

ABSTRACT
Rilpivirine (RPV), the latest nonnucleoside reverse transcriptase inhibitor active against HIV-1, is prescribed in a standard dosage of 25 mg once a day in combination with emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF). The aim of this observational study was to characterize the RPV pharmacokinetic profile, to quantify interpatient variability, and to identify potential factors that could influence drug exposure. RPV concentration data were collected from HIV-infected patients as part of routine therapeutic drug monitoring performed in our center (Laboratory of Clinical Pharmacology). A population pharmacokinetic analysis was performed with NONMEM by comparing various structural models. The influence of demographic and clinical covariates, as well as frequent genetic polymorphisms in 5 genes (CYP3A4*22, CYP3A5*3, CYP2C19*2, CYP2C19*17, UGT1A1*28, and UGT1A4*2), on RPV elimination was explored. A total of 325 plasma concentration measurements were obtained from 249 HIV-positive patients. Plasma concentrations ranged from 12 to 255 ng/ml. A one-compartment model with zero-order absorption best characterized RPV pharmacokinetics. The average RPV clearance (CL) was 11.7 liters/h, the average volume of distribution was 401 liters, and the mean absorption time was 4 h. The interinterindividual variability (IIV) for CL was estimated to be 33%. None of the available demographic or genetic covariates showed any influence on RPV pharmacokinetics, but 29% of the patients were predicted to present minimal concentrations below the recently identified target cutoff value of 50 ng/ml. The variability in RPV pharmacokinetics appears to be lower than that for most other antiretroviral drugs. However, under the standard regimen of 25 mg daily, a significant number of patients might be underdosed. It remains to be investigated whether the underexposure has an impact on the development of resistance while patients are on maintenance therapy.
KEYWORDS: rilpivirine, population pharmacokinetics, nonnucleoside reverse transcriptase, therapeutic drug monitoring
INTRODUCTION
Rilpivirine (RPV), the latest nonnucleoside reverse transcriptase inhibitor (NNRTI) active against HIV-1, is prescribed at 25 mg once daily (QD) in a fixed dose in combination with emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF). It is recommended for the first-line treatment of HIV-1 infection in naive patients with a viral load (VL) of less than 100,000 copies/mm3, since the impact of the baseline VL on the treatment response has been identified to be an issue in RPV efficacy (1, 2). Owing to its good tolerability and the convenience of the single-tablet regimen (3, 4), RPV is increasingly prescribed in virologically suppressed treatment-experienced patients when an antiretroviral regimen simplification is considered (5).
RPV solubility and systemic absorption are pH dependent, as demonstrated by an increased bioavailability in an acidic environment (6). Moreover, RPV should be administered with food to optimize bioavailability (7). It undergoes oxidative metabolism in the liver that is mainly mediated by CYP3A4 and to a lesser extent by CYP2C19 (1, 2). Recently, in vitro observations suggested that UGT1A1 and UGT1A4 are also involved in RPV metabolism (8).
In the phase III clinical studies ECHO and THRIVE, a clear relationship between the RPV minimum (trough) concentration (Cmin) and efficacy was established (1). In patients with a baseline viral load of less than 100,000 copies/ml, a Cmin of less than 44 ng/ml (quartile 1) was associated with twice as many virological failures than a Cmin of greater than 123 ng/ml (quartile 3) (1). It was therefore concluded that a minimal concentration of 50 ng/ml be maintained to increase the probability of a therapeutic response (9). This clinically established exposure target is probably more appropriate than the 90% inhibitory concentration (IC90) for viral growth adjusted for the protein concentration (12 ng/ml) derived from in vitro data, which was suggested as an alternate drug level target (2).
At the standard dosage, RPV is well tolerated. Nevertheless, a phase I clinical study reported that the QTc prolongation is dose dependent. At high dosages (3 to 12 times the approved dose), when plasma concentrations are higher than 500 ng/ml, a significant prolongation of the QTc interval (>10 ms) was observed (1). Caution is thus required when RPV is administered together with drugs known to be associated with torsades de pointes (1, 2, 10), as well as in the case of coadministration of potent CYP3A inhibitors interfering with RPV elimination. These are only limited pharmacokinetic data apart from those obtained within the framework of rigorous clinical trials (3, 4, 10, 11). As a consequence, the inter- and intrasubject variabilities in drug exposure, response, and toxicity are poorly characterized in the general population of patients receiving the treatment. In addition, the potential impact of the pharmacogenetic background has not yet been studied. The aim of this study was thus to build a population pharmacokinetic model with data from a cohort of unselected patients in order to describe the pharmacokinetics of RPV, to quantify the variability in drug levels, and to identify the potential contribution of demographic factors as well as functional variants of relevant genes that may be involved in the disposition of RPV. Finally, simulations were performed in order to compare the plasma concentrations at the time of the trough concentration after the administration of 50 mg QD with those after the administration of 25 mg twice daily (BID).
RESULTS
Study data.
A total of 325 plasma RPV concentration measurements were obtained from 249 HIV-infected individuals and included in the population pharmacokinetic analysis. The trivial number of samples per patient was between 1 and 4. The samples were drawn between 1.25 and 36 h after the last drug intake under steady-state conditions. The plasma concentrations were measured to be between 12 and 255 ng/ml.
Information regarding the HIV infection treatment background indicated that 87% of the patients had previously received antiretroviral treatments and only 13% were newly diagnosed with HIV infection and antiretroviral therapy naive. The demographic and genetic characteristics of the study population are presented in Table 1.
TABLE 1.
Demographic and genetic characteristics of study populationa
| Characteristic | Values |
|---|---|
| No. (%) of male subjects | 181 (72.7) |
| Median (range): | |
| Age (yr) | 46 (22–80) |
| Body wt (kg) | 74 (42–112) |
| Ht (cm) | 175 (150–198) |
| No. (%) of patients of the following ethnicity: | |
| Caucasian | 128 (51.4) |
| African | 29 (11.6) |
| Asian | 1 (0.4) |
| Other | 3 (1.2) |
| Unknown | 88 (35.3) |
| No. (%) of patients with the following ART status: | |
| Naive | 21 (8.4) |
| Experienced | 127 (51.0) |
| Unknown | 101 (40.6) |
| Median (range) CD4+ cell count (no. of cells/mm3) | 597 (6–30,000) |
| Median (range) HIV RNA Level (log10 no. of copies/ml) | 0 (0–17,200) |
| No. (%) of patients with the following genotype: | |
| rs35599367 C → T (CYP3A4*22) | |
| Ref-Hom | 111 (44.6) |
| Het-LOF | 8 (3.2) |
| Unknown | 130 (52.2) |
| rs776746 T → C (CYP3A5*3) | |
| Ref-Hom | 11 (4.4) |
| Het-LOF | 23 (9.2) |
| Hom-LOF | 85 (34.1) |
| Unknown | 130 (52.2) |
| rs4244285 G → A (CYP2C19*2) | |
| Ref-Hom | 83 (33.3) |
| Het-LOF | 36 (14.5) |
| Unknown | 130 (52.2) |
| rs12248560 C → T (CYP2C19*17) | |
| Ref-Hom | 67 (26.9) |
| Het-LOF | 44 (17.7) |
| Hom-GOF | 8 (3.2) |
| Unknown | 130 (52.2) |
| rs8175347 (UGT1A1*28) | |
| Ref-Hom | 36 (14.5) |
| Het-LOF | 68 (27.3) |
| Hom-LOF | 15 (6.0) |
| Unknown | 130 (52.2) |
| rs6755571 C → A (UGT1A4*2) | |
| Ref-Hom | 111 (44.6) |
| Het-LOF | 8 (3.2) |
| Unknown | 130 (52.2) |
Abbreviations: ART, antiretroviral treatment; Ref-Hom, homozygous for the reference allele; Het-LOF, heterozygous for a loss of function; Hom-LOF, homozygous for a loss of function.
Genotyping.
Valid results were obtained for all genotyped samples from the 119 participants. The minor allelic frequencies (MAFs) found in our Caucasian study population (n = 99) were 0.04, 0.90, 0.16, 0.24, 0.39, and 0.04 for CYP3A4*22 rs35599367, CYP3A5*3 rs776746, CYP2C19*2 rs4244285, CYP2C19*17 rs1224856, UGT1A1*28 rs8175347, and UGT1A4*2 rs6755571, respectively. All variants studied respected the Hardy-Weinberg equilibrium (P > 0.05). Resequencing results for the six individuals with extreme RPV levels (very low or very high plasma concentrations) are displayed in Table S1c in the supplemental material. Subject 1 presented a double loss of function (LOF) in CYP2C19 (CYP2C19*2/*4) and was heterozygous for a loss of function (Het-LOF) for the allele CYP3A4*22. Since CYP2C19*4 is a very rare allele in Caucasians (MAF, 0.004), we confirmed this finding with another PCR (12). Subject 2 was Het-LOF for the allele CYP2C192 and UGT1A1*28. Subject 3 and subject 4 were heterozygous for a gain of function (Het-GOF) for CYP2C19*17, with the latter subject also being Het-LOF for CYP3A5*3. Subject 5 did not present any variants of CYP2C19 but was Het-LOF for UGT1A1*28 and CYP3A4*22. Subject 6 was homozygous for a gain of function (Hom-GOF) for CYP2C19*17, Het-LOF for CYP3A5*3, and Het-LOF for UGT1A1*28.
Population pharmacokinetic analysis.
A one-compartment model with zero-order absorption from the gastrointestinal tract best fit the data. The use of first-order absorption or sequential zero- and first-order absorptions did not improve the description of the data (change in the objective function value [ΔOFV] = 0.0). No further improvement was observed upon assignment of the lag time (ΔOFV = −0.0). In addition to clearance (CL), an interindividual variability on the volume of distribution (V) or on the mean absorption time (D1) did not improve the description of the data (ΔOFV = −0.0). A combined proportional and additive error model adequately described the residual error.
Among the covariates tested, none appeared to significantly influence RPV CL. A 13% decrease (95% confidence interval [CI], 3 to 25%) in CL in females compared to that in males was observed, but the difference did not reach statistical significance (ΔOFV = −5.3). Application of none of the genetic polymorphisms tested showed any improvement in the model fit (ΔOFV < −3.05). The final population parameter values were a CL of 11.7 liters · h−1 (coefficient of variation [CV], 33%), a V of 401 liters, and a D1 of 4 h. Additive and proportional residual errors were 9.8 ng/ml and 21.6%, respectively. The derived elimination half-life was 24 h, and the mean absorption time was 2 h.
The concentration-versus-time plot with the population prediction and 95% prediction interval is illustrated in Fig. 1.
FIG 1.
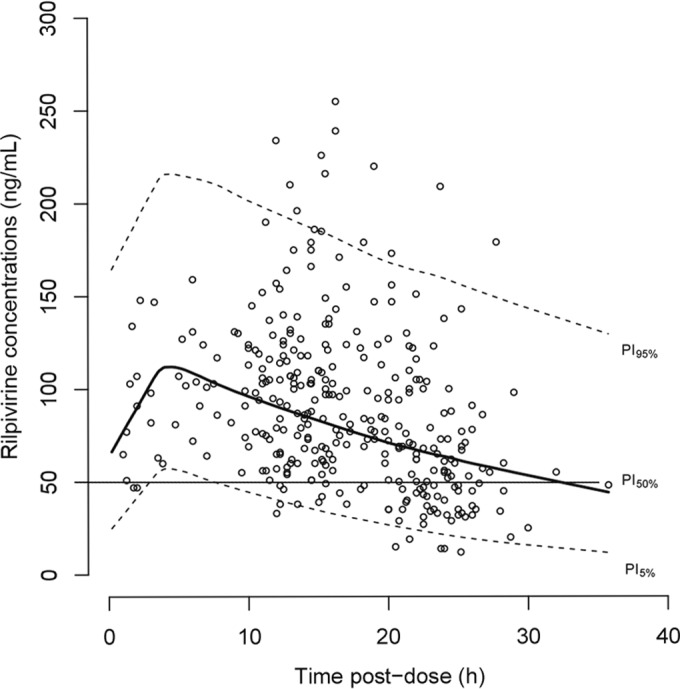
Observed plasma rilpivirine concentrations (open circles) with the median population predicted concentration (continuous heavy black line) and the 95% prediction interval (dashed lines). Continuous black line, the proposed target at 50 ng/ml.
Model evaluation and assessment.
The median parameter estimates obtained from bootstrap analysis and their 95% CIs are presented in Table 2. The model was acceptable, since the parameter estimates of the final population pharmacokinetic model lay within the 95% CI of the bootstrap analysis and differed by less than 11% from the median bootstrap analysis estimates. The external validation analysis showed a nonsignificant bias of 4% (95% CI, −2 to 12%) with an imprecision of 22% for the population prediction. A similar bias for individual predictions of −2% (95% CI, −5 to 8%) with an imprecision of 12% was calculated.
TABLE 2.
Pharmacokinetic parameters obtained in the final model with bootstrap analysis resultsa
| RPV parameter | Final population PK parameters |
Bootstrap resultb |
||
|---|---|---|---|---|
| Estimate (shrinkage [%]) | RSE (%) | Median | 95% CI | |
| CL/F (liters/h) | 11.7 | 2.8 | 11.7 | 10.9–12.5 |
| V/F (liters) | 401 | 14.1 | 393 | 291–525 |
| D1 (h) | 4 | 24.7 | 4 | 2.1–9.5 |
| IIV CL/F (%) | 33 (15) | 6. | 35 | 28–36 |
| Additive residual error (ng/ml) | 9.8 (30) | 13.6 | 9.6 | 7.3–13 |
| Proportional residual error (%) | 21.6 (30) | 7 | 21.2 | 17.6–24.2 |
Abbreviations: CL/F, mean apparent clearance; V/F, mean apparent volume of distribution; D1, mean absorption time; F, bioavailability; IIV, estimate of interindividual variability; RSE, relative standard error of the estimates defined as the standard error/estimate, with the standard error being directly retrieved from NONMEM.
Bootstrap results are for 2,000 samples.
Simulation.
The average predicted minimal concentration (Cmin) at 24 h under a standard 25-mg daily dosage regimen at steady state was 69 ng/ml (95% prediction interval [PI], 33.5 to 125.6 ng/ml). Cmin increased to a median of 140 ng/ml (95% PI, 39 to 446 ng/ml) and 167 ng/ml (95% PI, 59 to 406 ng/ml) after administration of RPV at 50 mg QD and 25 mg BID, respectively.
Accounting for the variability in RPV clearance, 29% of the patients receiving the standard regimen (25 mg QD) would be expected to present concentrations below the suggested target of 50 ng/ml, which is associated with a decrease in the rate of virologic success (2, 9). Increasing the dosage to 50 mg QD would decrease the proportion to 2.4%, and no patients receiving 25 mg BID would be expected to present a concentration below this cutoff. On the other hand, all concentrations were predicted to remain above 12 ng/ml, the target value derived from protein-adjusted in vitro data (2). In addition, no alternative dosage regimens would result in maximum concentrations above 500 ng/ml. Plots of the average concentrations with 95% PIs are presented in Fig. 2 for the 25-mg QD, 50-mg QD, and 25-mg BID regimens.
FIG 2.

Model-based simulations of rilpivirine at 25 mg QD (A), 50 mg QD (B), and 25 mg BID (C) in HIV-positive individuals. Solid lines, population median prediction (PI50%) from the model; shaded areas, 95% prediction intervals; dashed line, proposed target value of 50 ng/ml; dotted line, high concentration that could lead to a significant QTc prolongation.
DISCUSSION
The present study characterizes the population pharmacokinetics of RPV in a cohort of unselected HIV-1-infected adult patients. The estimates of CL, mean absorption time, and steady-state plasma concentrations were close to previously published results (1, 2). Our estimated half-life of 24 h was shorter than the 45 h reported by the manufacturer (1, 2). This difference most probably relates to differences in the study design, since most of our data were collected over a 28-h period, preventing estimation of the terminal redistribution of the drug from peripheral tissues. Accordingly, the estimated volume of distribution of 401 liters is lower than the previously reported one of 1,064 liters derived from a two-compartment model (1), which can explain the difference in the RPV elimination half-life. The clinical relevance of this difference is, however, uncertain.
The interpatient variability of rilpivirine pharmacokinetics was modest and in good accordance with values from phase III clinical trials (1, 13). Additionally, the absence of an impact of demographic, environmental, and clinical factors supports previous results (1, 13). A small and nonsignificant influence of gender was observed, which is similar to the findings in other reports and which appears to be clinically unimportant (1, 13, 14). Adherence and food intake are the two factors known to markedly influence RPV exposure (7); however, information on these two factors was not collected, so the influence of these two factors could not be investigated, which represents a limitation of the study.
None of the selected genetic variations in CYP3A4, CYP3A5, CYP2C19, UGT1A1, and UGT1A4 showed a significant impact on RPV CL, probably because the multiple enzymatic pathways involved in RPV metabolism might compensate for the loss of function of the gene for a particular enzyme. Another possible explanation is that most individuals carried the variants studied in the heterozygous form, so at least one functional allele continued to be functional, thus limiting the decrease in RPV clearance. At the same time, one of the six genotyped individuals (subject 1; see Table S1c in the supplemental material) with high RPV plasma levels at two different occasions (220 ng/ml 19 h after the last drug intake and 174 ng/ml as a trough concentration) carried two LOF CYP2C19 variants, providing a feasible, although not definite, explanation for the decreased RPV metabolism. Conversely, both subjects 3 and 4 were heterozygous carriers of the gain-of-function variant CYP2C19*17 homozygote, but they presented opposite extreme values for plasma rilpivirine concentrations. Subject 6, who was a CYP2C19*17 homozygote, also presented very high rilpivirine levels on one occasion. All other subjects were heterozygous carriers of an LOF allele in at least one of the genes studied. These results are insufficient to establish a correlation with extremely low and high rilpivirine levels. Moreover, the impact of CYP1A2 variability on rilpivirine CL was not evaluated in our study.
The CYP1A2 gene is reported to be involved in RPV metabolism (8). However, the CYP1A2 gene is highly inducible by many environmental factors, and functional polymorphisms have mostly been described in Asian populations (15). The two CYP1A2 variants that fulfilled our criterion of an MAF of greater than 0.05 in Caucasians (CYP1A2*1C and CYP1A2*1F) were not included in the study because of the limited evidence that they have a significant influence on RPV pharmacokinetics (15–17).
The average minimal concentration predicted by our model was 69 ng/ml, a value comparable to the 74 ng/ml observed at week 48 in the clinical studies ECHO and THRIVE (10). However, considering intersubject variability, our finding predicts that 29% of patients would present concentrations below the suggested target of 50 ng/ml recently associated with virological success (1) These results seem to be in accordance with those of a previous controlled study conducted with healthy volunteers, where 11% and 33% of the participants had concentrations below 50 ng/ml at 24 and 36 h, respectively (18).
Subtherapeutic drug exposure in a significant proportion of patients receiving standard RPV dosage regimens may explain the considerable difference in the rates of virological failure between patients receiving RPV and patients receiving efavirenz in a comparative clinical trial (11). The use of RPV at 50 mg QD or 25 mg BID would allow the concentrations to be increased above this cutoff value with a minimal risk of QTc prolongation. Increasing the dosage in some critical cases under QTc monitoring could be envisaged to avoid virological failure, yet it is not known whether drug underexposure has an impact on the development of resistance during long-term maintenance therapy.
This study suffers from some limitations that should be acknowledged. First, only one sample for RPV concentration determination was available for most patients, thus limiting our capacity to well differentiate intersubject variability from intrasubject variability. Regarding genetic testing, the size of the study population was powered to detect only a marked genetic influence, and the absence of genetic findings in this study is inconclusive. Only a few comedications were recorded to have been taken by our study population, and no moderate to potent CYP3A4 or CYP2C19 inhibitors/inductors were coadministered. Because this was an observational study, we cannot rule out the possibility that other confounding factors might influence RPV exposure. Comedications potentially affecting gastric pH and CYP3A4 or CYP2C19 inhibitors/inductors interacting with RPV were not reported to have been taken by our study population, and adherence to the treatment was not evaluated.
In conclusion, this study showed that RPV exhibits a moderate interindividual variability, but the standard 25-mg daily dosage regimen may lead to concentrations below the 50-ng/ml proposed therapeutic target in a significant proportion of patients, producing a risk of suboptimal antiretroviral efficacy. While the current evidence is insufficient to recommend routine therapeutic drug monitoring (TDM) for RPV, monitoring of plasma concentrations under specific conditions, such as treatment failure or a suspected drug-drug interaction, could be a useful adjunct to identify patients at risk of low RPV concentrations and to individualize their dosing regimen.
MATERIALS AND METHODS
Population and study design.
RPV concentration data were collected from 249 adult HIV-infected patients as part of routine TDM performed in our several Swiss centers between April 2013 and January 2015. At the usual follow-up visit, a blood sample (4.9 ml) was collected from patients receiving rilpivirine-based therapy. Time and date information for the last dose intake and blood sampling were recorded. All plasma samples were taken at steady state (at least 4 weeks after RPV regimen initiation).
In all participants, RPV was administered once daily at a fixed dose (25 mg) in combination with FTC and TDF in a single-tablet regimen. Of these patients, 119 were included within the framework of the Swiss HIV Cohort Study (www.shcs.ch) and gave written informed consent for genetic testing. The project was approved by the ethics committees of all participating centers (Centre hospitalier universitaire vaudois [Lausanne], Universitätsspital Zürich [Zurich], Universitätsspital Basel [Basel], Hôpitaux Universitaires de Genève [Geneva], Kantonsspital St. Gallen [Saint Gall], and Universitätsklinik für Infektiologie [Bern]).
Sampling and bioanalysis.
Plasma samples obtained from HIV-infected individuals were isolated by centrifugation and stored at −20°C until batch analysis in our laboratory (Laboratory of Clinical Pharmacology). On the day of the analysis, the viruses in the samples were inactivated by heating at 60°C for 60 min. Plasma RPV levels were determined by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) after protein precipitation with acetonitrile according to our previously reported analytical method (19). The calibration curves are linear and have a lower limit of quantification of 5 ng/ml. The method is precise (interday CV, 3 to 6.3%) and accurate (interday CV, 3.8 to 7.2%). Our laboratory participates in an international external quality assurance program for antiretroviral drug analysis (Stichting Kwaliteitsbewaking Klinische Geneesmiddelanalyse en Toxicologie [KKGT], Association for Quality Assessment in TDM and Clinical Toxicology, The Hague, The Netherlands [http://www.kkgt.nl/]).
Genotyping.
We tested 5 genes (CYP3A4, CYP2C19, CYP3A5, UGT1A1, and UGT1A4) involved in RPV metabolism based on the information provided by the manufacturer (1, 2) and the existing literature (8). We selected 6 single nucleotide polymorphisms (SNPs) with a proven functional effect and a minor allelic frequency (MAF) of greater than 0.05 in Caucasians: rs35599367, a marker of loss-of-function (LOF) allele CYP3A4*22; rs776746, a marker of LOF allele CYP3A5*3; rs4244285, a marker of LOF allele CYP2C19*2; rs12248560, a marker of gain-of-function (GOF) allele CYP2C19*17; rs8175347, a marker of LOF allele UGT1A1*28; and rs6755571, a marker of LOF allele UGT1A4*2) (http://www.cypalleles.ki.se/) (20–27).
Genotyping was performed by TaqMan allelic discrimination (Applied Biosystems, Foster City, CA, USA) for all alleles with the exception of rs8175347, which was genotyped by fragment size-based analysis as previously described (28). In the case of six individuals with extreme RPV levels (one subject with <50 ng/ml and five subjects with >170 ng/ml), we resequenced CYP2C19, covering 1,504 bp of the promoter region; 2,970 bp of the coding region, including exon-intron boundaries; and 1,434 bp of the 3′ untranslated region (3′ UTR). The primers, conditions, and TaqMan assays used are described in Table S1 in the supplemental material.
Population pharmacokinetic model. (i) Basic model.
The population PK analysis was performed using a nonlinear mixed effect modeling approach with NONMEM (version 7.1.0; ICON Development Solutions, Ellicott City, MD, USA). A stepwise procedure was used to identify the model that best fit the RPV data. One- and two-compartment models with first- and zero-order absorption from the gastrointestinal tract were tested. The best structural model appeared to be a one-compartment model with zero-order absorption. The estimated parameters were clearance (CL), volume of distribution (V), and duration of absorption (D1). Since RPV was only administered orally, CL and V represent apparent values (CL/F and V/F, respectively, where F is the absolute oral bioavailability).
(ii) Statistical model.
Exponential errors following a log-normal distribution were assumed for the description of the interindividual variability in the pharmacokinetic parameters, as shown by the equation θj = θ × eηj, where θj is the individual pharmacokinetic parameter of the jth individual, θ is the geometric average population value, and ηj is the subject-specific random effect value, which is an independent, normally distributed random variable with a mean of zero and a variance of ω2. A combined proportional and additive error model was used to describe the intraindividual (residual) variability.
(iii) Covariate model.
Relevant covariates investigated were sex, body weight, height, age, race, aspartate aminotransferase (AST) concentration, alanine aminotransferase (ALT) concentration, chronic hepatitis C virus (HCV) infection, hepatitis B virus (HBV) coinfection, and concomitant medication. The relationships between individual pharmacokinetic estimates and demographic and clinical covariates were first explored graphically. All potential and physiologically plausible relationships were tested in the models using linear or nonlinear functions as appropriate (categorical covariates were coded as 0 and 1, continuous covariates were centered on their median value, and a cutoff of 1.5 times the upper limit of normal was used to recode liver function tests into dichotomous variables). Then, the covariate selection was performed using a stepwise insertion/deletion approach. Missing values were imputed as the population median value.
Minimal concentrations (24 h after the last drug intake) were extrapolated for each patient on the basis of the individual parameters derived from the final model.
For each SNP analysis, individuals were classified into 3 genetic groups: homozygous for the reference allele (Ref), heterozygous for the variant allele (Het), and homozygous for the variant allele (Hom). The influence of selected genetic variants on RPV CL was first explored graphically. Only 3 SNPs (CYP3A4*22 rs35599367, UGT1A1*28 rs8175347, and CYP2C19*17 rs12248560) were retained and tested as potential covariates in the model. Individuals with missing genotype results were treated as a fourth group. Rich and reduced models were tested to estimate the impact of the genetic variants on individual rilpivirine pharmacokinetic parameters. Rich models allowed each genetic group (Ref, Het, Hom, and missing genotype) to have different values for the parameter. In the reduced models, genetic groups with similar parameter estimates were grouped in a unique variable.
Parameter estimation and selection.
All models were fit using the first-order conditional estimation method with interaction (FOCEI). The log-likelihood ratio test, based on the change of the objective function value (ΔOFV) provided by NONMEM, was used to discriminate between nested models. A decrease in the OFV of greater than 3.84 (P < 0.05) for one additional parameter in the structural model building was considered statistically significant, assuming that the ΔOFV between any two covariates in a nested model approximated a χ2 distribution. To account for multiple testing in the covariate model (Bonferroni correction), a change in the OFV of greater than 7.88 (P < 0.005) for one additional covariate was considered statistically significant during the backward deletion steps. Goodness-of-fit plots, pharmacokinetic parameter precision, and the interindividual variability decrease were further criteria considered for model selection.
Model evaluation and assessment.
The stability of the final model was assessed by the bootstrap method using the PsN-Toolkit (29) (version 3.5.3; Uppsala, Sweden). Median parameter values and their 95% confidence intervals (CIs) estimated from 2,000 resampled data sets were compared with the original model estimates.
In addition, a visual predictive check (VPC) was performed by simulating 1,000 individuals on the basis of the final pharmacokinetic estimates to calculate the 95% prediction interval (PI) for concentrations. Observed rilpivirine concentrations were visually compared with the 2.5th, 50th, and 97.5th percentiles of the simulated results. Figures were generated with R (version 2.15.1; R Development Core Team, Foundation for Statistical Computing, Vienna, Austria [http://www.r-project.org/]).
Finally, an external validation was performed using data for 33 samples collected from a separate group of 31 HIV-infected individuals treated with rilpivirine between February and October 2015, for whom complete dosing records and demographic characteristics were retrieved. Rilpivirine concentrations were predicted on the basis of the final models using the NONMEM option with a MAXEVAL value of 0. The predictive performance of the model was assessed in terms of bias (mean prediction error [MPE]) and precision (root mean square prediction error [RMSE]) and the associated 95% CI, as follows (30):
where Cpred is the population or individual predicted concentration, Cobs is the observed concentration for the validation group, and N corresponds to the number of observations.
Simulations.
Simulations were performed on the basis of the average values of the pharmacokinetic parameters and the variability obtained from the final model for 1,000 individuals, using rilpivirine at 50 mg QD or 25 mg BID. The mean and 95% prediction interval of the concentrations at the trough (i.e., at 12 and 24 h) were derived for both regimens. Simulations were performed in NONMEM.
Supplementary Material
ACKNOWLEDGMENTS
This work received support from the Swiss National Science Foundation (grant no. SNF 324730_141234 and 324730-165956 to L.A.D. and 20NA21-145955/Nano-IsyPeM2). We thank Janssen-Cilag AG (Switzerland) and Gilead Science Switzerland, which provided research grants. C. Barcelo is supported by an aFundaward from the Fundación Alfonso Martínez Escudero, Spain (grant no. MAD-1-2-105).
Janssen-Cilag AG (Switzerland) and Gilead Science Switzerland were not involved in the design or the conduct of analytical development, analysis of the results, or the writing of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00899-16.
REFERENCES
- 1.U.S. Food and Drug Administration. 2011. Edurant. NDA 202-022/N-000 for TMC278 (rilpivirine) IR tablet, 25 mg. U.S. Food and Drug Administration, Rockville, MD. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202022Orig1s000ClinPharmR.pdf.
- 2.European Medicines Agency. 2013. Eviplera. Assessment report (procedure no. EMEA/H/C/002312/II/0021). European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/002312/WC500158840.pdf. [Google Scholar]
- 3.Cohen CJ, Andrade-Villanueva J, Clotet B, Fourie J, Johnson MA, Ruxrungtham K, Wu H, Zorrilla C, Crauwels H, Rimsky LT, Vanveggel S, Boven K, THRIVE Study Group. 2011. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet 378:1–10. doi: 10.1016/S0140-6736(11)60983-5. [DOI] [PubMed] [Google Scholar]
- 4.Cohen CJ, Molina JM, Cassetti I, Chetchotisakd P, Lazzarin A, Orkin C, Rhame F, Stellbrink HJ, Li T, Crauwels H, Rimsky L, Vanveggel S, Williams P, Boven K, ECHO, THRIVE Study Groups. 2013. Week 96 efficacy and safety of rilpivirine in treatment-naive, HIV-1 patients in two phase III randomized trials. AIDS 27:939–950. doi: 10.1097/QAD.0b013e32835cee6e. [DOI] [PubMed] [Google Scholar]
- 5.Palella FJ Jr, Fisher M, Tebas P, Gazzard B, Ruane P, Van Lunzen J, Shamblaw D, Flamm J, Ebrahimi R, Porter D, White K, Hindman J, Elbert E, De-Oertel S, Fralich T. 2014. Simplification to rilpivirine/emtricitabine/tenofovir disoproxil fumarate from ritonavir-boosted protease inhibitor antiretroviral therapy in a randomized trial of HIV-1 RNA-suppressed participants. AIDS 28:335–344. doi: 10.1097/QAD.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 6.Sharma M, Saravolatz LD. 2013. Rilpivirine: a new non-nucleoside reverse transcriptase inhibitor. J Antimicrob Chemother 68:250–256. doi: 10.1093/jac/dks404. [DOI] [PubMed] [Google Scholar]
- 7.Crauwels HM, van Heeswijk RP, Buelens A, Stevens M, Boven K, Hoetelmans RM. 2013. Impact of food and different meal types on the pharmacokinetics of rilpivirine. J Clin Pharmacol 53:834–840. doi: 10.1002/jcph.107. [DOI] [PubMed] [Google Scholar]
- 8.Lade JM, Avery LB, Bumpus NN. 2013. Human biotransformation of the nonnucleoside reverse transcriptase inhibitor rilpivirine and a cross-species metabolism comparison. Antimicrob Agents Chemother 57:5067–5079. doi: 10.1128/AAC.01401-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yapa HJA, Moyle G, Else L, Khoo S, Back D, Karolia Z, Higgs C, Boffito M. 2013. Pharmacokinetics (PK) of tenofovir (TFV), emtricitabine (FTC), and rilpivirine (RPV) over 10 days following drug cessation, abstr PE10/6. Abstr Eur AIDS Conf, Brussels, Belgium. [Google Scholar]
- 10.Pozniak AL, Morales-Ramirez J, Katabira E, Steyn D, Lupo SH, Santoscoy M, Grinsztejn B, Ruxrungtham K, Rimsky LT, Vanveggel S, Boven K, TMC278-C204 Study Group. 2010. Efficacy and safety of TMC278 in antiretroviral-naive HIV-1 patients: week 96 results of a phase IIb randomized trial. AIDS 24:55–65. doi: 10.1097/QAD.0b013e32833032ed. [DOI] [PubMed] [Google Scholar]
- 11.Molina JM, Clumeck N, Redant K, Rimsky L, Vanveggel S, Stevens M, ECHO Study Group, THRIVE Study Group. 2013. Rilpivirine vs. efavirenz in HIV-1 patients with baseline viral load 100,000 copies/ml or less: week 48 phase III analysis. AIDS 27:889–897. doi: 10.1097/QAD.0b013e32835e1554. [DOI] [PubMed] [Google Scholar]
- 12.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crauwels H, van Schaick E, van Heeswijk RPG, Vanveggel S, Boven K, Vis P.. 2010. Effect of intrinsic and extrinsic factors on the pharmacokinetics of TMC278 in antiretroviral-naive, HIV-1-infected patients in ECHO and THRIVE. J Int AIDS Soc 13(Suppl 4):P186. [Google Scholar]
- 14.Hodder S, Arasteh K, De Wet J, Gathe J, Gold J, Kumar P, Mohapi L, Short W, Crauwels H, Vanveggel S, Boven K. 2012. Effect of gender and race on the week 48 findings in treatment-naive, HIV-1-infected patients enrolled in the randomized, phase III trials ECHO and THRIVE. HIV Med 13:406–415. doi: 10.1111/j.1468-1293.2012.00991.x. [DOI] [PubMed] [Google Scholar]
- 15.Gunes A, Dahl ML. 2008. Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms. Pharmacogenomics 9:625–637. doi: 10.2217/14622416.9.5.625. [DOI] [PubMed] [Google Scholar]
- 16.Song L, Du Q, Jiang X, Wang L. 2014. Effect of CYP1A2 polymorphism on the pharmacokinetics of agomelatine in Chinese healthy male volunteers. J Clin Pharm Ther 39:204–209. doi: 10.1111/jcpt.12118. [DOI] [PubMed] [Google Scholar]
- 17.Olsson E, Edman G, Bertilsson L, Hukic DS, Lavebratt C, Eriksson SV, Osby U. 2015. Genetic and clinical factors affecting plasma clozapine concentration. Prim Care Companion CNS Disord 17:14m01704. doi: 10.4088/PCC.14m01704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickinson L, Yapa HM, Jackson A, Moyle G, Else L, Amara A, Khoo S, Back D, Karolia Z, Higgs C, Boffito M. 2015. Plasma tenofovir, emtricitabine, and rilpivirine and intracellular tenofovir diphosphate and emtricitabine triphosphate pharmacokinetics following drug intake cessation. Antimicrob Agents Chemother 59:6080–6086. doi: 10.1128/AAC.01441-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aouri M, Calmy A, Hirschel B, Telenti A, Buclin T, Cavassini M, Rauch A, Decosterd LA. 2013. A validated assay by liquid chromatography-tandem mass spectrometry for the simultaneous quantification of elvitegravir and rilpivirine in HIV positive patients. J Mass Spectrom 48:616–625. doi: 10.1002/jms.3200. [DOI] [PubMed] [Google Scholar]
- 20.de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. 1994. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 269:15419–15422. [PubMed] [Google Scholar]
- 21.Ferguson RJ, De Morais SM, Benhamou S, Bouchardy C, Blaisdell J, Ibeanu G, Wilkinson GR, Sarich TC, Wright JM, Dayer P, Goldstein JA. 1998. A new genetic defect in human CYP2C19: mutation of the initiation codon is responsible for poor metabolism of S-mephenytoin. J Pharmacol Exp Ther 284:356–361. [PubMed] [Google Scholar]
- 22.Monaghan G, Ryan M, Seddon R, Hume R, Burchell B. 1996. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet 347:578–581. doi: 10.1016/S0140-6736(96)91273-8. [DOI] [PubMed] [Google Scholar]
- 23.Ehmer U, Vogel A, Schutte JK, Krone B, Manns MP, Strassburg CP. 2004. Variation of hepatic glucuronidation: novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology 39:970–977. doi: 10.1002/hep.20131. [DOI] [PubMed] [Google Scholar]
- 24.Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M. 2006. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113. doi: 10.1016/j.clpt.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. 2011. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 11:274–286. doi: 10.1038/tpj.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiener D, Fang JL, Dossett N, Lazarus P. 2004. Correlation between UDP-glucuronosyltransferase genotypes and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone glucuronidation phenotype in human liver microsomes. Cancer Res 64:1190–1196. doi: 10.1158/0008-5472.CAN-03-3219. [DOI] [PubMed] [Google Scholar]
- 27.Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E. 2001. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet 27:383–391. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 28.Baudhuin LM, Highsmith WE, Skierka J, Holtegaard L, Moore BE, O'Kane DJ. 2007. Comparison of three methods for genotyping the UGT1A1 (TA)n repeat polymorphism. Clin Biochem 40:710–717. doi: 10.1016/j.clinbiochem.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Lindbom L, Pihlgren P, Jonsson EN. 2005. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Sheiner LB, Beal SL. 1981. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 9:503–512. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.