosphorylation at T58 (Fig. 6 and 7) (9), EYA1 increased the levels of cMyc but reduced the levels of pT58-cMyc in both input and IP samples (Fig. 10). While EYA1 was abundantly coprecipitated by cMyc in cMyc-transfected cells, weak EYA1 was also coprecipitated by endogenous cMyc in non-His-cMyc-transfected 293 cells (Fig. 7C and 10A). As reported previously (13, 21, 22), cMyc efficiently coprecipitated endogenous FBW7 (Fig. 10A). Interestingly, in correlation with the reduction of pT58 levels, EYA1 greatly decreased the amount of endogenous FBW7 coprecipitated by cMyc and reduced cMyc ubiquitination (Fig. 10A). These data are in agreement with the previous finding that phospho-T58-cMyc binds to FBW7, which promotes cMyc ubiquitination and subsequent degradation. When lysates of cells expressing cMyc or EYA1 alone, or both, were immunoprecipitated using anti-Flag-EYA1, cMyc and endogenous FBW7 were also coprecipitated (Fig. 10B), suggesting that FBW7 and EYA1 form a complex in vivo, consistent with the report that FBW7 binds to EYA1 in vivo (24). In sum, these results suggest that EYA1 regulates cMyc stability by affecting the degradation machinery through binding to cMyc and regulation of the phospho-T58-Myc levels, which in turn impairs the recruitment of FBW7 to cMyc, subsequently leading to reduced ubiquitination of Myc.
FIG 10.
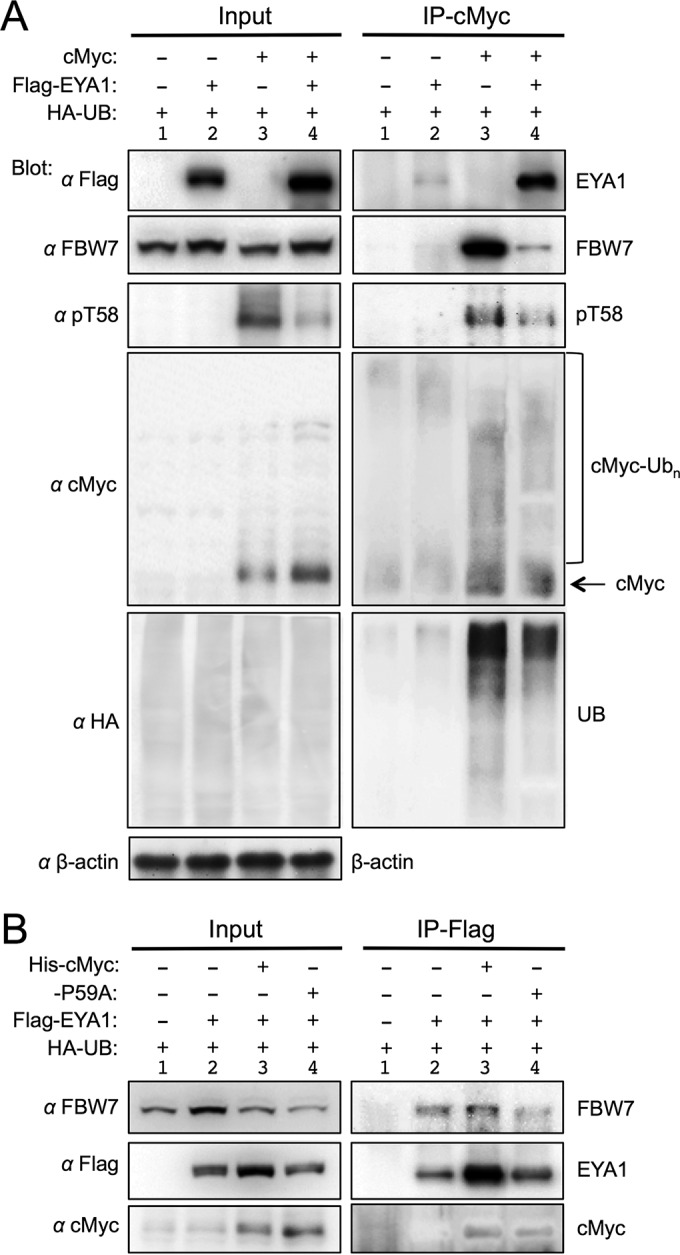
EYA1 affects cMyc-FBW7 binding and cMyc ubiquitination. (A) The physical interaction between cMyc and FBW7 is reduced in the presence of EYA1. For coIP analysis, lysates from 293 cells (lanes 1 and 3) or Flag-Eya1-overexpressing stable 293 cells (lanes 2 and 4) transfected with HA-Ub alone or together with His-cMyc were immunoprecipitated with anti-cMyc beads and analyzed by Western blotting with anti-FBW7, -cMyc, -pT58, -HA, -Flag, or -β-actin. Input, 5% of the amount used for IP. (B) CoIP analysis. Lysates from 293 cells (lane 1) or Flag-Eya1-overexpressing stable 293 cells (lanes 2 to 4) transfected with HA-Ub alone or together with His-cMyc or the His-P59A mutant were immunoprecipitated with anti-Flag M2 beads and analyzed on Western blots with anti-FBW7, -cMyc, or -Flag. Input, 5% of the amount used for IP.
DISCUSSION
EYA was initially reported to possess both tyrosine and threonine/serine phosphatase activities, and the serine/threonine phosphatase activity was assigned to the C-terminal haloacid dehalogenase ED of EYA based on the existence of the conserved motifs that are found in the aspartate-based phosphoserine/threonine phosphatase family (7). However, the previous studies did not provide direct experimental evidence demonstrating that the ED acts as a Ser/Thr phosphatase, as they assayed only the full-length EYA protein. In contrast, the ED alone is generally accepted to be a tyrosine phosphatase, as the D327N mutation abolishes EYA's tyrosine phosphatase activity (1, 5, 8). A recent follow-up study from another group that specifically characterized the catalytic activities of the NT or ED of EYA4, another member of the EYA family, led to the conclusion that the NT of EYA4 has threonine phosphatase activity but the ED of EYA4 has only tyrosine phosphatase activity (8). The catalytic motif for EYA4's threonine phosphatase activity was mapped to a set of four conserved tyrosine residues (Y4) in the NT, since replacement of the four tyrosine residues with alanine (Y4A) in the full-length protein abolished EYA4's threonine phosphatase activity (8). In this study, we showed that the NT alone has only threonine phosphatase activity and the ED alone is a dual-specificity phosphatase, but both domains interact with substrates to cooperatively regulate EYA1's phosphatase activity.
In contrast to EYA4, replacement of the set of four conserved tyrosine residues with alanine (Y4A) did not completely block EYA1's threonine phosphatase activity when full-length protein was used for the assay. This discrepancy could be due to different phosphothreonine peptides used in the two different studies and the structural divergence of the NT domains between these two family members, as we found that EYA1 shows striking conformation specificity for its substrates and the overall similarity of the NT domains between EYA1 and EYA4 is only 54.5%. Our observation that the Y4A mutation in the NT of full-length EYA1 led to a decrease in EYA1's threonine phosphatase activity in dephosphorylating pT58 suggests that EYA1 is likely to contain a regulatory residue(s) other than Y4 to increase its threonine phosphatase activity. Given that the Y4A mutation in the NT alone almost abolished the NT's threonine phosphatase activity, the additional residue(s) important for EYA1's threonine phosphatase activity is likely to be located in the ED. In support of this conclusion, we found that the ED alone also has threonine phosphatase activity and is a dual-specificity phosphatase domain, as discussed below.
When the C-terminal haloacid dehalogenase ED of EYA1 alone was assayed, in addition to tyrosine phosphatase activity, the domain also possessed threonine phosphatase activity. This is consistent with the structural feature of the domain, which shares the signature catalytic motifs of the serine/threonine protein phosphatase family (7). While it is clear that the ED acts as a catalytic domain for EYA1's tyrosine phosphatase activity, as demonstrated by multiple groups, the NT is likely to contain a regulatory residue(s) that may increase binding of EYA1 to its phosphotyrosine substrates, as the ED alone acts more weakly as a tyrosine phosphatase than the full-length EYA1 (Fig. 1 and 3). However, both domains can bind to the Myc protein, and either domain alone possesses an active site(s) for threonine phosphatase catalysis and substrate binding. Since the D327N mutation in the ED alone still retains some threonine phosphatase activity, there is likely another active/regulatory residue(s) in the ED for catalytic binding of EYA1 to the phosphothreonine-Myc substrate. Given the existence of another signature motif, CXXXXXR (aa 418 to 424), conserved in dual-specificity phosphatases (25–27), it is plausible that the motif may also contribute to EYA1's threonine phosphatase activity. Nonetheless, both the ED and NT appear to interact with substrates to cooperatively increase catalytic binding of EYA1 to regulate EYA1's threonine phosphatase activity, as either domain alone had weaker threonine phosphatase activity.
In contrast to its tyrosine or threonine phosphatase activity, EYA1 is much weaker as a serine phosphatase (Fig. 1) (8), and serine is the predominant target for phosphorylation in eukaryotic cells (7, 28). Thus, EYA may represent a new subgroup of dual-specificity phosphatases. Our results, combined with the recent observation that the NT of EYA possesses threonine phosphatase activity in cytoplasm (8), indicate a critical function as a threonine phosphatase in cells. We speculate that EYA may select its substrates for catalytic binding via multiple different sites in either domain to cross talk with diverse intracellular pathways in distinct cellular compartments to transduce signals promoting survival and growth. This also explains why the ED was previously reported to have no threonine phosphatase activity (8). EYA1 is a transcriptional coactivator, and it interacts with DNA-binding proteins and chromatin regulators to regulate gene expression (29–31). Therefore, it is also possible that EYA1 ensures specificity in cell signaling through association with other interacting proteins. Future studies on isolating physiological substrates and interacting proteins and characterizing enzyme-substrate specificity and regulation of EYA activity are necessary to better understand how EYA could serve multiple functions in response to the formation of a wide variety of distinct complexes in distinct cellular processes.
Our results, together with previous studies, indicate that PP2A and EYA1 are critical in regulating Myc turnover, but with distinct substrate conformation specificities. EYA1 has much weaker activity on pT58-P Myc wild-type peptide than on pT58-P59A mutant peptide. Our NMR analysis suggests a potential role of EYA1 in selective recognition of substrates, as the P59A mutation causes pronounced local backbone conformational changes at pT58 from anti (∼97% of the wild-type peptide) to syn (∼73% of the mutant peptide), resulting in markedly increased dephosphorylation by EYA1 (Fig. 5). This conformation preference of EYA1 in dephosphorylating Myc also explains previous observations that the ED of EYA4 does not possess threonine phosphatase activity when assayed on different peptides or that full-length EYA has weaker phosphatase activity than the NT or ED alone (1, 3, 8).
We found that EYA1-dependent dephosphorylation on T58 or EYA1-mediated stabilization of Myc was inhibited by PIN1, which is known to enhance PP2A's activity in dephosphorylating Myc (12, 13). PP2A is functionally inactivated in many cancers, and it is considered a tumor suppressor (32). In contrast, EYA has cancer-promoting activity, including sustained proliferative signaling, resistance to cell death, angiogenesis, metastasis, and replicative immortality, and is overexpressed in many cancers (19, 33–35). Our observation that depletion of Eya1 by shRNAs (∼80% loss) in human MDA-MB-231 breast cancer cells results in destabilization of Myc (∼40% of that in control cells) and accumulation of pT58-Myc (∼194% of that in control cells) suggests a novel mechanism regulating Myc stability and pT58 levels. In contrast to the increased pT58 levels, pS62 levels are only slightly reduced in EYA1 shRNA-KD MDA-MB-231 cells (∼15% reduction compared to the scramble shRNA control cells) (Fig. 8A). This is consistent with the observation that EYA1 does not appear to dephosphorylate Myc at S62 (Fig. 6B) (9). Since Myc is known to regulate many cellular activities important for tumorigenesis, destabilization of Myc by depletion of EYA1 in breast cancer cells might in turn contribute to reduced cyclin D1 levels, an increase in the doubling time, and growth arrest (Fig. 8). Deletion of Eya1 has similar effects on MEFs (Fig. 9). These results are in agreement with previous reports showing that the Myc dosage is critical for normal cell cycle progression (36).
The cMyc protein is targeted for degradation by FBW7 subsequent to phosphorylation (21, 22). Phosphorylation of cMyc at T58 by GSK3 during mitogenic response (37, 38) regulates binding of FBW7 to cMyc, as well as FBW7-mediated cMyc degradation and ubiquitination (22, 39). In agreement with this, we found that in correlation with EYA1-mediated cMyc stabilization and reduced levels of pT58, EYA1 greatly reduced cMyc-FBW7 binding and cMyc ubiquitination (Fig. 10). It is therefore likely that EYA1 may act to affect the FBW7-mediated cMyc degradation machinery to exert its oncogenic activity. In conclusion, our findings reported here provide novel insight into how EYA1 as a threonine phosphatase acts to regulate Myc stability in breast cancer cells. EYA1 may also selectively discriminate its substrates through its conformational specificity to regulate Myc stability in cell growth. Subtle quantitative differences in substrate conformation and in activities of phosphatases and kinases may be crucial for maintaining cell homeostasis in normal development, and deregulation of this balance may lead to unrestrained proliferation or growth arrest. EYA may also exert its oncogenic activity by counteracting PP2A. Given that Myc is stabilized in ∼70% of human cancers, this EYA-mediated mechanism is a potential therapeutic target for cancer.
MATERIALS AND METHODS
Constructs, peptides, and shRNAs.
His-C-Myc was from Addgene (ID45597), His-C-MycT58A was from Addgene (ID45598), Flag-Pin1 was from R. C. Sears, and Flag-cMyc and Flag-cMycT58A were from A. M. Kenney. HA-Flag-Eya1D327N, HA-Flag-Eya1, Flag-Eya1, Flag-Eya1NT, and Flag-Eya1ED were constructed in our laboratory. HA-Flag-Eya1-Y4A, HA-Flag-Eya1-Y4AD32N7, Flag-Eya1NT-Y4A, and Flag-Eya1ED-D327N were generated by site-directed mutagenesis using the QuikChange Lightning kit (catalog no. 210515; Agilent Technologies) according to the manufacturer's instructions and confirmed by sequencing.
The peptides were custom synthesized by Biomatik. All the peptides were purified by high-performance liquid chromatography (HPLC), and the amino acid sequences were confirmed by amino acid analysis and mass spectroscopy. The purity of each peptide was greater than 95%.
shRNA expression plasmids for human EYA1 in a pRFP-C-RS vector and its control scramble shRNA were purchased from OriGene (TR304716). They have been tested in multiple cell lines and were able to effectively deplete EYA1 (23). Target sequences for the Eya1 shRNAs were as follows: 1, 5′-CCAGCATTGGCGAAAGTCCTGCTGTATGG-3′; 2, 5′-ATCTGGCATCACCAGCCAAGCAGTTACAG-3′; 3, 5′-CACCATGCCTTGGAACTGGAGTACCTGTA-3′; 4, 5′-GCAACAAGCTACAGCCTATGCCACGTACC-3′.
We also used four EYA1 shRNAs (HSH005041-31-34-LVRU6GP) and control scramble shRNA (CSHCTR001-LVRU6GP) from Genecopoeia for knockdown experiments and obtained similar data.
Protein purification.
The Flag-tagged proteins were purified using anti-FLAG M2 affinity gel (A2220; Sigma) according the manufacturer's instructions. Briefly, the cells were lysed by incubating them for 1 h at 4°C in 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, and 1× protease and phosphatase inhibitor cocktail (78440; Thermo Scientific). After removing the cell debris by centrifugation at 12,000 rpm for 10 min at 4°C, the cell extracts were affinity purified with anti-FLAG M2 affinity gel and the protein was eluted with 50 mM Tris-HCl buffer (pH 7.5) containing 150 μg/ml 3× FLAG peptide (F4799; Sigma), 150 mM NaCl, and 10% glycerol. The purified proteins were confirmed by the standard Coomassie staining method.
In vitro phosphatase assay.
For peptide assays, ∼75 ng, 150 ng, and 300 ng of purified FLAG-EYA1 or its mutant proteins was incubated in a final volume of 50 μl with different concentrations of peptide (for most of them, 400 μM, unless otherwise specified) in a buffer containing 50 mM Tris-HCl (pH 7.5), 25 mM MgCl2, 0.5 mM EDTA, 1 mM dithiothreitol (DTT) at 37°C for 90 min. The reactions were stopped by addition of 100 μl Biomol green reagent (BML-AK111; Enzo Life Science). After ∼30 min incubation at room temperature, the released phosphate was quantified by measuring the optical density at 620 nm (OD620) on a microtiter plate reader (Spectra Max250; Molecular Devices).
NMR study.
The Myc peptide sample (1 to 2 mM) was prepared in a phosphate-buffered saline (PBS) buffer, pH 7.4, with 1% deuterated dimethyl sulfoxide (DMSO). All one-dimensional proton (1H) and two-dimensional 1H-13C heteronuclear single quantum coherence (HSQC) NMR spectra were collected at 25°C on a 500-MHz NMR spectrometer.
Computational study of the Myc peptides.
We performed computational molecular modeling analysis for three Myc peptides comprising residues ELLP-T-PPLSP, among which the peptide contains either a nonphosphorylated T5 (T58; WT), phosphorylated T5 (pT or pT58), or phosphorylated T5 and P6-to-Ala mutation (pTA or pT58P59A).
MD simulations.
The three linear peptides were built using the LEaP module within the AMBER (40) program. We performed a 1-μs MD simulation for each peptide. During the MD simulations, the titratable side chain carboxylic acid of the E residue was treated as polar negatively charged. The all-atom AMBER99SB force field (40) was used in all calculations. The three systems were initially minimized using the steepest descent and conjugates gradients methods to remove all the possible unfavorable interactions from the built peptide models. Then, by using a simulated annealing protocol, the systems were heated from 0 K to 3,000 K and then cooled slowly to 300 K. They were heated to 3,000 K for 150 ps with a temperature coupling parameter of 0.2 ps, cooled down slowly for 250 ps with a temperature coupling parameter of 4.0 ps, cooled down faster for another 200 ps with a temperature coupling parameter of 1.0 ps, then cooled down much faster for another 25 ps with a temperature coupling parameter of 0.5 ps, and finally cooled down even faster for another 50 ps with a temperature coupling parameter of 0.05 ps. A large nonbonded cutoff of 999 Å was used, since a nonperiodic boundary unit cell simulation was run. The generalized Born model was used to estimate the total solvation free energy of the three peptides. The maximum distance between atom pairs that were considered to carry out the pairwise summation involved in calculating the effective Born radii was set to a cutoff of 999 Å. This cutoff was sufficiently large never to truncate any of our nonbonded interactions during the simulation. The production MD simulation was carried out using the NVT (i.e., constant number of particles, volume, and temperature) ensemble. In the production stage, the temperature was maintained using the Langevin dynamics with a collision frequency of 5.0. The lengths of all bonds involving hydrogen atoms were kept fixed with the SHAKE algorithm (41). The pressure was not controlled or scaled. The equations of motion were integrated with a time step of 2 fs. The coordinates were saved every 5 ps. All MD simulations and analyses were performed using the AMBER (40) and Simulaid programs (42). Physiological conditions of 0.15 M concentration were kept during the minimization, the simulated annealing, and the MD production simulations.
Antibodies.
The antibodies used were as follows: anti-EYA1 (42-258; ProSci Inc.), anti-FLAG (F3165; Sigma), anti-His (SAB1306085; Sigma), antihemagglutinin (anti-HA) (H3663; Sigma), anti-cMyc (sc-40 or sc-764; Santa Cruz), anti-cMyc–agarose beads for IP (sc-40 AC; Santa Cruz), anti-phospho-T58 cMyc (sc-135647; Santa Cruz), anti-phospho-S62 cMyc (ab51156; Abcam), anti-cyclin D1 (sc-753; Santa Cruz), anti-cyclin A (sc-751; Santa Cruz), anti-FBW7 (ab109617; Abcam), anti-pH 3 (sc-8656R; Santa Cruz), anti-γH2AX (clone N1-431; BD Biosciences), anti-p27Kip1 (610242; BD Biosciences), anti-β-actin (sc-47778; Santa Cruz), and normal rabbit IgG (sc-2763; Santa Cruz).
Analysis of PIN1 on EYA1-mediated dephosphorylation.
Purified cMyc was incubated with ∼300 ng purified EYA1 in the same buffer used for the peptide assay. About 0.5 μM purified PIN1 was added to the samples, as indicated, and a similar amount of BSA was used as a control. The reaction was carried out at 30°C for different times, as indicated. The reaction samples were analyzed by Western blotting in triplicate and detected with anti-pT58, anti-pS62, and anti-Myc antibodies, respectively.
Cell culture, transfection, coIP, and Western blotting.
293 cells or Flag-Eya1-transfected stable 293 cells that stably express Flag-EYA1 (23) were cultured in normal Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and transfected using the calcium phosphate method. For coimmunoprecipitation, cells were lysed in 20 mM Tris-HCl (pH 7.5), 137 mM NaCl, 0.5% Triton, 2 mM EDTA, 10% glycerol, and 1× protease and phosphatase inhibitors at 4°C for 1 h. After removal of cell debris by centrifugation, the lysates were suspended in IP buffer (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 1.25 mM MgCl2, 0.17 mM EDTA, and 10% glycerol) and immunoprecipitated with anti-FLAG M2 beads or antibodies overnight. The protein A or A/G beads were added if antibodies were used for IP, and IgG was used as a negative control. The immunoprecipitated proteins were separated by SDS-PAGE and analyzed by Western blotting.
Cell growth analysis of scramble shRNA (shControl)- or shEYA1-transfected MDA-MB-231 cells and wild-type and Eya1−/− MEFs.
For scramble shRNA- or shEYA1-transfected MDA-MB-231 cells, 2 days after transfection, the cells were selected with puromycin to remove nontransfected cells and then amplified. Passage 3 after selection was used for analysis.
To isolate MEFs from mouse embryos, the liver, heart, and head were removed from the embryo, and the remaining embryo was digested with 0.25% trypsin in 2.21 mM EDTA to isolate single-cell suspensions of MEFs using standard procedures. All procedures involving living mice were approved by the Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai (06-822).
To measure cell growth speed, cells were trypsinized, and then 2 × 105 cells were replated in a 60-mm plate (in triplicate) and fed with DMEM containing 10% FBS. Following each day of growth at 37°C and 5% CO2, we counted the cells for 4 days.
Cycloheximide treatment and cell synchronization.
shControl- or shEYA1-transfected MDA-MB-231, wild-type, or Eya1−/− MEFs were grown in normal DMEM with 10% FBS. Scramble shRNA- or shEYA1-transfected MDA-MB-231 cells were treated with 100 μg/ml cycloheximide and harvested at the indicated time points for Western blot analysis. For wild-type and Eya1−/− MEFs, the cells were starved in DMEM with 0.1% FBS for 48 to 72 h before treating with cycloheximide (13).
MDA-MB-231 cells were synchronized with 100 ng/ml nocodazole for 16 h. Then, the roundup cells (defined as M phase cells) were collected by mechanical shake-off, released into fresh medium, and harvested at the indicated time points.
Flow cytometry assays of cell cycle distribution.
Analysis of the cell cycle profile was performed by flow cytometry. Nocodazole-treated cells, after release into medium at different time points, were harvested, fixed in 70% ethanol, washed with PBS, and then treated with propidium iodide (PI) staining solution (0.1% [vol/vol] Triton X-100, 10 μg/ml PI, and 100 μg/ml DNase-free RNase A in PBS). The stained cells were then subjected to flow cytometry. The cell cycle distribution was analyzed with Flow Jo software.
Quantitative real-time RT-PCR (RT-qPCR).
Total RNA was isolated from MDA-MB-231 cells, HEK293 cells, or MEFs with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. A QuantiTect reverse transcription (RT) kit (Qiagen) was used to synthesize first-strand cDNA from a total RNA template. Quantitative PCR was performed using a StepOnePlus PCR system and SYBR green detector (Qiagen). Normalization was performed using specific amplification of β-actin, and PCRs were performed in triplicate for each biological-duplicate experiment. The relative amounts of mRNA were calculated using the comparative threshold cycle (CT) method. The following primers were used: hEYA1, 5′-TTTCAACTTGGCAGACACACA-3′ and 5′-ATGTGCTTAGGTCCTGTCCG-3′; hβ-actin, 5′-GCACAGAGCCTCGCCTT-3′ and 5′-GTTGTCGACGACGAGCG-3′; mC-Myc, 5′-ACGCTTGGCGGGAAAAAGAAG-3′ and 5′-TGATGTTGGGTCAGTCGCAGG-3′; mβ-actin, 5′-AACGGCTCCGGCATGTGCAAAG-3′ and 5′-ACACGCAGCTCATTGTAGAAG-3′.
ACKNOWLEDGMENTS
We thank A. M. Kenney, R. S. Hegde, and R.C. Sears for kindly providing plasmids and Z. Karoulia for technical assistance.
This work was supported by NIH RO1DK064640 and RO1DC014718 (P.-X.X.) and RO1CA154809 and RO1CA087658 (M.-M.Z.).
We declare no competing financial interests.
REFERENCES
- 1.Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, Selengut JD, Parlikar BE, Rebay I. 2003. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 426:299–302. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- 2.Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. 2003. Eyes absent represents a class of protein tyrosine phosphatases. Nature 426:295–298. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 3.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. 2003. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426:247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 4.Xu PX. 2013. The EYA-SO/SIX complex in development and disease. Pediatr Nephrol 28:843–854. doi: 10.1007/s00467-012-2246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. 2009. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan B, Cheng L, Chiang HC, Xu X, Han Y, Su H, Wang L, Zhang B, Lin J, Li X, Xie X, Wang T, Tekmal RR, Curiel TJ, Yuan ZM, Elledge R, Hu Y, Ye Q, Li R. 2014. A phosphotyrosine switch determines the antitumor activity of ERbeta. J Clin Invest 124:3378–3390. doi: 10.1172/JCI74085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Okabe Y, Sano T, Nagata S. 2009. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 460:520–524. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Wong EY, Cheng C, Li J, Sharkar MT, Xu CY, Chen B, Sun J, Jing D, Xu PX. 2014. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell 31:434–447. doi: 10.1016/j.devcel.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrell AS, Sears RC. 2014. MYC degradation. Cold Spring Harb Perspect Med 4:a014365. doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu KP, Liou YC, Zhou XZ. 2002. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol 12:164–172. doi: 10.1016/S0962-8924(02)02253-5. [DOI] [PubMed] [Google Scholar]
- 12.Liou YC, Zhou XZ, Lu KP. 2011. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci 36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. 2004. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 14.Dominguez-Sola D, Dalla-Favera R. 2004. PINning down the c-Myc oncoprotein. Nat Cell Biol 6:288–289. doi: 10.1038/ncb0404-288. [DOI] [PubMed] [Google Scholar]
- 15.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. 2000. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell 6:873–883. doi: 10.1016/S1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 16.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. 2007. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Lu KP, Finn G, Lee TH, Nicholson LK. 2007. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol 3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 18.Stukenberg PT, Kirschner MW. 2001. Pin1 acts catalytically to promote a conformational change in Cdc25. Mol Cell 7:1071–1083. doi: 10.1016/S1097-2765(01)00245-3. [DOI] [PubMed] [Google Scholar]
- 19.Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, Hu J, Sun Y, Li X, Ertel A, Pestell RG. 2013. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer Res 73:4488–4499. doi: 10.1158/0008-5472.CAN-12-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Farrell AS, Daniel CJ, Arnold H, Scanlan C, Laraway BJ, Janghorban M, Lum L, Chen D, Troxell M, Sears R. 2012. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc Natl Acad Sci U S A 109:2790–2795. doi: 10.1073/pnas.1100764108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. 2004. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun J, Karoulia Z, Wong EY, Ahmed M, Itoh K, Xu PX. 2013. The phosphatase-transcription activator EYA1 is targeted by anaphase-promoting complex/Cdh1 for degradation at M-to-G1 transition. Mol Cell Biol 33:927–936. doi: 10.1128/MCB.01516-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Li X. 2014. The canonical wnt signal restricts the glycogen synthase kinase 3/fbw7-dependent ubiquitination and degradation of eya1 phosphatase. Mol Cell Biol 34:2409–2417. doi: 10.1128/MCB.00104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NP. 2004. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J 18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 26.Tautz L, Critton DA, Grotegut S. 2013. Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol Biol 1053:179–221. doi: 10.1007/978-1-62703-562-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso A, Pulido R. 2016. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J 283:1404–1429. doi: 10.1111/febs.13600. [DOI] [PubMed] [Google Scholar]
- 28.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed M, Xu J, Xu PX. 2012. EYA1 and SIX1 drive the neuronal developmental program in cooperation with the SWI/SNF chromatin-remodeling complex and SOX2 in the mammalian inner ear. Development 139:1965–1977. doi: 10.1242/dev.071670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmed M, Wong EY, Sun J, Xu J, Wang F, Xu PX. 2012. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev Cell 22:377–390. doi: 10.1016/j.devcel.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong EY, Ahmed M, Xu PX. 2013. EYA1-SIX1 complex in neurosensory cell fate induction in the mammalian inner ear. Hear Res 297:13–19. doi: 10.1016/j.heares.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seshacharyulu P, Pandey P, Datta K, Batra SK. 2013. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, Hegde RS, Cripe TP, Cancelas JA, Collins MH, Ratner N. 2010. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene 29:368–379. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, Hegde RS. 2010. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene 29:3715–3722. doi: 10.1038/onc.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, Vezzani C, O'Brien-Jenkins A, Wang J, Ward MR, Courreges MC, Fracchioli S, Medina A, Katsaros D, Weber BL, Coukos G. 2005. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res 65:925–932. [PubMed] [Google Scholar]
- 36.Schmidt EV. 1999. The role of c-myc in cellular growth control. Oncogene 18:2988–2996. doi: 10.1038/sj.onc.1202751. [DOI] [PubMed] [Google Scholar]
- 37.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. 2000. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregory MA, Qi Y, Hann SR. 2003. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 39.Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 40.Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. 2005. The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryckaert JP, Ciccotti G, Berendsen HJ. 1977. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular dynamics of N-alkanes. J Comput Phys 23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 42.Mezei M. 2010. Simulaid: a simulation facilitator and analysis program. J Comput Chem 31:2658–2668. doi: 10.1002/jcc.21551. [DOI] [PubMed] [Google Scholar]