

ABSTRACT
Optimizing antibiotic combinations is promising to combat multidrug-resistant Pseudomonas aeruginosa. This study aimed to systematically evaluate synergistic bacterial killing and prevention of resistance by carbapenem and aminoglycoside combinations and to rationally optimize combination dosage regimens via a mechanism-based mathematical model (MBM). We studied monotherapies and combinations of imipenem with tobramycin or amikacin against three difficult-to-treat double-resistant clinical P. aeruginosa isolates. Viable-count profiles of total and resistant populations were quantified in 48-h static-concentration time-kill studies (inoculum, 107.5 CFU/ml). We rationally optimized combination dosage regimens via MBM and Monte Carlo simulations against isolate FADDI-PA088 (MIC of imipenem [MICimipenem] of 16 mg/liter and MICtobramycin of 32 mg/liter, i.e., both 98th percentiles according to the EUCAST database). Against this isolate, imipenem (1.5× MIC) combined with 1 to 2 mg/liter tobramycin (MIC, 32 mg/liter) or amikacin (MIC, 4 mg/liter) yielded ≥2-log10 more killing than the most active monotherapy at 48 h and prevented resistance. For all three strains, synergistic killing without resistance was achieved by ≥0.88× MICimipenem in combination with a median of 0.75× MICtobramycin (range, 0.032× to 2.0× MICtobramycin) or 0.50× MICamikacin (range, 0.25× to 0.50× MICamikacin). The MBM indicated that aminoglycosides significantly enhanced the imipenem target site concentration up to 3-fold; achieving 50% of this synergistic effect required aminoglycoside concentrations of 1.34 mg/liter (if the aminoglycoside MIC was 4 mg/liter) and 4.88 mg/liter (for MICs of 8 to 32 mg/liter). An optimized combination regimen (continuous infusion of imipenem at 5 g/day plus a 0.5-h infusion with 7 mg/kg of body weight tobramycin) was predicted to achieve >2.0-log10 killing and prevent regrowth at 48 h in 90.3% of patients (median bacterial killing, >4.0 log10 CFU/ml) against double-resistant isolate FADDI-PA088 and therefore was highly promising.
KEYWORDS: imipenem, tobramycin, amikacin, synergy, mathematical modeling, Monte Carlo simulations, population pharmacokinetics and pharmacodynamics
INTRODUCTION
Antimicrobial resistance in Gram-negative pathogens and the shortage of new antibiotics in the discovery-and-development pipeline are causing a serious global health crisis (1–4). Multidrug-resistant Pseudomonas aeruginosa is an opportunistic nosocomial pathogen and is prevalent in bloodstream, wound, and respiratory tract infections (5, 6). Resistant isolates of P. aeruginosa have been found in hospitals worldwide and are associated with high rates of morbidity and mortality (5, 6). The high rates of resistant P. aeruginosa isolates present a substantial clinical challenge, especially for antibiotic therapies in severe infections (7). Among the β-lactam antibiotics, carbapenems have been considered a monotherapy treatment of choice due to their rapid bacterial killing and good activity against susceptible P. aeruginosa and other Gram-negative bacteria (6, 8, 9). However, in the last 10 to 15 years, carbapenem resistance has become a major challenge, and numbers of infections by resistant isolates have increased worldwide (6, 9, 10). An aminoglycoside (AGS) can cause substantial bacterial killing in monotherapy against P. aeruginosa, but rapid emergence of resistance occurs in vitro and in patients and is associated with treatment failure in 85% of patients (11, 12).
The extensive decline in new approved antibiotics, especially against Gram-negative pathogens, and the rapid emergence of resistance to all existing antibiotics in monotherapies cause significant challenges (1, 3). Synergistic combination therapy using available antibiotics offers an attractive and tangible option to treat infections by multidrug-resistant P. aeruginosa. In the past, in vitro studies of the combination of β-lactams with aminoglycosides have been conducted. However, these studies were limited in that they usually assessed one β-lactam plus one aminoglycoside and in that the studied isolates were susceptible to both antibiotics tested or were intermediate to one and susceptible to the other antibiotic (8, 13–15). Other studies did not evaluate synergy (16).
Our primary objective was to identify the imipenem (IPM) and aminoglycoside concentrations required to achieve substantial bacterial killing and prevent resistance via 48-h static-concentration time-kill (SCTK) experiments with three double-resistant P. aeruginosa isolates. Second, we aimed to characterize the extent and time course of synergistic bacterial killing and prevention of resistance by a new mechanism-based mathematical model (MBM). For our third objective, this MBM enabled us to rationally optimize combination dosage regimens for critically ill patients with bacteremia via novel, MBM-based Monte Carlo simulations that were based on previously reported human population pharmacokinetic (PK) models in critically ill patients.
(Part of this work was presented as a poster presentation at ASM Microbe, 16 to 20 June 2016, Boston, MA [74].)
RESULTS
All three studied clinical isolates were carbapenem resistant (Table 1). Isolate FADDI-PA088 was tobramycin resistant (MIC, 32 mg/liter) and amikacin susceptible (MIC, 4 mg/liter), whereas isolate FADDI-PA001 was tobramycin susceptible, with an MIC of 4 mg/liter (i.e., the highest MIC deemed susceptible by European Committee on Antimicrobial Susceptibility Testing [EUCAST] breakpoints), and amikacin resistant (MIC, 32 mg/liter). Isolate FADDI-PA022 was resistant to both tobramycin and amikacin. The log10 mutation frequencies on 3× MIC agar plates (Table 1) indicated the presence of an aminoglycoside-resistant population for FADDI-PA088 and an imipenem-resistant population for FADDI-PA001 before the initiation of antibiotic therapy.
TABLE 1.
MICs and log10 mutation frequencies in cation-adjusted Mueller-Hinton II brotha
| Strain | MIC (mg/liter) |
Log10 mutation frequency at 3× MIC |
||||
|---|---|---|---|---|---|---|
| Imipenem | Tobramycin | Amikacin | Imipenem | Tobramycin | Amikacin | |
| FADDI-PA088 | 16 | 32 | 4 | <−7.6 | −5.5 | −5.4 |
| FADDI-PA001 | 32 | 4 | 32 | −4.75 | <−7.8 | <−7.8 |
| FADDI-PA022 | 16 | 8 | >32 | <−7.3 | <−7.3 | NS |
NS, not studied.
Extensive and synergistic killing of isolate FADDI-PA088 was observed with imipenem-plus-aminoglycoside combinations. In monotherapy, the highest clinically achievable concentrations of imipenem, tobramycin, and amikacin yielded at least 3.0-log10 killing by 6 h, which was, however, followed by regrowth. Excitingly, as little as 1 mg/liter tobramycin (i.e., 1/32 of the MIC) achieved synergy with imipenem at 24 mg/liter and prevented regrowth over 48 h (Table 2 and see Fig. 2A1). Amikacin at 1.0 mg/liter demonstrated synergy in combination with 24 mg/liter imipenem and prevented regrowth over 48 h (Table 2 and see Fig. 2A2).
TABLE 2.
IPM concentrations required in combination with tobramycin or amikacin to achieve synergistic bacterial killing and prevention of regrowth of three P. aeruginosa isolatesb
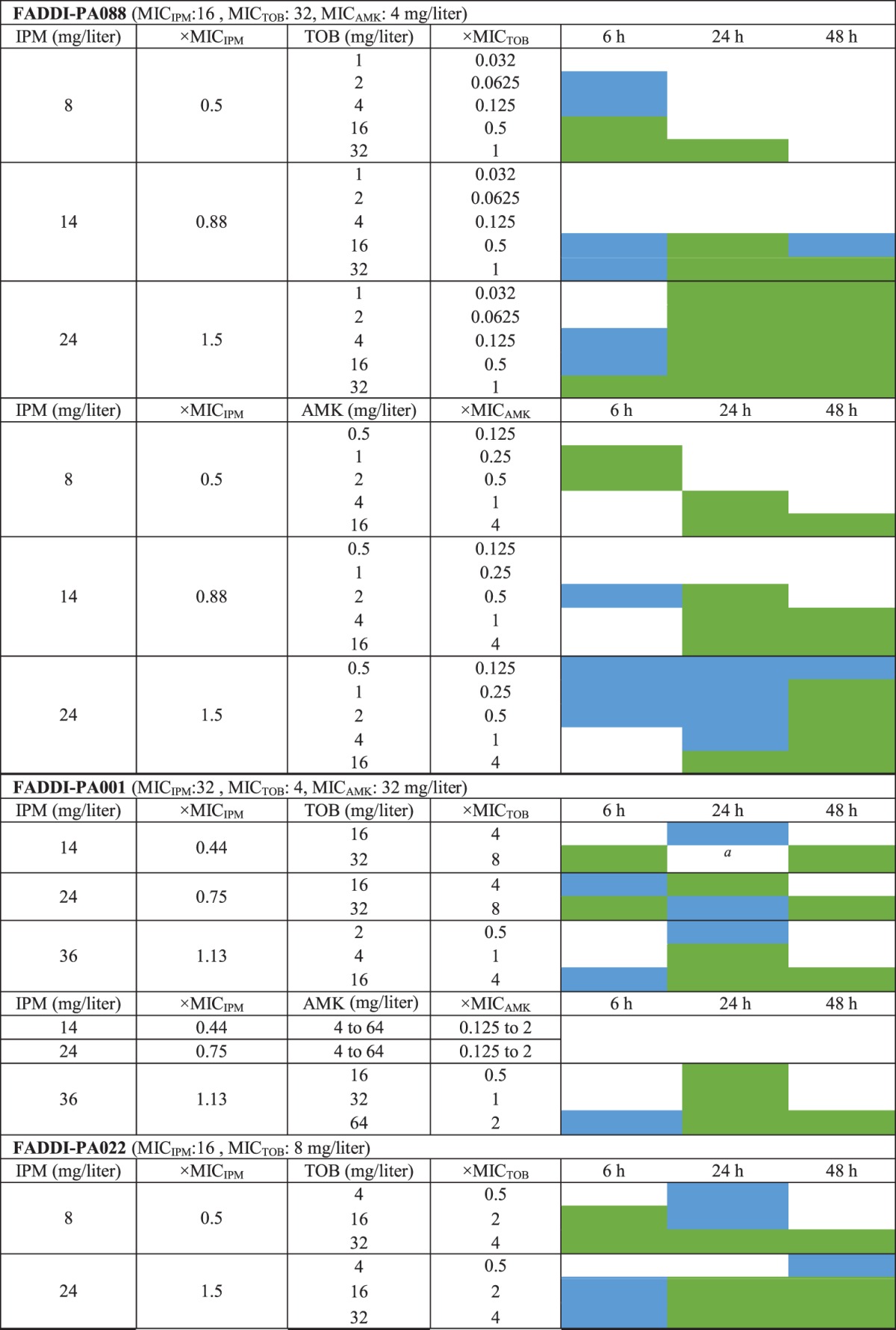
aTobramycin monotherapy yielded considerable killing. Thus, this combination was not synergistic.
IPM and AGS concentrations are shown as fold MICs. Cells shaded in green represent ≥2-log10 more killing than the most active monotherapy (i.e., meeting the empirical definition of synergy), and cells shaded in blue represent 1.0- to 2.0-log10 more killing than the most active monotherapy.
FIG 2.

Observed (markers) and individual fitted (lines) viable counts for imipenem combined with an aminoglycoside (tobramycin or amikacin) against three P. aeruginosa isolates. Observed viable counts below the limit of detection (i.e., below 1.0 log10 CFU/ml) were plotted as zero.
Against FADDI-PA001, imipenem at 36 mg/liter and tobramycin at 32 mg/liter achieved 2-log10 killing by 6 h in monotherapy, followed by extensive regrowth. No killing was observed with amikacin monotherapy. Imipenem at 14 mg/liter plus tobramycin at 32 mg/liter, imipenem at 24 mg/liter plus tobramycin at 16 to 32 mg/liter, and imipenem at 36 mg/liter plus tobramycin at 4 to 16 mg/liter yielded synergy (i.e., ≥2-log10 more killing than the most active monotherapy) over 24 to 48 h (Table 2 and see Fig. 2B1). Imipenem at 36 mg/liter was required to achieve synergy at 24 h with 16 to 64 mg/liter amikacin. Imipenem at 36 mg/liter combined with 64 mg/liter amikacin prevented regrowth over 48 h (Table 2 and see Fig. 2B2).
In SCTK experiments with FADDI-PA022, 24 mg/liter imipenem and 32 mg/liter tobramycin were the most active monotherapies, with 3.2-log10 and 2.3-log10 killing at 6 h. However, initial killing was followed by extensive regrowth at 48 h. Imipenem at 8 mg/liter plus tobramycin at 32 mg/liter and imipenem at 24 mg/liter plus 16 to 32 mg/liter tobramycin yielded at least 5-log10 killing and prevented regrowth over 48 h (Table 2 and see Fig. 2C).
Imipenem combinations suppressing resistance.
Emergence of resistance was defined as viable counts of resistant bacteria higher than those of the growth control (see Fig. 3, broken black horizontal line). All monotherapies (except imipenem monotherapies for FADDI-PA088) demonstrated, in part extensive, resistance emergence quantified on antibiotic-containing agar plates (3× MIC) for all three isolates (see Fig. 3; see also Fig. S2 in the supplemental material). The emergence of resistance was very extensive for aminoglycoside monotherapies, primarily for higher aminoglycoside concentrations in broth (tested range, 0.5 to 64 mg/liter).
FIG 3.

Resistant subpopulations of imipenem and aminoglycosides (tobramycin and amikacin) on 3× MIC agar plates at 48 h for treatment and control arms. All monotherapies resulted in the amplification of resistant subpopulations that grew on 3× MIC drug plates. The highest achievable clinically relevant concentrations of imipenem combined with tobramycin or amikacin suppressed the amplification of resistance over 48 h.
Clinically relevant concentrations for the combination of imipenem with tobramycin or amikacin suppressed resistance over 48 h against FADDI-PA088 and FADDI-PA022. For isolate FADDI-PA088, despite the presence of a preexisting aminoglycoside-resistant subpopulation (2.1 to 2.3 log10 CFU/ml at 0 h), 14 mg/liter imipenem plus ≥16 mg/liter tobramycin or ≥4 mg/liter amikacin and 24 mg/liter imipenem plus 1 to 16 mg/liter tobramycin or amikacin suppressed resistance to both imipenem and aminoglycosides (see Fig. 3A). Against isolate FADDI-PA022, imipenem at 8 mg/liter plus 32 mg/liter tobramycin and imipenem at 24 mg/liter in combination with 16 and 32 mg/liter tobramycin displayed suppression of resistance to both imipenem and aminoglycosides (see Fig. 3C). Isolate FADDI-PA001 harbored a preexisting imipenem-resistant subpopulation (3.1 log10 CFU/ml at 0 h); imipenem at 24 mg/liter plus 32 mg/liter tobramycin and imipenem at 36 mg/liter in combination with 16 mg/liter tobramycin (or 64 mg/liter amikacin) suppressed resistance to both imipenem and the aminoglycosides over 48 h (see Fig. 3B).
Mechanism-based modeling.
Our MBM contained three preexisting bacterial populations with different susceptibilities to imipenem and the aminoglycosides (Fig. 1). This MBM simultaneously described the effects of imipenem plus tobramycin and of imipenem plus amikacin and yielded unbiased and precise curve fits for all monotherapies and combinations (Fig. 2; see also Fig. S1 in the supplemental material). The coefficient of correlation for the observed versus individual fitted log10 viable counts was ≥0.976 against all three isolates.
FIG 1.

The IPMS/AGSS population is susceptible to both imipenem and the aminoglycoside, the IPMR/AGSI population is imipenem resistant and has intermediate susceptibility to the aminoglycoside, and the IPMI/AGSR population is aminoglycoside resistant and has intermediate susceptibility to imipenem. The maximum killing rate constants (Kmax) and the antibiotic concentrations causing 50% of Kmax (KC50) are explained in Table 3. The effect of aminoglycosides on the outer membrane (i.e., the aminoglycoside enhancing the target site penetration of imipenem) was applied to all three strains. The outer membrane effect terms Imax,OM and IC50,OM are explained in Table 3. A life cycle growth model (61, 64) was utilized for each of the three populations to describe bacterial growth and replication.
Mechanistic synergy due to the aminoglycoside enhancing the target site concentration of imipenem (i.e., the effect of the aminoglycoside on the outer membrane) occurred for all three populations of FADDI-PA088. Synergy was present for two of the three bacterial populations (SS, i.e., IPMS/AGSS, and IR, i.e., IPMI/AGSR) for FADDI-PA001 and FADDI-PA022 (Fig. 1). The inclusion of mechanistic synergy for the RI (i.e., IPMR/AGSI) population did not improve the model performance for these two isolates. Mechanistic synergy was expressed as a decrease in the KC50,IPM (the imipenem concentration causing 50% of Kmax) of the respective bacterial populations with increasing aminoglycoside concentrations (see Fig. 4). A decrease in the KC50,IPM in the MBM is equivalent to an increase of the imipenem target site concentration.
FIG 4.

The outer membrane effect of the aminoglycosides (i.e., the aminoglycoside enhancing the target site penetration of imipenem) resulted in a decrease in the imipenem concentration required for half-maximal bacterial killing of two or three bacterial populations (KC50,IPM). Results are shown for three P. aeruginosa isolates. The decrease in the KC50,IPM was dependent on the aminoglycoside (tobramycin or amikacin) concentration. Parameters are explained in Table 3.
Mechanistic synergy was most pronounced for isolate FADDI-PA088: the imipenem concentration required to provide half-maximal killing for all three populations was decreased by 2.4- to 2.9-fold in the presence of 32 mg/liter tobramycin and 32 mg/liter amikacin compared to the situation without an aminoglycoside being present (see Fig. 4A). There was a lesser decrease in the KC50,IPM noted for FADDI-PA001 (see Fig. 4B). For isolate FADDI-PA022, the imipenem concentration yielding half-maximal killing of the aminoglycoside-resistant population (KC50,IR,IPM) decreased 1.7-fold in the presence of 32 mg/liter of tobramycin compared to no tobramycin (see Fig. 4C). This decrease in the KC50,IPM in the presence of an aminoglycoside contributed to the killing of the bacterial populations by imipenem. For all three isolates, we estimated the aminoglycoside concentration required for half-maximal permeabilization of the outer membrane (IC50,OM,AGS), i.e., the concentration required for 50% mechanistic synergy (Table 3 and see Fig. 4). Similarly, the aminoglycoside concentration required for 80% mechanistic synergy is also illustrated in Fig. 4.
TABLE 3.
Population mean parameter estimates for the imipenem-plus-aminoglycoside (tobramycin or amikacin) combination models against three P. aeruginosa isolates
| Parameter | Abbreviation | Population mean value (SE [%]) for strain, treatment |
||
|---|---|---|---|---|
| FADDI-PA088, IPM + AGS | FADDI-PA001, IPM + AGS | FADDI-PA022, IPM + TOB | ||
| Log10 CFU, initial inoculum | LogCFU0 | 7.54 (3.8)a | 7.92 (1.2)a | 6.99 (1.3) |
| 7.46 (1.8)b | 7.78 (1.0)b | |||
| Log10 CFU, maximum population size | CFUmax | 9.56 (1.8) | 9.23 (0.7) | 9.54 (2.9) |
| Replication rate constant (h−1) | k21 | 50 (fixed) | 50 (fixed) | 50 (fixed) |
| Mean generation time (min) | ||||
| Population 1 (CFUSS) | k12S−1 | 26.9 (6.2) | 45.3 (10.9) | 63.8 (10.8) |
| Population 2 (CFURI) | k12I−1 | 873 (8.5) | 481 (15.2) | 565 (19.9) |
| Population 3 (CFUIR) | k12R−1 | 26.9 (6.2) | 45.3 (10.9) | 63.8 (10.8) |
| Log10 mutation frequencies | ||||
| IPM | LogMUT,IPM | −4.73 (5.8) | −4.99 (4.0) | −3.90 (9.4) |
| AGS | LogMUT,AGS | −7.00 (5.9)a | −7.33 (4.2)a | −7.55 (4.0) |
| −6.68 (3.4)b | −6.66 (4.0)b | |||
| Killing by IPM | ||||
| Maximum killing rate constant (h−1) | Kmax,IPM | 3.34 (5.5) | 3.21 (18.5) | 2.23 (10) |
| Imipenem concn causing 50% Kmax,IPM (mg/liter) | ||||
| Population 1 (CFUSS) | KC50,SS,IPM | 0.992 (33) | 33.2 (11.1) | 15.2 (25.7) |
| Population 2 (CFURI) | KC50,RI,IPM | 264 (9.7) | 118 (17.8) | 67.3 (24.2) |
| Population 3 (CFUIR) | KC50,IR,IPM | 23.5 (11) | 61.8 (9.0) | 52.7 (33.8) |
| Killing by AGS | ||||
| Maximum killing rate constants (h−1) | ||||
| Population 1 | Kmax,SS,AGS | 11.8 (7.9) | 2.84 (15.8) | 2.93 (11.8%) |
| Population 2 | Kmax,RI,AGS | 3.28 (26.3) | 3.14 (15.9) | 2.80 (21.2%) |
| Population 3 | Kmax,IR,AGS | 11.8 (7.9) | 2.84 (15.8) | 2.93 (11.8%) |
| AGS concn causing 50% Kmax,AGS (mg/liter) | ||||
| Population 1 | KC50,SS,AGS | 18.6 (39.6)a | 16.6 (17)a | 54.9 (8.1)a |
| 4.88 (25.3)b | 153 (12.7)b | |||
| Population 2 | KC50,RI,AGS | 849 (5.7)a | 228 (16.7)a | 568 (10.9)a |
| 280 (15.7)b | 738 (8.3)b | |||
| Population 3 | KC50,IR,AGS | 615 (4.1)a | 46.7 (9.9)a | 159 (34.7)a |
| 329 (10.8)b | 170 (10.5)b | |||
| Mean turnover time for hypothetical signal molecules (=1/kout,sig) (h) | MTTsig | 1 (fixed) | 1 (fixed) | 0.087 (31.3) |
| Maximum inhibition by hypothetical signal molecules | Imax,sig12 | 0.997 (13.7) | 0.996 (11.3) | 0.881 (56.2) |
| Log10 hypothetical signal molecule concn at 50% max effect | Log10,IC50,Sig | 10.2 (4.3) | 9.63 (2.3) | 8.95 (26) |
| Permeabilization of the outer membrane by AGS | ||||
| Maximum fractional decrease of KC50,IPM by AGS via outer membrane disruption | Imax,OM,AGS | 0.678c (31.8) | 0.130c (11.1) | 0.604c (126) |
| AGS concn causing 50% Imax,OM,AGS (mg/liter) | IC50,OM,AGS | 5.55 (29.3)a | 1.54 (17.5)a | 4.29 (21.6)a |
| 1.13 (49.0)b | 4.80 (14.2)b | |||
| Hill coefficient for bacterial killing | ||||
| IPM | HillIPM | 3.00 (13.8) | 3.09 (14.2) | 3.08 (27.4) |
| AGS | HillAGS | 1.11 (5.5) | 2.04 (10) | 0.998 (11.9)a |
| SD of residual error on log10 scale | SDCFU | 0.378 (6.0) | 0.290 (5.2) | 0.475 (10.4) |
Parameter estimates for tobramycin.
Parameter estimates for amikacin.
These estimates mean that the imipenem target site concentrations increased up to 3.11-fold for FADDI-PA088, 1.15-fold for FADDI-PA001, and 2.53-fold for FADDI-PA022 in the presence of high AGS concentrations (≫IC50,OM,AGS).
We extended the MBM shown in Fig. 1 by simultaneously fitting the time courses of both the total population and the populations resistant to imipenem or the aminoglycoside (quantified on 3× MIC agar plates) for isolate FADDI-PA088. This extended MBM adequately captured the growth and killing of resistant bacteria, provided unbiased and reasonably precise curve fits (see Fig. S3 in the supplemental material), and thus supported the proposed structural model (Fig. 1).
In this extended MBM, the fraction of the aminoglycoside-resistant population that was quantifiable on aminoglycoside-containing agar plates increased with higher aminoglycoside concentrations. This fraction was previously defined as the subpopulation fraction by Ly et al. (17). The extended MBM suggested inducible aminoglycoside resistance for the aminoglycoside-resistant population (i.e., IR) (Fig. 1). For FADDI-PA088, an amikacin concentration of 1.08 mg/liter and a tobramycin concentration of 11.7 mg/liter yielded half-maximal induction of the subpopulation fraction for the aminoglycoside. The other populations (i.e., SS and RI) (Fig. 1) had constant, i.e., noninducible, subpopulation fractions for imipenem and the aminoglycosides, suggesting that there was no inducible resistance.
Monte Carlo simulations.
A continuous infusion of 5 g/day imipenem with a 1-g loading dose was predicted to achieve median (5th to 95th percentiles) unbound steady-state concentrations of 16.7 mg/liter (9.63 to 29.1 mg/liter) (see Fig. 5A). The 1-g imipenem loading dose was included in the simulation to achieve steady-state concentrations more rapidly. Following a 0.5-h infusion of 7 mg/kg of body weight tobramycin, the predicted concentrations (medians [5th to 95th percentiles]) were 17.7 mg/liter (12.8 to 23.8 mg/liter) at 0.5 h and 1.43 mg/liter (0.584 to 2.78 mg/liter) at 24 h. Against the double-resistant isolate (FADDI-PA088) (initial inoculum of 107.53 ± 0.3 CFU/ml), the simulated imipenem monotherapies given as intermittent short-term infusions yielded 1.56-log10 (1.1- to 2.4-log10) bacterial killing, followed by rapid regrowth to ≥107.5 CFU/ml at 8 to 14 h (see Fig. 5B). A continuous infusion of imipenem at 5 g/day in monotherapy was predicted to achieve 3.3-log10 (2.8- to 4.0-log10) bacterial killing, followed by rapid regrowth to ≥107.5 CFU/ml for the majority of the simulated patients. Monotherapy with tobramycin at 7 mg/kg was predicted to achieve 3.1-log10 (2.9- to 3.6-log10) bacterial killing, followed by rapid and extensive regrowth to ≥107.5 CFU/ml at 8 to 14 h (see Fig. 5B).
FIG 5.

Simulated plasma concentrations and viable-count profiles of double-resistant (MICIPM of 16 mg/liter and MICTOB of 32 mg/liter) P. aeruginosa isolate FADDI-PA088 for monotherapies and combination dosage regimens with an initial inoculum of 107.5 CFU/ml. The Monte Carlo simulations included 10,000 critically ill virtual patients with bacteremia. These patients were assumed to have normal renal function and to completely lack any effect of the immune system. The success rate (Success), defined as ≤5.5 log10 CFU/ml at 48 h, is indicated for each simulated dosage regimen. All imipenem infusions were given over 1 h (except for the continuous infusion). Tobramycin was infused over 0.5 h.
The intermittent-dosage regimens of imipenem (1 g every 8 h [q8h] and q6h as 1-h infusions) combined with 7 mg/kg tobramycin were predicted to lead to bacteriological failure with extensive regrowth of resistant bacteria at 48 h; only 7.0% to 11.2% of the simulated patients were predicted to succeed (see Fig. 5C). For a continuous infusion of imipenem at 4 g/day (with a 1-g loading dose) combined with 7 mg/kg tobramycin, the predicted probability of success was 80.9%. Excitingly, the combination of a continuous infusion of imipenem at 5 g/day (with a 1-g loading dose) plus 7 mg/kg tobramycin (0.5-h infusion) was predicted to achieve ≥2.0-log10 bacterial killing without regrowth in 90.3% of the patients at 48 h and ≥3.5-log10 bacterial killing in 75% of patients (see Fig. 5C). The 50th percentile for 10,000 hypothetical patients was predicted to achieve ≥2-log10 killing over 7 days without regrowth for the regimen with 5 g/day imipenem plus tobramycin (data not shown).
DISCUSSION
This study presents the first systematic evaluation of synergy for carbapenem-plus-aminoglycoside combinations against carbapenem- and aminoglycoside-resistant clinical P. aeruginosa isolates using in vitro time-kill studies. Monte Carlo simulations based on the latest MBM were utilized to rationally optimize combination dosage regimens against a double-resistant isolate. Even at the highest clinically achievable unbound concentration, monotherapy with either imipenem or an aminoglycoside yielded limited bacterial killing, followed by rapid bacterial regrowth and emergence of resistance in a difficult-to-treat high bacterial inoculum.
Imipenem combined with either tobramycin or amikacin demonstrated substantially enhanced bacterial killing and suppression of resistance at clinically relevant concentrations. Against the double-resistant isolate FADDI-PA088, synergistic bacterial killing at 24 h occurred with aminoglycoside concentrations as low as 1/32 of the MIC (i.e., 1 mg/liter tobramycin with a tobramycin MIC [MICTOB] of 32 mg/liter) (Table 2 and Fig. 2). Resistance suppression at 48 h was achieved at 1/32 of the MIC for tobramycin and 1/8 of the MIC for amikacin (i.e., 0.5 mg/liter amikacin with an amikacin MIC [MICAMK] of 4 mg/liter) (Fig. 3). Our MBM indicated that the aminoglycoside enhanced the target site concentration of imipenem (Fig. 4). For our three isolates, we estimated that an aminoglycoside concentration (IC50,OM,AGS) of 1.13 or 1.54 mg/liter half-maximally permeabilized the outer membrane of isolates with an aminoglycoside MIC of 4 mg/liter; ∼4.29 to 5.55 mg/liter aminoglycoside was needed for this synergistic effect in isolates with an aminoglycoside MIC of 8 or 32 mg/liter (Table 3). In agreement with our results, Loh et al. (18) found a correlation between the aminoglycoside MIC and the affinity of various aminoglycosides for binding to the outer membrane of P. aeruginosa; strain-to-strain variability in the outer membrane affinity was considerable. To our knowledge, this is the first mechanism-based time course model that quantified the aminoglycoside concentration required to enhance outer membrane permeability of P. aeruginosa (19). Overall, these MBM results demonstrated that clinically achievable aminoglycoside concentrations can achieve synergistic killing and prevent resistance in combination with imipenem against carbapenem-resistant and double-resistant P. aeruginosa isolates.
While monotherapies failed with limited bacterial killing followed by extensive regrowth, combinations containing ≥0.88× MIC of imipenem were much more likely to prevent resistance over 48 h than were combinations with lower imipenem concentrations (Table 2 and Fig. 2 and 3). The extent of synergy (measured as viable counts that were ≥2 log10 CFU/ml lower than those of the most active monotherapy) tended to be greater at 24 and 48 h than at 6 h (Table 2). For combinations in the lower range of clinically relevant concentrations, resistance emergence after 24 h was most likely due to the amplification of preexisting resistant mutants, as we found in our clinical isolates (Fig. 3; see also Fig. S2 in the supplemental material).
Our findings substantially extend the results from previous studies on β-lactams in combination with aminoglycosides against P. aeruginosa. In the past, studies that used checkerboard methods showed that carbapenem-plus-aminoglycoside combinations were synergistic against P. aeruginosa; however, limited or no information on suppression of resistance and the type of synergy was provided (20–26). A few SCTK combination studies demonstrated that combinations (a β-lactam plus an aminoglycoside) are beneficial (14, 27, 28) but also reported extensive regrowth at 24 h (28). Synergistic bacterial killing, suppression of resistance, or both were found for the combination of meropenem plus an aminoglycoside in the hollow-fiber infection model and in murine pneumonia and septicemia models (8, 15, 29). Louie et al. evaluated the combination of meropenem plus tobramycin in a murine pneumonia model against a double-susceptible (meropenem MIC of 0.5 to 1 mg/liter and tobramycin MIC of 1 mg/liter) P. aeruginosa isolate (8). None of these studies evaluated or optimized dosage regimens against P. aeruginosa isolates that were resistant to carbapenems, aminoglycosides, or both. Furthermore, the mechanism of synergy has not been characterized based on a mathematical PK/pharmacodynamic (PD) model that incorporates bacterial killing, regrowth, and resistance.
Our MBM for the time course of synergistic bacterial killing and prevention of resistance had the same model structure and yielded consistent parameter estimates across three clinical P. aeruginosa isolates (Fig. 1 and Table 3). Mechanistic synergy (19, 30) was successfully incorporated with the aminoglycoside disrupting the outer membrane, thereby enhancing penetration and the concentrations of imipenem at its target site. This was expressed as a considerable decrease in the KC50,IPM against the relevant bacterial populations of all three isolates (Fig. 4). This mechanism of synergy is in close agreement with data from studies that showed outer membrane disruption of P. aeruginosa by albumin-conjugated aminoglycosides (31, 32). The outer membrane of P. aeruginosa presents a major penetration barrier (33, 34), and its disruption is likely to enhance the target site penetration of imipenem (19, 35).
We performed Monte Carlo simulations against a high inoculum of a double-resistant clinical isolate of P. aeruginosa, FADDI-PA088, which represented the 98th percentile of the MIC distribution of both antibiotics according to the EUCAST database. The targeted critically ill patients were assumed to lack any effect of the immune system and to have normal renal function and bacteremia. The predicted between-patient variability of the unbound plasma concentrations in critically ill patients was large (Fig. 5) due to the large variability in clearance (34% coefficient of variation [CV] for imipenem and 31% CV for tobramycin) and in the volume of distribution of the central compartment for imipenem (81% CV) (36, 37). All monotherapies failed to prevent regrowth over 48 h (Fig. 5B).
The rationally optimized combination of imipenem at 5 g/day as a continuous infusion (with a 1-g loading dose) plus tobramycin at 7 mg/kg every 24 h was predicted to achieve ≥2-log10 killing at 24 h and 48 h without regrowth in 90.3% of patients (i.e., regrowth in only 9.7% of patients) (Fig. 5C). The success rate for a continuous infusion of 4 g/day imipenem (with a 1-g loading dose) plus 7 mg/kg tobramycin every 24 h was 80.9%. These simulations were performed with a high inoculum of 107.5 CFU/ml of an extremely difficult-to-treat (double-resistant) isolate, and most of the regimens provided a benefit over 24 to 48 h. While our Monte Carlo simulations for bacteremia used a relatively high initial inoculum, which harbored preexisting resistant mutants, the predicted success rates are conservative and would have been higher for lower initial inocula.
These simulations represent a near-worst-case scenario with a highly tobramycin-resistant isolate (MIC of 32 mg/liter). Against FADDI-PA088, the combination of amikacin plus imipenem is expected to be more efficacious than that of tobramycin plus imipenem (Fig. 2A2), since this isolate had an amikacin MIC of 4 mg/liter and a tobramycin MIC of 32 mg/liter. The selected target endpoint for successful therapy (≥2 log10 units at 24 h and 48 h) was based on data from a study by Drusano et al. (38). This study suggested that achieving a target of ≥2-log10 bacterial killing at 24 h allows the granulocytes to contribute a few additional log10 CFU/ml of bacterial killing in an immunocompetent murine pneumonia model and minimizes the emergence of resistance.
Studies by Kumar et al. (39, 40) found that rapid and early antibiotic treatment significantly increases survival in an immunocompetent murine infection model of septic shock. These studies and our simulations suggest that a continuous infusion of imipenem at 5 g/day (with a loading dose) plus tobramycin at 7 mg/kg every 24 h may provide a beneficial effect in reducing the bacterial burden to an extent where the immune system in immunocompetent patients can achieve bacterial clearance. Therefore, prospective validation of this optimized combination dosage regimen in dynamic infection models is warranted.
Our results demonstrate the importance of evaluating the robustness of combination dosage regimens for effective early therapy via Monte Carlo simulations in the presence of the high between-patient variability in PK that is typically observed in critically ill patients (41–43). Our most robust regimen, which contained imipenem at 5 g/day as a continuous infusion, was promising with regard to success rate of therapy; however, this dose may slightly increase the risk of seizures compared to that with 2 to 4 g imipenem per day (44, 45). Following imipenem treatment, nausea and vomiting were observed in one patient (out of 45 patients in the study); these symptoms were considered to be probably due to rapid infusion, as they disappeared after the duration of infusion was increased (46). Several studies demonstrated that therapeutic drug monitoring (TDM) may be a valuable strategy to optimize antibiotic exposure and minimize toxicity in critically ill patients (42, 47, 48). This approach may help to attain effective imipenem concentrations using 2 or 4 g imipenem per day.
In summary, clinically relevant concentrations of imipenem plus an aminoglycoside provided synergistic bacterial killing and suppression of resistance against a high inoculum of clinical isolates resistant to a carbapenem or to both a carbapenem and an aminoglycoside. Approximately 1.34 mg/liter aminoglycoside was sufficient to enhance the imipenem target site concentration half-maximally if the aminoglycoside MIC was 4 mg/liter; ∼4.88 mg/liter aminoglycoside was required if the aminoglycoside MIC was 8 or 32 mg/liter. Mechanism-based modeling suggested that disruption of the outer membrane by an aminoglycoside contributed to the synergy of combinations of imipenem plus an aminoglycoside. Against a double-resistant isolate of P. aeruginosa, Monte Carlo simulations predicted a 90.3% success rate in achieving ≥2-log10 killing at 24 h and 48 h for a clinically relevant imipenem-plus-tobramycin combination dosage regimen. Thus, future evaluation of this promising combination dosage regimen in dynamic infection models is warranted to provide guidance on effective early therapy against infections caused by extremely difficult-to-treat double-resistant P. aeruginosa isolates.
MATERIALS AND METHODS
Bacterial isolates and susceptibility testing.
We studied three carbapenem-resistant P. aeruginosa isolates (FADDI-PA088, FADDI-PA022, and FADDI-PA001) from the collection at Monash University (Table 1). These isolates were resistant to tobramycin, amikacin, or both aminoglycosides. All susceptibility testing and SCTK experiments were performed with cation-adjusted Mueller-Hinton II broth (CAMHB; BBL, BD, Sparks, MD). Counting of viable bacteria was conducted on cation-adjusted Mueller-Hinton II agar (CAMHA; Medium Preparation Unit, The University of Melbourne). Stock solutions of imipenem (Merck Sharp & Dohme Pty., NSW, Australia), tobramycin (AK Scientific, Inc., Union City, CA), and amikacin (Sigma-Aldrich, St. Louis, MO) were prepared in sterile, distilled water and filter sterilized with a Millex-GV 0.22-μm polyvinylidene difluoride (PVDF) syringe filter (Merck Millipore Ltd., Cork, Ireland). The MICs were determined in triplicate according to Clinical and Laboratory Standards Institute guidelines (49). EUCAST breakpoints were used to define resistance to carbapenem and the aminoglycosides (50).
Static-concentration time-kill experiments.
Clinically relevant imipenem concentrations ranging from 8 to 36 mg/liter were studied as monotherapies and in combination with an aminoglycoside. The evaluated aminoglycosides were tobramycin (1 to 32 mg/liter) and amikacin (0.5 to 64 mg/liter). The selected antibiotic concentrations included the highest clinically achievable average unbound plasma concentrations at steady state (19, 36, 51). Higher and lower antibiotic concentrations were included to explore the range of synergistic concentrations and to consider the MICs for the isolates.
As previously described (30, 52, 53), the SCTK experiments were performed with a clinically relevant high initial inoculum (107.5 CFU/ml) to mirror more severe infections (54–59). Serial broth samples were taken within 5 to 10 min before dosing (i.e., at 0 h) and at 1, 3, 6, 24, 29, and 48 h. At 24 h, bacterial suspensions were centrifuged, the supernatant was carefully removed, and bacteria were resuspended in fresh, prewarmed broth with the targeted antibiotic concentration(s). Informed by our imipenem stability data using CAMHB at 35°C, an imipenem amount corresponding to 50% of the original dose was supplemented at 6 and 30 h to further offset the thermal degradation of imipenem, as previously described (19, 60). All bacterial samples were washed twice in sterile saline. Viable counts were determined by manual plating of 100 μl of an undiluted or an appropriately diluted suspension in saline onto CAMHA plates (19, 30, 52, 53). Serial dilution (1:10) was performed via the addition of 100 μl of an undiluted bacterial suspension to 900 μl of sterile saline. Aliquots of the undiluted or diluted sample were plated onto agar plates supplemented with imipenem, tobramycin, or amikacin at 3× MIC to quantify the resistant subpopulations.
Mechanism-based modeling of combinations of imipenem and an aminoglycoside.
An MBM was developed to describe and predict the time course of bacterial growth, killing, and resistance; to characterize the synergy for the studied carbapenem-plus-aminoglycoside combinations (19, 61–63); and to enable mechanism-based Monte Carlo simulations based on previously reported population PK models (36, 37).
Life cycle growth model.
The developed PD model contained a life cycle growth model that describes bacterial replication via two states (61, 64, 65): bacterial cells that are growing and preparing for replication are in state 1, and bacteria immediately before replication reside in state 2. The first-order growth rate constant (k12) determines the mean generation time (MGT). Bacterial replication was assumed to be rapid, with the replication rate constant k21 being fixed at 50 h−1 (64).
Model for combinations of imipenem and an aminoglycoside.
The model for the combinations of imipenem and an aminoglycoside includes three preexisting bacterial populations: population 1 was susceptible to both antibiotics (CFUSS), population 2 was imipenem resistant and aminoglycoside intermediate (CFURI), and population 3 was imipenem intermediate and aminoglycoside resistant (CFUIR) (Fig. 1). Each population was described by the two states (i.e., compartments) of the life cycle growth model. Thus, the composite model contained six bacterial compartments. The total concentration of all viable bacteria (CFUall) was
| (1) |
CFUNNx denotes the concentration of viable bacteria of population NN in state x. At high bacterial densities, high concentrations of hypothetical signal molecules (Csig) were assumed to inhibit the bacterial growth rate via the inhibition term Inhk12, as defined below and described previously (61):
| (2) |
Imax,sig12 is the maximum extent of inhibition of the rate of replication at high Csig, and IC50,sig is the signal molecule concentration associated with 50% Imax,sig12.
In most cases, the rates of bacterial killing by high concentrations of imipenem or the aminoglycoside or by their synergistic combinations were higher than the bacterial growth rate. Thus, we applied a direct-killing process with a Hill function to describe killing by each antibiotic. The time course of bacterial killing and, where applicable, regrowth (if it occurred) during treatment with imipenem, tobramycin, amikacin, or combinations thereof was fitted simultaneously for all three isolates. The differential equation for the concentration of bacteria belonging to population 1 (IPMS/AGSS) in state 1 (CFUSS1) comprised killing by imipenem (concentration of IPM [CIPM]) and an aminoglycoside (concentration of the aminoglycoside [CAGS]) (initial conditions are described below):
| (3) |
The plateau factor (PLAT) is defined as 1 − [CFUall/(CFUall + CFUmax)], with CFUmax being the maximum population size. The factor 2 represents the doubling of bacteria during replication (61). The maximum killing rate constant for imipenem (Kmax,IPM), the imipenem concentration causing 50% Kmax for the double-susceptible population (KC50,SS,IPM), and the Hill coefficient for IPM (HillIPM) affected both states 1 and 2 of the population. The maximum killing rate constant for the aminoglycoside was denoted Kmax,SS,AGS, and the aminoglycoside concentration causing 50% Kmax was denoted KC50,SS,AGS. The ability of the aminoglycosides to permeabilize the outer membrane (OM_effect) (i.e., the synergy term) is described in equation 5 below. The differential equation for state 2 of population 1 (CFUSS2) was
| (4) |
The differential equations for population 2 (i.e., CFURI) and population 3 (i.e., CFUIR) used the same structure as the one for double-susceptible population 1 but different parameters for KC50,IPM, k12, Kmax,AGS, and KC50,AGS, as described previously (19, 30).
Mechanism-based modeling of synergy.
The presence of mechanistic synergy (i.e., antibiotic A enhancing killing by antibiotic B of one or multiple bacterial populations) was evaluated previously (30). Mechanistic synergy was incorporated by assuming that disruption and, thus, permeabilization of the bacterial outer membrane by aminoglycosides (31, 32, 35) enhance the target site penetration of imipenem. This effect was implemented in the model via the term OM_effect by the aminoglycoside decreasing the KC50,IPM in a concentration-dependent manner. The effect of aminoglycosides on the outer membrane was described as follows (parameters are described in Table 3):
| (5) |
Initial conditions.
The total inoculum (log CFU0) and the log10 mutation frequencies for population 2 (RI) and population 3 (IR) were estimated (Fig. 1). Initial conditions were implemented as described previously (19, 61).
Observation model.
An additive residual-error model on a log10 scale was used to fit the log10 viable counts. For observations of viable counts below 100 CFU/ml (equivalent to <10 colonies per plate), a previously developed residual-error model was utilized to fit the number of colonies per plate (52). Observed viable counts below the limit of counting (i.e., 1 log10 CFU/ml, equivalent to 1 colony per agar plate) were plotted as zero.
Estimation.
The importance sampling algorithm (S-ADAPT setting, pmethod = 4) was used for the simultaneous estimation of all PD parameters in the parallelized S-ADAPT software (version 1.57) (66) facilitated by the SADAPT-TRAN tool (67, 68). The between-curve variability of the PD parameters was fixed to a final, small coefficient of variation (52). Competing models were evaluated by the objective function (−1 × log likelihood in S-ADAPT), the biological plausibility of the parameter estimates, standard diagnostic plots, and visual predictive checks, as described previously (69–72).
Monte Carlo simulations.
For each dosage regimen, we performed Monte Carlo simulations for 10,000 adult critically ill patients with normal renal function. We combined the final MBM with previously reported human population PK models for imipenem (37) and tobramycin (36) in critically ill patients using Berkeley Madonna software (version 8.3.18). Tobramycin and imipenem population PK models, parameter estimates, and between-subject variability were implemented as reported previously (19).
The simulated patients were assumed to have bacteremia caused by a multidrug-resistant (MDR) P. aeruginosa isolate (FADDI-PA088) and lacked any aspect of the immune system. This isolate had an imipenem MIC of 16 mg/liter and a tobramycin MIC of 32 mg/liter, which reflect the 98th percentiles of the EUCAST MIC distributions of both antibiotics for P. aeruginosa (last accessed 22 April 2016) (50).
Unbound plasma concentrations were simulated by assuming unbound fractions of 0.91 for imipenem and 1.0 for tobramycin (36, 37). Viable-count profiles were predicted for imipenem and tobramycin in monotherapy and in combination by using the combined MBM. The dosage regimens simulated included 1 g imipenem given every 6 or 8 h as a 1-h infusion and 4 or 5 g imipenem given as a continuous infusion with a 1-g loading dose (dosed as a 1-h infusion). The simulated tobramycin regimen was 7 mg/kg given as a 0.5-h infusion every 24 h. A successful therapeutic outcome was defined as ≥2-log10 bacterial killing compared to the initial inoculum, i.e., viable counts of the total population of 105.5 CFU/ml or lower, at both 24 and 48 h (38–40, 73). We chose the 48-h time point for our double-resistant isolates and assumed that the immune system (38, 73) can clear the bacteria remaining at 48 h, if antibiotic combination therapy achieves at least 2-log10 killing without regrowth over 48 h. The success rate was calculated as the fraction of all simulated patients who were predicted to achieve this target.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly supported by Australian National Health and Medical Research Council (NHMRC) project grants (APP1045105 to J.B.B., R.L.N., and C.B.L., APP1101553 to C.B.L., J.B.B., and R.L.N., and APP1062040 to C.B.L. and J.B.B.). R.Y. is thankful to the Monash Institute of Graduate Research for providing a Monash graduate scholarship and a Monash international postgraduate research scholarship. C.B.L. is the recipient of a NHMRC career development fellowship (APP1062509).
We have no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01011-16.
REFERENCES
- 1.Boucher HW, Talbot GH, Benjamin DK Jr, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D. 2013. 10 × ‘20 progress—development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 56:1–17. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Infectious Diseases Society of America, Spellberg B, Blaser M, Guidos RJ, Boucher HW, Bradley JS, Eisenstein BI, Gerding D, Lynfield R, Reller LB, Rex J, Schwartz D, Septimus E, Tenover FC, Gilbert DN. 2011. Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis 52(Suppl 5):S397–S428. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 4.Walker B, Barrett S, Polasky S, Galaz V, Folke C, Engstrom G, Ackerman F, Arrow K, Carpenter S, Chopra K, Daily G, Ehrlich P, Hughes T, Kautsky N, Levin S, Maler KG, Shogren J, Vincent J, Xepapadeas T, de Zeeuw A. 2009. Environment. Looming global-scale failures and missing institutions. Science 325:1345–1346. doi: 10.1126/science.1175325. [DOI] [PubMed] [Google Scholar]
- 5.Dantas RC, Ferreira ML, Gontijo-Filho PP, Ribas RM. 2014. Pseudomonas aeruginosa bacteraemia: independent risk factors for mortality and impact of resistance on outcome. J Med Microbiol 63:1679–1687. doi: 10.1099/jmm.0.073262-0. [DOI] [PubMed] [Google Scholar]
- 6.Woodford N, Turton JF, Livermore DM. 2011. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 7.Partridge SR. 2011. Analysis of antibiotic resistance regions in Gram-negative bacteria. FEMS Microbiol Rev 35:820–855. doi: 10.1111/j.1574-6976.2011.00277.x. [DOI] [PubMed] [Google Scholar]
- 8.Louie A, Liu W, Fikes S, Brown D, Drusano GL. 2013. Impact of meropenem in combination with tobramycin in a murine model of Pseudomonas aeruginosa pneumonia. Antimicrob Agents Chemother 57:2788–2792. doi: 10.1128/AAC.02624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zavascki AP, Bulitta JB, Landersdorfer CB. 2013. Combination therapy for carbapenem-resistant Gram-negative bacteria. Expert Rev Anti Infect Ther 11:1333–1353. doi: 10.1586/14787210.2013.845523. [DOI] [PubMed] [Google Scholar]
- 10.Lee CS, Doi Y. 2014. Therapy of infections due to carbapenem-resistant Gram-negative pathogens. Infect Chemother 46:149–164. doi: 10.3947/ic.2014.46.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kresken M, Korber-Irrgang B, Lauffer J, Decker-Burgard S, Davies T. 2011. In vitro activities of ceftobiprole combined with amikacin or levofloxacin against Pseudomonas aeruginosa: evidence of a synergistic effect using time-kill methodology. Int J Antimicrob Agents 38:70–75. doi: 10.1016/j.ijantimicag.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 12.Milatovic D, Braveny I. 1987. Development of resistance during antibiotic therapy. Eur J Clin Microbiol 6:234–244. doi: 10.1007/BF02017607. [DOI] [PubMed] [Google Scholar]
- 13.Drusano GL, Liu W, Fregeau C, Kulawy R, Louie A. 2009. Differing effects of combination chemotherapy with meropenem and tobramycin on cell kill and suppression of resistance of wild-type Pseudomonas aeruginosa PAO1 and its isogenic MexAB efflux pump-overexpressed mutant. Antimicrob Agents Chemother 53:2266–2273. doi: 10.1128/AAC.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam VH, Schilling AN, Lewis RE, Melnick DA, Boucher AN. 2004. Novel approach to characterization of combined pharmacodynamic effects of antimicrobial agents. Antimicrob Agents Chemother 48:4315–4321. doi: 10.1128/AAC.48.11.4315-4321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tam VH, Schilling AN, Neshat S, Poole K, Melnick DA, Coyle EA. 2005. Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:4920–4927. doi: 10.1128/AAC.49.12.4920-4927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim TP, Lee W, Tan TY, Sasikala S, Teo J, Hsu LY, Tan TT, Syahidah N, Kwa AL. 2011. Effective antibiotics in combination against extreme drug-resistant Pseudomonas aeruginosa with decreased susceptibility to polymyxin B. PLoS One 6:e28177. doi: 10.1371/journal.pone.0028177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ly NS, Bulitta JB, Rao GG, Landersdorfer CB, Holden PN, Forrest A, Bergen PJ, Nation RL, Li J, Tsuji BT. 2015. Colistin and doripenem combinations against Pseudomonas aeruginosa: profiling the time course of synergistic killing and prevention of resistance. J Antimicrob Chemother 70:1434–1442. doi: 10.1093/jac/dku567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loh B, Grant C, Hancock RE. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob Agents Chemother 26:546–551. doi: 10.1128/AAC.26.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadav R, Landersdorfer CB, Nation RL, Boyce JD, Bulitta JB. 2015. Novel approach to optimize synergistic carbapenem-aminoglycoside combinations against carbapenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 59:2286–2298. doi: 10.1128/AAC.04379-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anan N, Toba S, Ito A, Nakamura R, Tsuji M. 2011. In vitro combination effects of doripenem with aminoglycoside or ciprofloxacin against Pseudomonas aeruginosa. Jpn J Antibiot 64:203–216. [PubMed] [Google Scholar]
- 21.Balke B, Hogardt M, Schmoldt S, Hoy L, Weissbrodt H, Haussler S. 2006. Evaluation of the E test for the assessment of synergy of antibiotic combinations against multiresistant Pseudomonas aeruginosa isolates from cystic fibrosis patients. Eur J Clin Microbiol Infect Dis 25:25–30. doi: 10.1007/s10096-005-0076-9. [DOI] [PubMed] [Google Scholar]
- 22.Gerceker AA, Gurler B. 1995. In-vitro activities of various antibiotics, alone and in combination with amikacin against Pseudomonas aeruginosa. J Antimicrob Chemother 36:707–711. doi: 10.1093/jac/36.4.707. [DOI] [PubMed] [Google Scholar]
- 23.Giamarellou H. 1986. Aminoglycosides plus beta-lactams against gram-negative organisms. Evaluation of in vitro synergy and chemical interactions. Am J Med 80:126–137. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura A, Hosoda M, Kato T, Yamada Y, Itoh M, Kanazawa K, Nouda H. 2000. Combined effects of meropenem and aminoglycosides on Pseudomonas aeruginosa in vitro. J Antimicrob Chemother 46:901–904. doi: 10.1093/jac/46.6.901. [DOI] [PubMed] [Google Scholar]
- 25.Tasaka K, Ishida A, Chinzei T. 2002. Antimicrobial activity of carbapenems and the combined effect with aminoglycoside against recent clinical isolates of Pseudomonas aeruginosa. Jpn J Antibiot 55:181–186. doi: 10.7164/antibiotics.55.181. [DOI] [PubMed] [Google Scholar]
- 26.Yamashiro Y, Ogake N, Takahata M, Minami S. 2000. In vitro interaction of piperacillin and imipenem/cilastatin combined with aminoglycosides against Pseudomonas aeruginosa. Jpn J Antibiot 53:194–200. [PubMed] [Google Scholar]
- 27.Niida M, Sakakibara S, Kawabata T, Maebashi K, Takata T, Hikida M. 2004. Combined effects of arbekacin with biapenem against in vitro and in vivo model of a mixture of MRSA and Pseudomonas aeruginosa. Jpn J Antibiot 57:288–293. [PubMed] [Google Scholar]
- 28.Wise R, Ashby JP, Andrews JM. 1989. The antibacterial activity of meropenem in combination with gentamicin or vancomycin. J Antimicrob Chemother 24(Suppl A):233–238. doi: 10.1093/jac/24.suppl_A.233. [DOI] [PubMed] [Google Scholar]
- 29.Ulrich E, Trautmann M, Krause B, Bauernfeind A, Hahn H. 1989. Comparative efficacy of ciprofloxacin, azlocillin, imipenem/cilastatin and tobramycin in a model of experimental septicemia due to Pseudomonas aeruginosa in neutropenic mice. Infection 17:311–315. doi: 10.1007/BF01650716. [DOI] [PubMed] [Google Scholar]
- 30.Landersdorfer CB, Ly NS, Xu H, Tsuji BT, Bulitta JB. 2013. Quantifying subpopulation synergy for antibiotic combinations via mechanism-based modeling and a sequential dosing design. Antimicrob Agents Chemother 57:2343–2351. doi: 10.1128/AAC.00092-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadurugamuwa JL, Clarke AJ, Beveridge TJ. 1993. Surface action of gentamicin on Pseudomonas aeruginosa. J Bacteriol 175:5798–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadurugamuwa JL, Lam JS, Beveridge TJ. 1993. Interaction of gentamicin with the A band and B band lipopolysaccharides of Pseudomonas aeruginosa and its possible lethal effect. Antimicrob Agents Chemother 37:715–721. doi: 10.1128/AAC.37.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obara M, Nakae T. 1991. Mechanisms of resistance to beta-lactam antibiotics in Acinetobacter calcoaceticus. J Antimicrob Chemother 28:791–800. doi: 10.1093/jac/28.6.791. [DOI] [PubMed] [Google Scholar]
- 34.Sato K, Nakae T. 1991. Outer membrane permeability of Acinetobacter calcoaceticus and its implication in antibiotic resistance. J Antimicrob Chemother 28:35–45. doi: 10.1093/jac/28.1.35. [DOI] [PubMed] [Google Scholar]
- 35.Bulitta JB, Ly NS, Landersdorfer CB, Wanigaratne NA, Velkov T, Yadav R, Oliver A, Martin L, Shin BS, Forrest A, Tsuji BT. 2015. Two mechanisms of killing of Pseudomonas aeruginosa by tobramycin assessed at multiple inocula via mechanism-based modeling. Antimicrob Agents Chemother 59:2315–2327. doi: 10.1128/AAC.04099-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conil JM, Georges B, Ruiz S, Rival T, Seguin T, Cougot P, Fourcade O, Houin G, Saivin S. 2011. Tobramycin disposition in ICU patients receiving a once daily regimen: population approach and dosage simulations. Br J Clin Pharmacol 71:61–71. doi: 10.1111/j.1365-2125.2010.03793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakka SG, Glauner AK, Bulitta JB, Kinzig-Schippers M, Pfister W, Drusano GL, Sorgel F. 2007. Population pharmacokinetics and pharmacodynamics of continuous versus short-term infusion of imipenem-cilastatin in critically ill patients in a randomized, controlled trial. Antimicrob Agents Chemother 51:3304–3310. doi: 10.1128/AAC.01318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drusano GL, Liu W, Fikes S, Cirz R, Robbins N, Kurhanewicz S, Rodriquez J, Brown D, Baluya D, Louie A. 2014. Interaction of drug- and granulocyte-mediated killing of Pseudomonas aeruginosa in a murine pneumonia model. J Infect Dis 210:1319–1324. doi: 10.1093/infdis/jiu237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Haery C, Paladugu B, Kumar A, Symeoneides S, Taiberg L, Osman J, Trenholme G, Opal SM, Goldfarb R, Parrillo JE. 2006. The duration of hypotension before the initiation of antibiotic treatment is a critical determinant of survival in a murine model of Escherichia coli septic shock: association with serum lactate and inflammatory cytokine levels. J Infect Dis 193:251–258. doi: 10.1086/498909. [DOI] [PubMed] [Google Scholar]
- 40.Vazquez-Grande G, Kumar A. 2015. Optimizing antimicrobial therapy of sepsis and septic shock: focus on antibiotic combination therapy. Semin Respir Crit Care Med 36:154–166. doi: 10.1055/s-0034-1398742. [DOI] [PubMed] [Google Scholar]
- 41.Matar KM, Al-Lanqawi Y, Abdul-Malek K, Jelliffe R. 2013. Amikacin population pharmacokinetics in critically ill Kuwaiti patients. Biomed Res Int 2013:202818. doi: 10.1155/2013/202818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts JA, Lipman J. 2009. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit Care Med 37:840–851. doi: 10.1097/CCM.0b013e3181961bff. [DOI] [PubMed] [Google Scholar]
- 43.Tod M, Padoin C, Petitjean O. 2000. Clinical pharmacokinetics and pharmacodynamics of isepamicin. Clin Pharmacokinet 38:205–223. doi: 10.2165/00003088-200038030-00002. [DOI] [PubMed] [Google Scholar]
- 44.Cannon JP, Lee TA, Clark NM, Setlak P, Grim SA. 2014. The risk of seizures among the carbapenems: a meta-analysis. J Antimicrob Chemother 69:2043–2055. doi: 10.1093/jac/dku111. [DOI] [PubMed] [Google Scholar]
- 45.Koppel BS, Hauser WA, Politis C, van Duin D, Daras M. 2001. Seizures in the critically ill: the role of imipenem. Epilepsia 42:1590–1593. [DOI] [PubMed] [Google Scholar]
- 46.Szczygiel B, Pertkiewicz M, Meszaros J, Otto M, Grochowiecki T, Rudnicka J, Hacel I, Solecka-Lasak M. 1992. Imipenem in the treatment of patients with severe surgical infection. Pol Tyg Lek 47:638–641. [PubMed] [Google Scholar]
- 47.Patel BM, Paratz J, See NC, Muller MJ, Rudd M, Paterson D, Briscoe SE, Ungerer J, McWhinney BC, Lipman J, Roberts JA. 2012. Therapeutic drug monitoring of beta-lactam antibiotics in burns patients—a one-year prospective study. Ther Drug Monit 34:160–164. doi: 10.1097/FTD.0b013e31824981a6. [DOI] [PubMed] [Google Scholar]
- 48.Pea F, Cojutti P, Sbrojavacca R, Cadeo B, Cristini F, Bulfoni A, Furlanut M. 2011. TDM-guided therapy with daptomycin and meropenem in a morbidly obese, critically ill patient. Ann Pharmacother 45:e37. doi: 10.1345/aph.1P745. [DOI] [PubMed] [Google Scholar]
- 49.Clinical and Laboratory Standards Institute. 2015. Performance standards for antimicrobial susceptibility testing: 25th informational supplement. M100-S25. CLSI, Wayne, PA. [Google Scholar]
- 50.European Committee on Antimicrobial Susceptibility Testing. 2015. Breakpoint tables for interpretation of MICs and zone diameters, version 5.0. http://www.eucast.org/clinical_breakpoints/.
- 51.Taccone FS, Laterre PF, Spapen H, Dugernier T, Delattre I, Layeux B, De Backer D, Wittebole X, Wallemacq P, Vincent JL, Jacobs F. 2010. Revisiting the loading dose of amikacin for patients with severe sepsis and septic shock. Crit Care 14:R53. doi: 10.1186/cc8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bulitta JB, Yang JC, Yohonn L, Ly NS, Brown SV, D'Hondt RE, Jusko WJ, Forrest A, Tsuji BT. 2010. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother 54:2051–2062. doi: 10.1128/AAC.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nicasio AM, Bulitta JB, Lodise TP, D'Hondt RE, Kulawy R, Louie A, Drusano GL. 2012. Evaluation of once-daily vancomycin against methicillin-resistant Staphylococcus aureus in a hollow-fiber infection model. Antimicrob Agents Chemother 56:682–686. doi: 10.1128/AAC.05664-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konig C, Simmen HP, Blaser J. 1998. Bacterial concentrations in pus and infected peritoneal fluid—implications for bactericidal activity of antibiotics. J Antimicrob Chemother 42:227–232. doi: 10.1093/jac/42.2.227. [DOI] [PubMed] [Google Scholar]
- 55.LaPlante KL, Rybak MJ. 2004. Impact of high-inoculum Staphylococcus aureus on the activities of nafcillin, vancomycin, linezolid, and daptomycin, alone and in combination with gentamicin, in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 48:4665–4672. doi: 10.1128/AAC.48.12.4665-4672.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S, Kwon KT, Kim HI, Chang HH, Lee JM, Choe PG, Park WB, Kim NJ, Oh MD, Song DY, Kim SW. 2014. Clinical implications of cefazolin inoculum effect and b-lactamase type on methicillin-susceptible Staphylococcus aureus bacteremia. Microb Drug Resist 20:568–574. doi: 10.1089/mdr.2013.0229. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Echevarria KL, Hughes DW, Cadena JA, Bowling JE, Lewis JS II. 2014. Comparison of cefazolin versus oxacillin for treatment of complicated bacteremia caused by methicillin-susceptible Staphylococcus aureus. Antimicrob Agents Chemother 58:5117–5124. doi: 10.1128/AAC.02800-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rose WE, Leonard SN, Rossi KL, Kaatz GW, Rybak MJ. 2009. Impact of inoculum size and heterogeneous vancomycin-intermediate Staphylococcus aureus (hVISA) on vancomycin activity and emergence of VISA in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 53:805–807. doi: 10.1128/AAC.01009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.So W, Crandon JL, Zhanel GG, Nicolau DP. 2014. Comparison of in vivo and in vitro pharmacodynamics of a humanized regimen of 600 milligrams of ceftaroline fosamil every 12 hours against Staphylococcus aureus at initial inocula of 106 and 108 CFU per milliliter. Antimicrob Agents Chemother 58:6931–6933. doi: 10.1128/AAC.03652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louie A, Bied A, Fregeau C, Van Scoy B, Brown D, Liu W, Bush K, Queenan AM, Morrow B, Khashab M, Kahn JB, Nicholson S, Kulawy R, Drusano GL. 2010. Impact of different carbapenems and regimens of administration on resistance emergence for three isogenic Pseudomonas aeruginosa strains with differing mechanisms of resistance. Antimicrob Agents Chemother 54:2638–2645. doi: 10.1128/AAC.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bulitta JB, Ly NS, Yang JC, Forrest A, Jusko WJ, Tsuji BT. 2009. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:46–56. doi: 10.1128/AAC.00489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheah SE, Li J, Tsuji BT, Forrest A, Bulitta JB, Nation RL. 2016. Colistin and polymyxin B dosage regimens against Acinetobacter baumannii: differences in activity and the emergence of resistance. Antimicrob Agents Chemother 60:3921–3933. doi: 10.1128/AAC.02927-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacobs M, Gregoire N, Couet W, Bulitta JB. 2016. Distinguishing antimicrobial models with different resistance mechanisms via population pharmacodynamic modeling. PLoS Comput Biol 12:e1004782. doi: 10.1371/journal.pcbi.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maidhof H, Johannsen L, Labischinski H, Giesbrecht P. 1989. Onset of penicillin-induced bacteriolysis in staphylococci is cell cycle dependent. J Bacteriol 171:2252–2257. doi: 10.1128/jb.171.4.2252-2257.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsuji BT, Bulitta JB, Brown T, Forrest A, Kelchlin PA, Holden PN, Peloquin CA, Skerlos L, Hanna D. 2012. Pharmacodynamics of early, high-dose linezolid against vancomycin-resistant enterococci with elevated MICs and pre-existing genetic mutations. J Antimicrob Chemother 67:2182–2190. doi: 10.1093/jac/dks201. [DOI] [PubMed] [Google Scholar]
- 66.Bauer RJ, Guzy S, Ng C. 2007. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J 9:E60–E83. doi: 10.1208/aapsj0901007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bulitta JB, Bingolbali A, Shin BS, Landersdorfer CB. 2011. Development of a new pre- and post-processing tool (SADAPT-TRAN) for nonlinear mixed-effects modeling in S-ADAPT. AAPS J 13:201–211. doi: 10.1208/s12248-011-9257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bulitta JB, Landersdorfer CB. 2011. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J 13:212–226. doi: 10.1208/s12248-011-9258-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bulitta JB, Duffull SB, Kinzig-Schippers M, Holzgrabe U, Stephan U, Drusano GL, Sorgel F. 2007. Systematic comparison of the population pharmacokinetics and pharmacodynamics of piperacillin in cystic fibrosis patients and healthy volunteers. Antimicrob Agents Chemother 51:2497–2507. doi: 10.1128/AAC.01477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bulitta JB, Zhao P, Arnold RD, Kessler DR, Daifuku R, Pratt J, Luciano G, Hanauske AR, Gelderblom H, Awada A, Jusko WJ. 2009. Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients. Cancer Chemother Pharmacol 63:1049–1063. doi: 10.1007/s00280-008-0827-2. [DOI] [PubMed] [Google Scholar]
- 71.Tsuji BT, Okusanya OO, Bulitta JB, Forrest A, Bhavnani SM, Fernandez PB, Ambrose PG. 2011. Application of pharmacokinetic-pharmacodynamic modeling and the justification of a novel fusidic acid dosing regimen: raising Lazarus from the dead. Clin Infect Dis 52(Suppl 7):S513–S519. doi: 10.1093/cid/cir166. [DOI] [PubMed] [Google Scholar]
- 72.Landersdorfer CB, Kirkpatrick CM, Kinzig-Schippers M, Bulitta JB, Holzgrabe U, Drusano GL, Sorgel F. 2007. Population pharmacokinetics at two dose levels and pharmacodynamic profiling of flucloxacillin. Antimicrob Agents Chemother 51:3290–3297. doi: 10.1128/AAC.01410-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Drusano GL, Liu W, Kulawy R, Louie A. 2011. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother 55:5300–5305. doi: 10.1128/AAC.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yadav R, Bulitta JB, Nation RL, Landersdorfer CB. 2016. Optimization of synergistic combinations against carbapenem- and aminoglycoside-resistant clinical isolates of Pseudomonas aeruginosa via mechanism-based modelling, poster 504. ASM Microbe 2016, Boston, MA, USA, 16 to 20 June, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.