

ABSTRACT
Caged Garcinia xanthones (CGXs) constitute a family of natural products that are produced by tropical/subtropical trees of the genus Garcinia. CGXs have a unique chemical architecture, defined by the presence of a caged scaffold at the C ring of a xanthone moiety, and exhibit a broad range of biological activities. Here we show that synthetic CGXs exhibit antimalarial activity against Plasmodium falciparum, the causative parasite of human malaria, at the intraerythrocytic stages. Their activity can be substantially improved by attaching a triphenylphosphonium group at the A ring of the caged xanthone. Specifically, CR135 and CR142 were found to be highly effective antimalarial inhibitors, with 50% effective concentrations as low as ∼10 nM. CGXs affect malaria parasites at multiple intraerythrocytic stages, with mature stages (trophozoites and schizonts) being more vulnerable than immature rings. Within hours of CGX treatment, malaria parasites display distinct morphological changes, significant reduction of parasitemia (the percentage of infected red blood cells), and aberrant mitochondrial fragmentation. CGXs do not, however, target the mitochondrial electron transport chain, the target of the drug atovaquone and several preclinical candidates. CGXs are cytotoxic to human HEK293 cells at the low micromolar level, which results in a therapeutic window of around 150-fold for the lead compounds. In summary, we show that CGXs are potent antimalarial compounds with structures distinct from those of previously reported antimalarial inhibitors. Our results highlight the potential to further develop Garcinia natural product derivatives as novel antimalarial agents.
KEYWORDS: caged Garcinia xanthones, Giemsa stain-negative body, Plasmodium falciparum, malaria, mitochondria
Malaria remains a leading infectious disease in the tropical and subtropical regions of the world. In the past 15 years, the rates of morbidity and mortality due to malaria have decreased dramatically because of the wide use of artemisinin-based combination chemotherapy, indoor residual spraying, the distribution of insecticide-treated bed nets, and other malaria prevention and research efforts (1–3). However, malaria parasites have evolved mechanisms to adapt to various immunological and chemical pressures. They display genomic plasticity, readily accumulating mutations and rearrangements to overcome antimalarial drugs (4). Clinical isolates that are resistant to a majority of the antimalarial drugs available have spread widely in areas where malaria is endemic (5, 6). The facile development and spread of parasite drug resistance clearly threaten the achievements attained so far and impede efforts to eradicate malaria in the near future. The recent appearance and spread of artemisinin tolerance underscores the need for continued urgent efforts to develop new antimalarial reagents (7–9).
Plants of the genus Garcinia produce an intriguing family of caged-xanthone-derived natural products that have a documented value in traditional Eastern medicine (10). Collectively referred to as caged Garcinia xanthones (CGXs), these compounds are structurally defined by an unusual motif in which the C ring of an allylated xanthone has been converted into a tricyclic cage (Fig. 1). This motif is further decorated via A-ring substitutions and peripheral oxidations to produce a variety of natural products with a broad range of bioactivities (11). Gambogic acid (GBA), the archetype of this family, potently inhibits cancer cell proliferation in solid tumors (13–17) and hematological malignancies (18) and has entered clinical trials in China for patients with non-small-cell lung, colon, and renal cancers (19). In addition, the potent cytotoxicity of several CGXs at low micromolar concentrations has been well documented (20–23). Efforts to unveil the minimum structural motif of CGXs that is accountable for their observed anticancer activity led to the identification of cluvenone (CLV) (24, 25). This compound was found to have potency similar to that of GBA in inhibiting cancer cell growth against the NCI60 cell panel and a promising window of selectivity against nontumor cells (26). Although the detailed mechanism of action of GBA, CLV, and related compounds has not yet been delineated, several studies indicate that they localize to mitochondria and exhibit their bioactivity by affecting mitochondrial structure and function (27, 28). Along these lines, the hydroxylated CLVs MAD28 and MAD44 were recently found to bind to the mitoNEET family of iron-sulfur-containing proteins that are located at the outer mitochondrial membrane (29).
FIG 1.
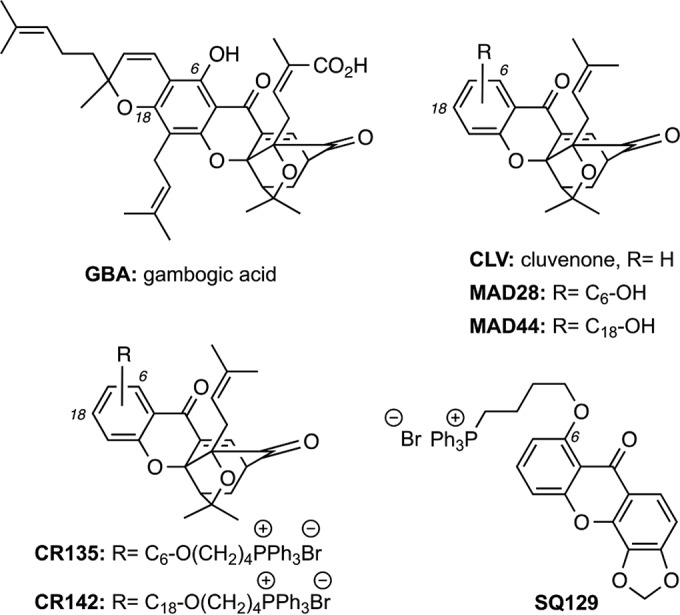
Chemical structures of GBA and related CGXs. CLV defines the structure of the common CGX motif. Hydroxylation at the C6 and C18 centers of CLV produces MAD28 and MAD44, respectively. Attachment of a triphenylphosphonium salt at these hydroxylated sites produces CR135 and CR142, respectively.
The mitochondrion of malaria parasites is an essential organelle that has been validated as an antimalarial drug target (30, 31). Compounds that disrupt essential mitochondrial functions within the parasite are either in clinical use or in clinical trials as potential antimalarial agents (32). For instance, atovaquone (a component of Malarone) is a clinically approved drug that selectively inhibits the parasite mitochondrial electron transport chain (mtETC) at the cytochrome bc1 complex, leading to collapse of the mitochondrial membrane potential (33). Moreover, the dihydroorotate dehydrogenase (DHODH) inhibitor DSM265 is currently undergoing phase II clinical trials (34). Inhibition of DHODH blocks pyrimidine biosynthesis, which is an essential pathway in malaria parasites (35). Since malaria parasites require mitochondrial functions for survival, we speculated that GBA and derivative compounds might exhibit antimalarial activity. In this study, we examined the antimalarial activities of GBA and synthetic CGXs against the human malaria parasite Plasmodium falciparum.
RESULTS
Effect of CGXs on the growth of the human malaria parasite P. falciparum.
The erythrocytic stage of malaria parasites causes all of the clinical symptoms associated with malaria and is the target of most antimalarial drugs. P. falciparum strains isolated in various geographic regions have different sensitivities to antimalarial drugs. Dd2 is a multidrug-resistant clone selected from an Indochina isolate by using mefloquine pressure (36). To test if GBA and related CGXs (Fig. 1) have activities against drug-resistant parasites, we performed growth inhibition assays based on [3H]hypoxanthine incorporation by Dd2 parasites (Fig. 2). Artemisinin was included as a control in this assay, which yielded a 50% effective concentration (EC50) of 12.4 ± 1.5 nM, similar to a previous report (37). As shown in Fig. 2, GBA and CLV exhibited moderate antimalarial activities with EC50s of 0.28 ± 0.03 and 0.75 ± 0.03 μM, respectively. Similar antimalarial activity was observed with MAD28, the C6-hydroxylated CLV, which exhibited an EC50 of 0.26 ± 0.02 μM. However, MAD44, the C18-hydroxylated CLV, was less effective, with an EC50 of 4.1 ± 0.3 μM. CR135 and CR142 were synthesized from MAD28 and MAD44, respectively, by conjugating a triphenylphosphonium group at C6 of MAD28 and C18 of MAD44 (38). Importantly, CR135 and CR142 exhibited remarkable antimalarial activities, with EC50s as low as 7.9 and 11.1 nM, respectively. Thus, conjugating the A ring of the caged xanthone structure with a triphenylphosphonium group drastically improves its antimalarial activity. Specifically, adding this group to the C6-hydroxyl group of MAD28 decreased the EC50 by about 30-fold from 267 nM (MAD28) to 7.9 nM (CR135). The same modification at the C18-hydroxyl group decreased the EC50 about 370-fold from 4,100 nM (MAD44) to 11.1 nM (CR142). To test if a caged xanthone is required for the robust activity of CR135, we replaced the caged xanthone structure of CR135 with a planar xanthone and obtained the compound SQ129. The antimalarial activity of SQ129 (EC50, 106.5 ± 13.1 nM) was much weaker than that of CR135 (Fig. 2), suggesting that a caged xanthone moiety is also needed for optimal antimalarial activity.
FIG 2.
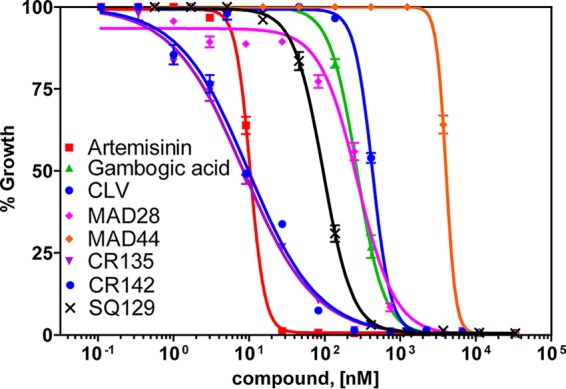
Antimalarial effects of CGX compounds on P. falciparum parasites. The antimalarial efficiency of CGXs in Dd2 parasites was measured by the [3H]hypoxanthine incorporation assay (see Materials and Methods). The x axis indicates the concentrations of a tested compound, and the y axis indicates the percentage of [3H]hypoxanthine incorporation compared to that in no-drug controls. The assays were set up with triplicate wells for each concentration of each compound tested. The averaged data of five independent experiments are shown (n = 5).
We repeated the growth inhibition assays several times with selected CGX compounds in drug-sensitive (3D7) and drug-resistant (Dd2) P. falciparum parasites, and the average EC50s are presented in Table 1. Collectively, these data show that GBA and related CGXs have moderate antimalarial potency but their efficacy increases dramatically upon conjugation of the caged xanthone motif with a triphenylphosphonium group.
TABLE 1.
EC50s of CR135, CR142, MAD28, and SQ129 in malaria parasites and human cells
| Parasite or human cell type | Avg EC50a ± SE of: |
|||
|---|---|---|---|---|
| CR135 | CR142 | MAD28 | SQ129 | |
| Dd2 (nM) | 10.2 ± 2.9 | 15.0 ± 4.6 | 267.5 ± 20.5 | 106.5 ± 13.1 |
| 3D7 (nM) | 12.3 ± 3.5 | 18.1 ± 3.7 | 312.4 ± 23.7 | 99.5 ± 10.2 |
| HEK293 (μM) | 1.45 ± 0.35 | 2.21 ± 0.42 | 1.83 ± 0.14 | 9.6 ± 0.45 |
The data shown are from three to five independent experiments. Note that concentrations are nanomolar for P. falciparum lines Dd2 and 3D7 but micromolar for HEK293 cells.
Effect of CGXs on the intraerythrocytic development cycle of P. falciparum.
Within red blood cells (RBCs), P. falciparum undergoes a 48-h intraerythrocytic development cycle during which the maturation process can be subdivided into ring, trophozoite, and schizont stages (39). Compounds CR135, CR142, and MAD28, which exhibited the best EC50s, were selected for this study, while dimethyl sulfoxide (DMSO) and SQ129 served as controls. We tightly synchronized Dd2 parasites and subsequently treated them at the trophozoite stage with CGX compounds at 10 times the EC50 (see Materials and Methods) and monitored them from 2 to 48 h postaddition. At each time point, thin blood smears were prepared for morphological studies and parasite aliquots were fixed and stained with SYBR green for determination of parasitemia by flow cytometry.
Parasite morphological changes and parasitemia during the treatment time course are presented in Fig. 3. In the DMSO control, parasites progressed normally throughout the 48-h life cycle (Fig. 3A, top). However, trophozoites treated with CR135 or CR142 displayed a characteristic morphological change, the appearance of a large Giemsa stain-resistant area (Fig. 3A, red arrows), giving the appearance of an empty volume that fills much of the parasite, as soon as 2 h after treatment. For future reference, we named this morphologically aberrant structure a Giemsa stain-negative body (GNB). The proportion of these parasites containing a large GNB increased from ∼20% (2 h of drug treatment) to ∼30% after 4 h of drug treatment (Fig. 3B). In samples treated with CR135 or CR142 for 8 h, the proportion of empty-looking parasites decreased to ∼5% because of the progression of the parasite growth cycle, producing a large population of newly invaded erythrocytes containing young ring stage parasites (Fig. 3B). These newly formed ring stage parasites seemed to be morphologically normal (Fig. 3A). During an additional 16 h of exposure to CR135 or CR142 (24 h total), these new rings progressed to late rings without any observable morphological defects (Fig. 3A). However, in the next 24 h (48 h total postaddition, Fig. 3A), growth appeared to become blocked at the early trophozoite stage, with the blocked parasites regaining the characteristic empty-looking appearance.
FIG 3.

Viability of P. falciparum parasites treated with CGX derivatives. Dd2 parasites were tightly synchronized and treated with compounds at 10 times the EC50 starting at the mid-trophozoite stage. Giemsa smears were made for morphological studies; representative images are shown in panel A. The red arrows indicate the specific morphological structures not Giemsa stained. In panel B, the percentages of GNB parasites in control and drug (CR135 and CR142)-treated cultures are quantified. In panel C, the parasitemia, as determined by SYBR green staining and flow cytometry, at each time point is plotted. For each sample, 1,000,000 events were collected and analyzed. The time course was repeated three times, and the error bars in panels B and C indicate the standard error of three biological replicates.
On the other hand, MAD28 seemed to kill parasites by a different mechanism(s). As shown in Fig. 3A, MAD28 did not cause the formation of a large empty area inside parasites during the treatment time course. Nevertheless, at the concentration tested (2.6 μM), MAD28 significantly delayed the progression of parasites. After 4 h of treatment, fewer trophozoites progressed to multinucleated schizonts. After 8 h of MAD28 treatment, there were very few new rings in the sample. SQ129 behaved differently from CR135/CR142 or MAD28 (Fig. 3A). It did not induce a large GNB in the parasites or inhibit parasite growth as dramatically as MAD28. In all, these data suggest that CR135 works similarly to CR142; however, CR135 and CR142 kill malaria parasites through a mechanism(s) distinct from that of MAD28.
Quantitative parasitemia data for the same 48-h treatment time course, determined by flow cytometry, are presented in Fig. 3C. CR135 and CR142 knocked down parasitemia significantly after drug treatment for 24 h. Even though there were some residual “parasites” present after drug treatment for 48 h with CR135 or CR142, these parasites were dead and did not progress (Fig. 3A). After 96 h of treatment with CR135 or CR142, parasitemia dropped to an undetectable level and there were just a few dead remnant parasites or purple dots within the host cells observed after staining (data not shown). MAD28, on the other hand, seemed to be a faster killer than CR135 or CR142. It reduced parasitemia significantly after 8 h of drug treatment (Fig. 3B). The kinetics of these parasite killing data suggest that CR135 and CR142 eliminate parasites more slowly than MAD28 does, and again, it appears that CR135 and CR142 work similarly, while MAD28 kills parasites through a different mechanism(s).
As shown in Fig. 3A, CR135 and CR142 caused the formation of a large, Giemsa stain-resistant area (GNB) inside the parasite. It was noticeable that this large empty area was adjacent to the hemozoin crystal. The intimate localization of this structure to hemozoin prompted us to investigate the integrity of the food vacuole under treatment with CR135 and CR142 by immunofluorescence assay (IFA) with a food vacuole marker. Dd2 parasites were synchronized and treated with CR135 and CR142 (10 times the EC50), respectively, for 8 h, from the early trophozoite stage to the mid-trophozoite stage. Compared to 4 h of treatment starting at the mid-trophozoite stage, we noticed that the percentage of GNB parasites increased significantly (up to 50%) when treatment started at a younger trophozoite stage and lasted longer (8 h) (data not shown). Posttreatment, the parasites were fixed with formaldehyde-glutaraldehyde. An anti-plasmepsin II polyclonal antibody was used to visualize the structure of the food vacuole; plasmepsin II, an aspartic protease for hemoglobin digestion, is a marker for this organelle (40, 41). As shown in Fig. 4, in the control parasite, one relatively large globular structure containing the hemozoin crystal was clearly visible, and a few small spots next to the main body were also present. This staining pattern represents the normal food vacuole architecture (40, 41). Under treatment with CR135 and CR142, however, the intactness of the food vacuole appeared to be lost. As shown in Fig. 4, the main globular staining disappeared; instead, there were many small granular fluorescing particles scattered throughout the cytosol, suggesting that the integrity of the food vacuole was damaged by treatment with the compounds. A correlation between the large vacant space in the GNB parasites (Fig. 3A) and the IFA images (Fig. 4) is, however, not evident. Thus, the nature of this feature remains unclear (see Discussion).
FIG 4.
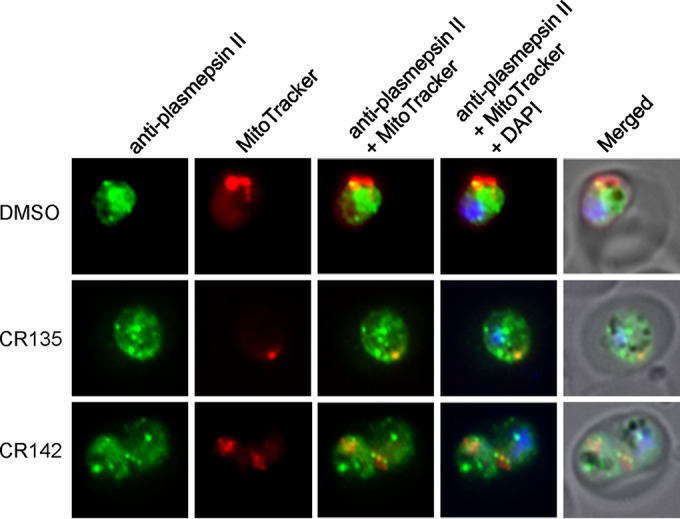
Food vacuole integrity of P. falciparum parasites treated with CR135 and CR142. Representative IFA images show the food vacuole morphology of parasites treated with the vehicle, CR135, and CR142, individually. Anti-plasmepsin II polyclonal antibody (rabbit) was diluted 1:1,000, and an Alexa Fluor 488-conjugated anti-rabbit secondary antibody was diluted 1:350. MitoTracker Red stains active mitochondria.
To further characterize the effects of these CGX compounds on parasites, we also treated P. falciparum with 10 times the EC50 starting with synchronized schizonts and synchronized rings. As shown in Fig. S1A in the supplemental material, when treatment was begun at the schizont stage, these compounds significantly inhibited parasite development. At 10 times the EC50, MAD28 was the most effective compound in this regard. As shown in Fig. S1B in the supplemental material, when a parasite culture at 5% schizont parasitemia was treated with MAD28, the parasitemia only declined to 3% after 8 h of treatment, indicating that ∼60% of the schizonts still remained inside the host cells. In control parasites treated with DMSO in parallel, only ∼10% of the schizonts had not egressed after 8 h of incubation. CR135 and CR142 also inhibited parasite growth but to a lesser extent than MAD28. Because of the strong inhibitory effect of MAD28, the parasitemia after 24 h of treatment dropped to a very low level (see Fig. S1C in the supplemental material). Again, CR135 and CR142 were slow killers and the parasitemia after 24 h of drug treatment remained much higher than that seen after MAD28 treatment (see Fig. S1C in the supplemental material). Collectively, these data suggest that CGX compounds inhibit parasite growth at the schizont stage, possibly by blocking schizont maturation and/or parasite egress. MAD28 was a strong inhibitor at the concentration used (2.6 μM). However, because of its narrow therapeutic window, MAD28 is quite toxic to mammalian cells at 10 times the EC50 (Table 1).
Different phenomena were observed when treatments were initiated at the ring stage. As shown in Fig. S2A in the supplemental material, the compounds did not inhibit the progression of young rings to mature rings significantly, as 8 h of drug treatment did not cause dramatic morphological changes. For all of the compounds tested, the parasitemia seen after 8 h of drug treatment was comparable to that seen after the control DMSO treatment (see Fig. S2B in the supplemental material). The antimalarial effects of these compounds became much more evident after 24 h of drug treatment. CR135 and CR142 arrested the parasites at early trophozoite stages and formed a large GNB inside the parasites; MAD28 blocked the parasites at an earlier stage but did not cause the formation of a large, empty-looking structure. The parasitemias measured after 24 h of treatment with each of the compounds are presented in Fig. S2C in the supplemental material.
Mitochondrial morphology of P. falciparum parasites treated with CGXs.
GBA and CGXs are pleiotropic compounds that kill cancer cells via multiple mechanisms (11). One important target is the mitochondrion, which undergoes apoptosis under drug treatment (27). MAD28 has recently been shown to inhibit an iron-sulfur cluster binding protein, mitoNEET, on the outer membrane of mitochondria in breast cancer cells (29). MitoNEET is involved in cellular iron homeostasis and various mitochondrial functions (42).
To determine the effects of these compounds on parasite mitochondria, we preloaded synchronized parasites at the early trophozoite stage with MitoTracker (see Materials and Methods), treated them with compounds at 10 times the EC50, and observed their mitochondrial morphologies over time. In parasites treated with CR135, CR142, or MAD28 for 4 h, the mitochondria were morphologically indistinguishable from those treated with the DMSO vehicle (data not shown). However, in samples treated with compounds for 8 h, we observed significant morphological changes in the mitochondria (Fig. 5). As shown in Fig. 5A, in the DMSO control-treated parasite, the mitochondrion formed a continuous tubular filament structure, indicating a healthy mitochondrion. However, in parasites treated with CR135, CR142, or MAD28, the mitochondria appeared fragmented and punctate. Each parasite contained one or several MitoTracker-stained dots but no long tubular structures. To further quantify this phenomenon, we examined 200 parasites from each treatment (drug or vehicle) and classified them as having either tubular or punctate mitochondria. As shown in Fig. 5B, after treatment with the DMSO control, >95% of the parasites had tubular structures; however, the percentage of tubular mitochondria decreased to <5% in drug-treated parasites. These data strongly suggest that these CGX compounds have a strong detrimental effect on the parasite mitochondria.
FIG 5.

Mitochondrial morphologies of P. falciparum parasites treated with CGX derivatives. (A) Representative images showing the mitochondrial morphology of parasites treated with the vehicle or compounds for 8 h. (B) Quantitation of the relative fraction of healthy tubular mitochondria in parasites treated with the vehicle or compounds for 8 h. For each treatment, mitochondrial morphology was assessed in 200 parasites. This experiment was repeated three times. Error bars indicate the standard error of three biological replicates.
Effect of CGXs on the mtETC.
During the asexual blood stages, the mtETC of malaria parasites is required to recycle ubiquinol to ubiquinone, which serves as the electron acceptor for DHODH, an essential enzyme in the pyrimidine biosynthesis pathway (35). Providing the parasite with a yeast DHODH (yDHODH) enzyme that utilizes a different electron acceptor establishes an alternate pathway for pyrimidine biosynthesis and renders the parasite resistant to all bc1 complex inhibitors (35). Thus, yDHODH transgenic lines have become a convenient tool for determining if a compound targets the mtETC. Here, we utilized the 3D7attB-yDHODH line, which has the yDHODH gene integrated into the genome at a nonessential locus (43). As shown in Fig. 6, this transgenic line was fully resistant to atovaquone, as expected, but was still susceptible to all of the CGX compounds tested, suggesting that the target(s) of the CGXs does not reside in the parasite mtETC. Consequently, these drugs likely target another essential function(s) either inside or outside the mitochondrion. We note that the EC50s of these compounds against the 3D7attB-yDHODH parasites (Fig. 6) were slightly higher than those against the 3D7 parasites (Table 1). The reasons for the variation are undetermined, but it may arise during the genetic transfections and lengthy selection procedures required to generate the 3D7attB-yDHODH line.
FIG 6.
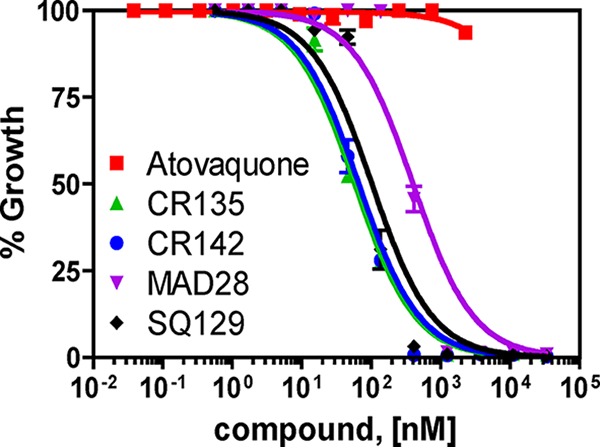
CGX compounds do not target the mETC of P. falciparum. The potency of CGX compounds against mtETC-independent strain 3D7attB-yDHODH was determined with the [3H]hypoxanthine incorporation assay. Calculated average EC50s of compounds (nanomolar) ± the standard errors against this strain from three independent experiments: CR135, 58.0 ± 5.7; CR142, 61.8 ± 6.4; MAD28, 383.6 ± 19.5; SQ129, 101.3 ± 8.1.
Toxicity of CGXs to human cells.
To test the toxicity of these CGXs to human cells, we performed the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay with HEK293 cells. The cells were exposed to a range of concentrations of each selected CGX compound for 24 and 48 h as described in Materials and Methods. As shown in Fig. 7, after 24 h of drug exposure, we found that the EC50s of CR135, CR142, and MAD28 fell in the low micromolar range. SQ129 was less toxic to HEK293 cells, with a significantly higher EC50. The average EC50s from three independent experiments are shown in Table 1. The EC50s for the 48-h drug exposure were quite similar to those for 24 h of treatment (data not shown). These data suggest that these CGX compounds are toxic to human cells at low micromolar concentrations. Accordingly, the therapeutic window is around 150-fold for CR135 or CR142, while it is much narrower for MAD28 (∼7-fold).
FIG 7.
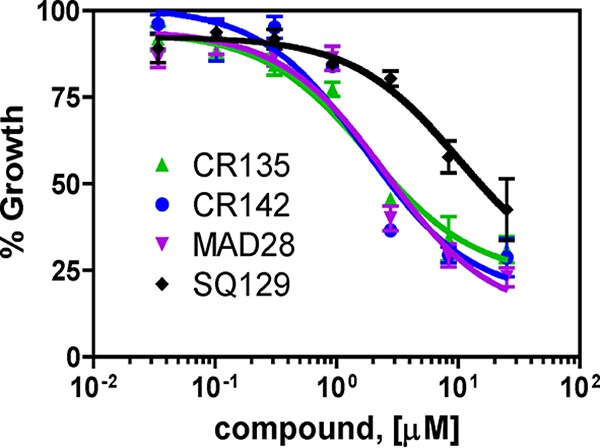
Cytotoxicity of CGX derivatives to HEK293 cells. The cytotoxicity of CGX compounds to human HEK293 cells treated for 24 h was determined by an MTT assay (see Materials and Methods). For each concentration of each compound tested, triplicate wells were set up. The y axis (percent growth) indicates the MTT signal in a drug-treated sample as a percentage of that in a no-drug control. The average of three biological replicates is shown. Calculated EC50s are listed in Table 1.
DISCUSSION
In this study, we tested the antimalarial activities of CGX compounds against P. falciparum malaria parasites at the intraerythrocytic stages. GBA and synthetic derivatives have been of great interest to medicinal chemistry because of their potent activities against cancer cells. For the first time, this study shows that these compounds also possess antimalarial activity against both drug-sensitive and drug-resistant parasite lines. Our data extend the biological functions of GBA and CGXs beyond their known anticancer (11), antibacterial (44), and antiviral (45) effects to include antiparasitic activity.
GBA and its derivatives showed moderate antimalarial activity, with submicromolar EC50s. The antimalarial activity of compounds CR135 and CR142, in which the caged xanthone motif was conjugated with a triphenylphosphonium group, resulting in low nanomolar EC50s, was greatly enhanced. It has been proposed that delocalized lipophilic cations, such as the triphenylphosphonium group, can specifically and efficiently drive their cargo to active mitochondria, which maintain a negative-inside transmembrane gradient (46, 47). Such a delivery strategy has led to the development of MitoQ, a ubiquinone-triphenylphosphonium conjugate that has entered clinical trials against neurodegenerative diseases (48, 49). Moreover, improvement of antimalarial potency has been reported for a series of 1,4-naphthoquinones (the chemical class that includes atovaquone) conjugated with a triphenylphosphonium group (50, 51). In infected RBCs, the parasite's mitochondrion is the organelle with the most negative membrane potential. Therefore, we reason that CR135 and CR142 accumulate in or at the parasite's mitochondrion in higher concentrations than in the rest of the parasite and the host. In turn, the parasite mitochondrion is likely a major action site for these compounds. In support of this hypothesis, we also observed that CR135 and CR142 caused mitochondrial fragmentation in the parasite 8 h after drug treatment (Fig. 4), indicating that the parasite mitochondrion contains at least one target of CR135 and CR142. Interestingly, in cancer cells, both CR135 and CR142 showed lower toxicity than their parent compounds, MAD28 and MAD44 (38), suggesting that structural modifications of the CGX motif could improve both potency and selectivity against malaria parasites. Besides the triphenylphosphonium group, the caged xanthone moiety of CR135 and CR142 is also critical for function, since SQ129, a synthetic derivative that contains a triphenylphosphonium group but lacks the caged xanthone, showed only moderate antimalarial activity.
We found that CGXs kill malaria parasites at multiple asexual stages. Metabolically active trophozoite and schizont stage parasites are more sensitive to these compounds than ring stage parasites are (Fig. 3). Our data also show that treatment with different CGXs caused distinct morphological changes in the parasites, suggesting that the various individual modifications of these compounds may result in differing modes of action. Particularly, CR135 and CR142 exhibited antimalarial activities at low nanomolar concentrations (Fig. 2). These compounds caused the formation of a large Giemsa stain-negative structure (GNB) inside the parasite that occurred during trophozoite development (Fig. 3). The appearance of a GNB seemed to be specific to CR135 or CR142 and not MAD28 (Fig. 3). Since this enlarged area was close to the hemozoin particle (Fig. 3), we thought that it might be an enlargement of the food vacuole. Indeed, the integrity of the food vacuole was damaged by CR135 and CR142 treatment, as determined by IFA with a food vacuole marker (Fig. 4). Under drug (CR135 or CR142) treatment, the globular structure of the food vacuole of a normal parasite was absent and the vacuole marker plasmepsin II was distributed among many punctate particles scattered throughout the parasite cytosol (Fig. 4). These small particles, however, did not appear to outline a large structure resembling the empty area seen in Giemsa-stained parasites after drug treatment (compare Fig. 3 and 4). Therefore, the nature of the GNB seen in thin blood smears remains unknown.
The mechanisms of action of CGXs against malaria parasites remain unknown. In cancer cells, GBA and other CGXs are known to cause mitochondrial damage and apoptosis (11). The mtETC is an essential process in malaria parasites and a known antimalarial drug target. We have shown that in the asexual blood stages, the critical function of the mtETC is to sustain the activity of the parasite DHODH for pyrimidine biosynthesis (35, 43). Provision of the parasites with the yDHODH, which does not rely on the mtETC, makes the parasites resistant to all mtETC inhibitors (35, 43). Importantly however, yDHODH transgenic parasites were found to be sensitive to CGXs (Fig. 5). This result rules out the possibility that CGXs target the mtETC. It is likely that CR135 and CR142 target another, as-yet-unknown, essential mitochondrial function(s) or other pathways beyond the mitochondrion.
CGX compounds seem to target multiple pathways. The integrity of the food vacuole is lost after 8 h of treatment with CR135 and CR142. We attempted to generate resistant clones by culturing 108 Dd2 parasites with CR135, CR142, and MAD28 at three and five times their EC50s, individually. However, none of these conditions yielded any resistant parasites over a 2-month period (data not shown), consistent with the assumption that CGX compounds have various targets. Targeting of multiple pathways can be beneficial to prevent or delay the appearance of drug resistance. Indeed, standard antimalarial chemotherapy involves the use of a combination of several agents, since monotherapy can often select resistant parasites rapidly. Therefore, it could be an advantage for CGX compounds to target multiple pathways. On the other hand, action against multiple targets can make it challenging to optimize parasite-specific inhibitors via a rational program of chemical modifications. CGX compounds kill mammalian cells at low micromolar concentrations (1.5 to 2.2 μM; Fig. 7), somewhat below the common concentration (>10 μM) of other antimalarial compounds under development. While the initial GCX compounds had a narrow selective window, addition of a triphenylphosphonium group to the xanthone moiety expanded the therapeutic window dramatically from 7-fold (MAD28) to 150-fold (CR135/CR142) (Table 1). Thus, it is possible to develop better CGX derivatives through medicinal chemistry. As the most efficient antimalarial compound of the series, CR135 could be a lead for future chemical optimization.
From a drug development point of view, GBA and related CGXs clearly have potential for further development as antimalarial drugs. Importantly, conjugation of the CGX motif with selective transporters and delivery systems can substantially improve potency and target selectivity. This was shown by CR135 and CR142, which exhibit strong antimalarial activity at low nanomolar concentrations. It is thus reasonable to predict that further chemical optimization of the caged xanthone backbone would yield compounds with more potent and selective antimalarial activities. Interestingly, CGX compounds were not included in the “malaria box,” a set of 400 antimalarial compounds made available to the research community by the Medicines for Malaria Venture and partner companies and organizations (52). The CGX compounds appear to represent a new and unexplored class of antimalarial agents with a chemical backbone totally different from those of other chemical scaffolds discovered by high-throughput screening or other methods.
GBA and gamboge have been used in Eastern medicine for hundreds of years. GBA has entered a phase II clinical trial in China as an anticancer agent in patients with non-small-cell lung, colon, and renal cancers (19). Studies of GBA toxicity for mice, dogs, and rats have indicated that GBA has decent therapeutic windows for cancer therapies (53, 54) without any toxic effects on blood pressure, heart rate, or respiratory frequency at pharmacologically relevant doses (55). An innocuous dose of GBA in rats was established to be 60 mg/kg after administration for a total of 13 weeks at a frequency of one administration every other day. This dose was more than 10 times as high as that used in human clinical trials (52, 53). In addition, bioavailability studies (40 and 80 mg/kg) with rats showed that GBA was rapidly accumulated in the liver, where it can be metabolized by various routes, including oxidation, hydration, glutathionylation, and glucosidation, and was excreted mainly in bile from 0 to 24 h postdosing (56, 57). These studies attest to the pharmacological potential of GBA and the CGX motif. Moreover, the developed synthetic strategies allow rapid and high-yield access to designed CGX analogs, thereby potentially paving the way for the development of CGX-based antimalarial agents.
In conclusion, we evaluated the effect of GBA and related synthetic analogues of CGXs as antimalarial agents. We found that these compounds were cytotoxic to P. falciparum malaria parasites at submicromolar concentrations. Importantly, conjugating these compounds with a phosphonium salt improved their efficacy by about 2 orders of magnitude, resulting in lead compounds with a promising therapeutic window. Further modification of the caged xanthone motif and/or the delivery subunit could further increase the selective cytotoxicity of the compound and lead to the development of a promising lead candidate.
MATERIALS AND METHODS
Parasite lines and parasite culture.
P. falciparum strains Dd2 (resistant to chloroquine, mefloquine, and pyrimethamine) and 3D7 (drug sensitive) are the wild-type lines used in this study. P. falciparum 3D7attB-yDHODH is a transgenic line bearing a copy of the yDHODH gene in its genome, rendering the parasite resistant to inhibitors targeting the mtETC (35, 43). Parasites were cultured in human O+ erythrocytes (Interstate Blood Bank) in complete RPMI 1640 medium supplemented with 0.5% AlbuMAX (Invitrogen), 15 mM HEPES, 10 mg/liter hypoxanthine, 25 mM NaHCO3, and 50 μg/liter gentamicin. Cultures were incubated at 37°C in an incubator filled with a low-oxygen gas mixture (89% N2, 5% CO2, and 6% O2).
Growth inhibition assay via [3H]hypoxanthine incorporation.
Antimalarial activity was determined by measuring [3H]hypoxanthine incorporation in parasites exposed to compounds in 96-well plates. The compounds tested were initially dissolved in DMSO to a 10 mM concentration. Parasite cultures at 1% parasitemia and a 1.5% hematocrit were exposed to serial dilutions of each compound or no-compound medium for 24 h. After 24 h, each well was pulsed with 0.5 μCi of [3H]hypoxanthine and incubated for another 24 h. Parasites were then frozen at −80°C overnight. Parasites were lysed by thawing, and nucleic acids were collected on filters with a cell harvester (PerkinElmer Life Sciences). Filters were air dried, and 30 μl of MicroScint O (PerkinElmer Life Sciences) was added to each well. Incorporation of [3H]hypoxanthine was quantified with a TopCount scintillation counter (PerkinElmer Life Sciences).
Flow cytometry assessment of parasitemia.
Dd2 parasites were tightly synchronized by multiple rounds of alanine treatment (0.5 M alanine, 10 mM HEPES, pH 7.6) of ring stage cultures (58). Upon reaching the mid-trophozoite stage, synchronized parasites were inoculated into a 24-well plate with each well harboring a 2-ml culture with a 2.5% hematocrit. Parasites were exposed to DMSO (0.5 μl/ml) or compounds at 10 times their EC50s. Specifically, the concentrations of CR135, CR142, MAD28, and SQ129 were 0.1, 0.15, 2.6, and 1.1 μM, respectively. At 2, 4, 8, 24, and 48 h posttreatment, a small aliquot was taken from each well and used to prepare a Giemsa-stained thin smear for morphological examination. At the same time points, another aliquot of each parasite culture (5 to 10 μl of pellet) was fixed with 4% formaldehyde at 37°C for 1 h or at 4°C overnight on a rotator. The samples were then washed three times with phosphate-buffered saline (PBS) and stained with SYBR green (Thermo Fisher Scientific) for 1 h at room temperature on a rotator. They were then washed three times, and ∼1 μl of the pellet was resuspended in 1 ml of sterile H2O. The samples were analyzed with a BD Accuri C6 flow cytometer. Uninfected RBCs, unstained and stained with SYBR green, and unstained infected trophozoites were used as controls. For each sample, 1,000,000 events were collected and cell debris was removed by proper gating strategy. The percentage of SYBR green-positive events was taken to be the parasitemia.
Microscopy.
Thin blood smears were fixed with 100% methanol, air dried, and stained with Giemsa dye solution for 10 min. Morphologies of infected RBCs were examined under a Leica microscope, and pictures were taken with a 16-megapixel camera. When parasitemia was determined microscopically, at least 1,000 RBCs were counted. For mitochondrial morphologies, parasites were stained with MitoTracker Red CMXRos (Invitrogen) at 60 nM for 30 min, washed three times with PBS, and then incubated with a test compound. After compound treatment, parasites were lightly fixed with 1% formaldehyde for 10 min and observed under an Olympus fluorescence microscope.
IFA.
Dd2 parasites were tightly synchronized with several rounds of alanine treatment. At the early trophozoite stage, parasites were exposed to DMSO or selected CGX compounds at 10 times their EC50s for 8 h. Thirty minutes before sample harvesting, 60 nM MitoTracker Red CMXRos (Invitrogen) was added to each culture. Parasites were then washed three times with PBS. Thin blood smears were made for each condition. The remaining cultures were fixed with 4% formaldehyde–0.0075% glutaraldehyde at 4°C overnight. The samples were then permeabilized with 0.1% Triton X-100, reduced with 0.1 mg/ml sodium borohydride, and blocked with 5% bovine serum albumin–PBS in accordance with our standard IFA procedure (59). To monitor the morphological changes in the food vacuole upon drug treatment, an anti-plasmepsin II polyclonal antibody (rabbit antiserum MRA-66, contributed by Daniel E. Goldberg) was obtained from BEI Resources, National Institute of Allergy and Infectious Diseases (NIAID), NIH (https://www.beiresources.org/). The antibody was diluted 1:1,000 and incubated with the samples at 4°C overnight. An Alexa Fluor 488-conjugated anti-rabbit secondary antibody (Molecular Probes) at a dilution of 1:350 was then added, and the mixture was incubated at 4°C overnight. All other steps followed the standard protocol (59). The parasites were then visualized under the Olympus fluorescence microscope.
MTT cell growth assay.
HEK293 is a noncarcinoma cell line stably transformed with adenovirus DNA that can grow indefinitely in vitro (60). These mammalian cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. The cytotoxicity of CGX compounds to HEK293 cells was determined by an MTT assay in 96-well plates in accordance with the manufacture's protocol (EMD Millipore Corporation). Briefly, 10,000 cells in 50 μl of DMEM were inoculated into each well. The plates were incubated in a 37°C incubator (5% CO2) for 2 h to allow the cells to attach. Compounds were then serially diluted in another 96-well plate, and 50-μl aliquots of various concentrations were added to wells previously seeded with cells. The final volume of medium in each well was 0.1 ml. After exposure to compounds for 24 and 48 h, 10 μl of MTT solution (5 mg/ml) was added to each well and the plates were incubated at 37°C for 4 h to allow the MTT to be reduced to purple formazan in live cells. After 4 h of incubation with MTT, 100 μl of isopropanol containing 0.04 N HCl was added to each well. Isopropanol dissolves formazan, yielding a homogeneous blue solution that can be measured colorimetrically. Absorbance was measured with an enzyme-linked immunosorbent assay plate reader (Tecan US) at 570 nm versus a reference wavelength of 630 nm.
Compound synthesis.
GBA was isolated from gamboge resin via its pyridine salt (27). CLV and the hydroxylated CLVs MAD28 and MAD44 were synthesized as previously reported (25, 61). The synthesis of the triphenylphosphonium salt conjugates is described in the supplemental material. CR135 and CR142 were synthesized from MAD28 and MAD44, respectively, by treating them with 1,4-dibromobutane (5 eq) and potassium carbonate (2 eq) in dimethylformamide (38). The resulting bromide was then converted to the triphenylphosphonium salt upon treatment with triphenylphosphine (Ph3P; 5 eq) in acetonitrile at 150°C under microwave irradiation. Preparation of SQ129 proceeded in three steps that included (i) protection of the catechol functionality of trihydroxylated xanthone (62, 63) with diiodomethane and sodium bicarbonate, (ii) bromination of the C6 phenol with 1,4-dibromobutane, and (iii) treatment of the resulting bromide with Ph3P. Detailed experimental procedures and spectroscopic and analytical data are provided in the supplemental material.
Data analysis.
For [3H]hypoxanthine incorporation and MTT assays, triplicate wells were set for each condition tested. The mean values of measurements of parasites or HEK293 cells treated with DMSO alone were set as 100%. All other measurements were compared to DMSO controls. The dose-response data were then analyzed with GraphPad Prism Version 4 to obtain curves fitted by nonlinear regression and the corresponding EC50s. The mean ± standard error of all biological replicates for each condition is reported in Results.
Supplementary Material
ACKNOWLEDGMENTS
The plasmepsin II antibody was obtained through BEI Resources (NIAID, NIH). The antibody was originally contributed by Daniel E. Goldberg of Washington University in St. Louis. We thank James M. Burns, Jr., of the Drexel University College of Medicine for help in arranging the delivery of the antibody. HEK293 cells were kindly provided by Sandhya Kortagere of the Drexel University College of Medicine. We thank Wei Xu for technical assistance in culturing HEK293 cells and Sihem Sassi-Gaha for help with MTT assays. We also thank Anthony Mrse (UCSD NMR Facility) and Yongxuan Su (UCSD MS Facility) for assistance with the spectroscopic characterizations.
This work was supported by grants AI028398 from the NIH to Akhil B. Vaidya, CRC-15-380737 from the Cancer Research Coordinating Committee to Emmanuel A. Theodorakis, and TRG5780085 from the Thailand Research Fund to Oraphin Chantarasriwong.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01220-16.
REFERENCES
- 1.WHO. 2015. World malaria report 2015. World Health Organization, Geneva, Switzerland: http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/. [Google Scholar]
- 2.Crompton PD, Moebius J, Portugal S, Waisberg M, Hart G, Garver LS, Miller LH, Barillas-Mury C, Pierce SK. 2014. Malaria immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious disease. Annu Rev Immunol 32:1–14. doi: 10.1146/annurev-immunol-032713-120220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. 2014. Malaria. Lancet 383:723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 4.Miller LH, Ackerman HC, Su XZ, Wellems TE. 2013. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med 19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui L, Mharakurwa S, Ndiaye D, Rathod PK, Rosenthal PJ. 2015. Antimalarial drug resistance: literature review and activities and findings of the ICEMR Network. Am J Trop Med Hyg 93(3 Suppl):57–68. doi: 10.4269/ajtmh.15-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takala-Harrison S, Laufer MK. 2015. Antimalarial drug resistance in Africa: key lessons for the future. Ann N Y Acad Sci 1342:62–67. doi: 10.1111/nyas.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor C. M, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. 2015. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, Chotivanich K, Imwong M, Pukrittayakamee S, Dhorda M, Nguon C, Lim P, Amaratunga C, Suon S, Hien TT, Htut Y, Faiz MA, Onyamboko MA, Mayxay M, Newton PN, Tripura R, Woodrow CJ, Miotto O, Kwiatkowski DP, Nosten F, Day NP, Preiser PR, White NJ, Dondorp AM, Fairhurst RM, Bozdech Z. 2015. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 347:431–435. doi: 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. 2010. Chemistry and biology of the caged Garcinia xanthones. Chemistry 16:9944–9962. doi: 10.1002/chem.201000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han QB, Xu HX. 2009. Caged Garcinia xanthones: development since 1937. Curr Med Chem 16:3775–3796. doi: 10.2174/092986709789104993. [DOI] [PubMed] [Google Scholar]
- 12.Reference deleted.
- 13.Gu HY, Wang XT, Rao SY, Wang J, Zhao J, Ren FL, Mu R, Yang Y, Qi Q, Liu W, Lu N, Ling H, You QD, Guo QL. 2008. Gambogic acid mediates apoptosis as a p53 inducer through down-regulation of mdm2 in wild-type p53-expressing cancer cells. Mol Cancer Ther 7:3298–3305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]
- 14.Huang GM, Sun Y, Ge X, Wan X, Li CB. 2015. Gambogic acid induces apoptosis and inhibits colorectal tumor growth via mitochondrial pathways. World J Gastroenterol 21:6194–6205. doi: 10.3748/wjg.v21.i20.6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li CL, Qi Q, Lu N, Dai QS, Li FN, Wang XT, You QD, Guo QL. 2012. Gambogic acid promotes apoptosis and resistance to metastatic potential in MDA-MB-231 human breast carcinoma cells. Biochem Cell Biol 90:718–730. doi: 10.1139/o2012-030. [DOI] [PubMed] [Google Scholar]
- 16.Wang YJ, Xiang W, Wang M, Huang T, Xiao XY, Wang L, Tao D, Dong LY, Zeng FQ, Jiang GS. 2014. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br J Pharmacol 171:618–635. doi: 10.1111/bph.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi T, Yi Z, Cho SG, Luo J, Pandey MK, Aggarwal BB, Liu M. 2008. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res 68:1843–1850. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang LJ, Chen Y. 2013. New targets for the antitumor activity of gambogic acid in hematologic malignancies. Acta Pharmacol Sin 34:191–198. doi: 10.1038/aps.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi Y, Zhan XK, Yu H, Xie GR, Wang ZZ, Xiao W, Wang YG, Xiong FX, Hu JF, Yang L, Cui CX, Wang JW. 2013. An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin Med J (Engl) 126:1642–1646. [PubMed] [Google Scholar]
- 20.Asano J, Chiba K, Tada M, Yoshii T. 1996. Cytotoxic xanthones from Garcinia hanburyi. Phytochemistry 41:815–820. doi: 10.1016/0031-9422(95)00682-6. [DOI] [PubMed] [Google Scholar]
- 21.Thoison O, Fahy J, Dumontet V, Chiaroni A, Riche C, Tri MV, Sevenet T. 2000. Cytotoxic prenylxanthones from Garcinia bracteata. J Nat Prod 63:441–446. doi: 10.1021/np9903088. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Cao S, Goh S, Hsu A, Tan BK. 2002. Mitochondrial destabilisation and caspase-3 activation are involved in the apoptosis of Jurkat cells induced by gaudichaudione A, a cytotoxic xanthone. Planta Med 68:198–203. doi: 10.1055/s-2002-23142. [DOI] [PubMed] [Google Scholar]
- 23.Xu YJ, Yip SC, Kosela S, Fitri E, Hana M, Goh SH, Sim KY. 2000. Novel cytotoxic, polyprenylated heptacyclic xanthonoids from Indonesian Garcinia gaudichaudii (Guttiferae). Org Lett 2:3945–3948. doi: 10.1021/ol006730t. [DOI] [PubMed] [Google Scholar]
- 24.Batova A, Lam T, Wascholowski V, Yu AL, Giannis A, Theodorakis EA. 2007. Synthesis and evaluation of caged Garcinia xanthones. Org Biomol Chem 5:494–500. doi: 10.1039/B612903J. [DOI] [PubMed] [Google Scholar]
- 25.Chantarasriwong O, Cho WC, Batova A, Chavasiri W, Moore C, Rheingold AL, Theodorakis EA. 2009. Evaluation of the pharmacophoric motif of the caged Garcinia xanthones. Org Biomol Chem 7:4886–4894. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batova A, Altomare D, Chantarasriwong O, Ohlsen KL, Creek KE, Lin YC, Messersmith A, Yu AL, Yu J, Theodorakis EA. 2010. The synthetic caged Garcinia xanthone cluvenone induces cell stress and apoptosis and has immune modulatory activity. Mol Cancer Ther 9:2869–2878. doi: 10.1158/1535-7163.MCT-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guizzunti G, Batova A, Chantarasriwong O, Dakanali M, Theodorakis EA. 2012. Subcellular localization and activity of gambogic acid. Chembiochem 13:1191–1198. doi: 10.1002/cbic.201200065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guizzunti G, Theodorakis EA, Yu AL, Zurzolo C, Batova A. 2012. Cluvenone induces apoptosis via a direct target in mitochondria: a possible mechanism to circumvent chemo-resistance? Invest New Drugs 30:1841–1848. doi: 10.1007/s10637-011-9745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai F, Morcos F, Sohn YS, Darash-Yahana M, Rezende CO, Lipper CH, Paddock ML, Song L, Luo Y, Holt SH, Tamir S, Theodorakis EA, Jennings PA, Onuchic JN, Mittler R, Nechushtai R. 2015. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc Natl Acad Sci U S A 112:3698–3703. doi: 10.1073/pnas.1502960112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaidya AB, Mather MW. 2009. Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol 63:249–267. doi: 10.1146/annurev.micro.091208.073424. [DOI] [PubMed] [Google Scholar]
- 31.Mather MW, Henry KW, Vaidya AB. 2007. Mitochondrial drug targets in apicomplexan parasites. Curr Drug Targets 8:49–60. doi: 10.2174/138945007779315632. [DOI] [PubMed] [Google Scholar]
- 32.Wells TN, Hooft van Huijsduijnen R, Van Voorhis WC. 2015. Malaria medicines: a glass half full? Nat Rev Drug Discov 14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava IK, Rottenberg H, Vaidya AB. 1997. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272:3961–3966. doi: 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- 34.Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El Mazouni F, Lao Y, Kokkonda S, Tomchick DR, Deng X, Laird T, Bhatia SN, March S, Ng CL, Fidock DA, Wittlin S, Lafuente-Monasterio M, Benito FJ, Alonso LM, Martinez MS, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Haselden JN, Louttit J, Cui Y, Sridhar A, Zeeman AM, Kocken C, Sauerwein R, Dechering K, Avery VM, Duffy S, Delves M, Sinden R, Ruecker A, Wickham KS, Rochford R, Gahagen J, Iyer L, Riccio E, Mirsalis J, Bathhurst I, Rueckle T, Ding X, Campo B, Leroy D. 2015. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med 7:296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. 2007. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 36.Guinet F, Dvorak JA, Fujioka H, Keister DB, Muratova O, Kaslow DC, Aikawa M, Vaidya AB, Wellems TE. 1996. A developmental defect in Plasmodium falciparum male gametogenesis. J Cell Biol 135:269–278. doi: 10.1083/jcb.135.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alin MH, Bjorkman A, Ashton M. 1990. In vitro activity of artemisinin, its derivatives, and pyronaridine against different strains of Plasmodium falciparum. Trans R Soc Trop Med Hyg 84:635–637. doi: 10.1016/0035-9203(90)90129-3. [DOI] [PubMed] [Google Scholar]
- 38.Theodoraki MA, Rezende CO Jr, Chantarasriwong O, Corben AD, Theodorakis EA, Alpaugh ML. 2015. Spontaneously-forming spheroids as an in vitro cancer cell model for anticancer drug screening. Oncotarget 6:21255–21267. doi: 10.18632/oncotarget.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuteja R. 2007. Malaria—an overview. FEBS J 274:4670–4679. doi: 10.1111/j.1742-4658.2007.05997.x. [DOI] [PubMed] [Google Scholar]
- 40.Francis SE, Banerjee R, Goldberg DE. 1997. Biosynthesis and maturation of the malaria aspartic hemoglobinases plasmepsins I and II. J Biol Chem 272:14961–14968. doi: 10.1074/jbc.272.23.14961. [DOI] [PubMed] [Google Scholar]
- 41.Klemba M, Beatty W, Gluzman I, Goldberg DE. 2004. Trafficking of plasmepsin II to the food vacuole of the malaria parasite Plasmodium falciparum. J Cell Biol 164:47–56. doi: 10.1083/jcb200307147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamir S, Paddock ML, Darash-Yahana-Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F, Cabantchik IZ, Onuchic JN, Jennings PA, Mittler R, Nechushtai R. 2015. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim Biophys Acta 1853:1294–1315. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 43.Ke H, Morrisey JM, Ganesan SM, Painter HJ, Mather MW, Vaidya AB. 2011. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot Cell 10:1053–1061. doi: 10.1128/EC.05049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rukachaisirikul V, Phainuphong P, Sukpondma Y, Phongpaichit S, Taylor WC. 2005. Antibacterial caged-tetraprenylated xanthones from the stem bark of Garcinia scortechinii. Planta Med 71:165–170. doi: 10.1055/s-2005-837785. [DOI] [PubMed] [Google Scholar]
- 45.Reutrakul V, Anantachoke N, Pohmakotr M, Jaipetch T, Sophasan S, Yoosook C, Kasisit J, Napaswat C, Santisuk T, Tuchinda P. 2007. Cytotoxic and anti-HIV-1 caged xanthones from the resin and fruits of Garcinia hanburyi. Planta Med 73:33–40. doi: 10.1055/s-2006-951748. [DOI] [PubMed] [Google Scholar]
- 46.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. 2008. Targeting mitochondria. Acc Chem Res 41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 47.Yousif LF, Stewart KM, Kelley SO. 2009. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- 48.Mao P, Manczak M, Shirendeb UP, Reddy PH. 2013. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim Biophys Acta 1832:2322–2331. doi: 10.1016/j.bbadis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith RA, Murphy MP. 2010. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 50.Long TE, Lu X, Galizzi M, Docampo R, Gut J, Rosenthal PJ. 2012. Phosphonium lipocations as antiparasitic agents. Bioorg Med Chem Lett 22:2976–2979. doi: 10.1016/j.bmcl.2012.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu X, Altharawi A, Gut J, Rosenthal PJ, Long TE. 2012. 1,4-Naphthoquinone cations as antiplasmodial agents: hydroxy-, acyloxy-, and alkoxy-substituted analogues. ACS Med Chem Lett 3:1029–1033. doi: 10.1021/ml300242v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TN, Willis P. 2013. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo Q, Qi Q, You Q, Gu H, Zhao L, Wu Z. 2006. Toxicological studies of gambogic acid and its potential targets in experimental animals. Basic Clin Pharmacol Toxicol 99:178–184. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]
- 54.Qi Q, You Q, Gu H, Zhao L, Liu W, Lu N, Guo Q. 2008. Studies on the toxicity of gambogic acid in rats. J Ethnopharmacol 117:433–438. doi: 10.1016/j.jep.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 55.Zhao L, Zhen C, Wu Z, Hu R, Zhou C, Guo Q. 2010. General pharmacological properties, developmental toxicity, and analgesic activity of gambogic acid, a novel natural anticancer agent. Drug Chem Toxicol 33:88–96. doi: 10.3109/01480540903173534. [DOI] [PubMed] [Google Scholar]
- 56.Yang J, Ding L, Hu L, Qian W, Jin S, Sun X, Wang Z, Xiao W. 2011. Metabolism of gambogic acid in rats: a rare intestinal metabolic pathway responsible for its final disposition. Drug Metab Dispos 39:617–626. doi: 10.1124/dmd.110.037044. [DOI] [PubMed] [Google Scholar]
- 57.Zheng Z, Ou W, Zhang X, Li Y, Li Y. 2015. UHPLC-MS method for determination of gambogic acid and application to bioavailability, pharmacokinetics, excretion and tissue distribution in rats. Biomed Chromatogr 29:1581–1588. doi: 10.1002/bmc.3462. [DOI] [PubMed] [Google Scholar]
- 58.Kutner S, Breuer WV, Ginsburg H, Aley SB, Cabantchik ZI. 1985. Characterization of permeation pathways in the plasma membrane of human erythrocytes infected with early stages of Plasmodium falciparum: association with parasite development. J Cell Physiol 125:521–527. doi: 10.1002/jcp.1041250323. [DOI] [PubMed] [Google Scholar]
- 59.Balabaskaran Nina P, Morrisey JM, Ganesan SM, Ke H, Pershing AM, Mather MW, Vaidya AB. 2011. ATP synthase complex of Plasmodium falciparum: dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J Biol Chem 286:41312–41322. doi: 10.1074/jbc.M111.290973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16:869–871. [DOI] [PubMed] [Google Scholar]
- 61.Elbel KM, Guizzunti G, Theodoraki MA, Xu J, Batova A, Dakanali M, Theodorakis EA. 2013. A-ring oxygenation modulates the chemistry and bioactivity of caged Garcinia xanthones. Org Biomol Chem 11:3341–3348. doi: 10.1039/c3ob40395e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tisdale EJ, Slobodov I, Theodorakis EA. 2003. Biomimetic total synthesis of forbesione and desoxymorellin utilizing a tandem Claisen/Diels-Alder/Claisen rearrangement. Org Biomol Chem 1:4418–4422. doi: 10.1039/B311833A. [DOI] [PubMed] [Google Scholar]
- 63.Tisdale EJ, Slobodov I, Theodorakis EA. 2004. Unified synthesis of caged Garcinia natural products based on a site-selective Claisen/Diels-Alder/Claisen rearrangement. Proc Natl Acad Sci U S A 101:12030–12035. doi: 10.1073/pnas.0401932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.