

ABSTRACT
Adequate concentrations of efavirenz in the central nervous system (CNS) are necessary to suppress viral replication, but high concentrations may increase the likelihood of CNS adverse drug reactions. The aim of this investigation was to evaluate the efavirenz distribution in the cerebrospinal fluid (CSF) and the brain by using a physiologically based pharmacokinetic (PBPK) simulation for comparison with rodent and human data. The efavirenz CNS distribution was calculated using a permeability-limited model on a virtual cohort of 100 patients receiving efavirenz (600 mg once daily). Simulation data were then compared with human data from the literature and with rodent data. Wistar rats were administered efavirenz (10 mg kg of body weight−1) once daily over 5 weeks. Plasma and brain tissue were collected for analysis via liquid chromatography-tandem mass spectrometry (LC-MS/MS). The median maximum concentrations of drug (Cmax) were predicted to be 3,184 ng ml−1 (interquartile range [IQR], 2,219 to 4,851 ng ml−1), 49.9 ng ml−1 (IQR, 36.6 to 69.7 ng ml−1), and 50,343 ng ml−1 (IQR, 38,351 to 65,799 ng ml−1) in plasma, CSF, and brain tissue, respectively, giving a tissue-to-plasma ratio of 15.8. Following 5 weeks of oral dosing of efavirenz (10 mg kg−1), the median plasma and brain tissue concentrations in rats were 69.7 ng ml−1 (IQR, 44.9 to 130.6 ng ml−1) and 702.9 ng ml−1 (IQR, 475.5 to 1,018.0 ng ml−1), respectively, and the median tissue-to-plasma ratio was 9.5 (IQR, 7.0 to 10.9). Although it is useful, measurement of CSF concentrations may give an underestimation of the penetration of antiretrovirals into the brain. The limitations associated with obtaining tissue biopsy specimens and paired plasma and CSF samples from patients make PBPK modeling an attractive tool for probing drug distribution.
KEYWORDS: efavirenz, PBPK, CNS, toxicity
INTRODUCTION
Despite its widespread use, patients receiving efavirenz (EFV)-containing therapy frequently report central nervous system (CNS) disturbances. Symptoms of efavirenz-associated adverse drug reactions (ADRs) occur at a high frequency and can include depression, anxiety, abnormal dreams, and hallucinations (1). The majority of patients report development of CNS disorders shortly after commencing efavirenz therapy, with symptoms dissipating during the initial months of therapy. A minority of patients continue to experience symptoms for the duration of efavirenz use (2). More recently, efavirenz CNS ADRs have been shown to have more long-term effects (3).
In addition to the negative impact on the quality of the patient's life, CNS ADRs may also lead to a decrease in patient adherence. Poor patient adherence to antiretroviral medication is a major concern, in particular for drugs displaying a low genetic barrier to resistance, such as efavirenz (4). The impact of CNS side effects on patient adherence is not clearly defined. Some previous studies indicate that patients demonstrate tolerance to CNS side effects, with minimal impacts on patient adherence (5, 6). However, a recent study demonstrated that 60% of patients reported CNS side effects as the primary reason for discontinuation, versus 3% of patients receiving alternative antiretroviral therapies (3).
There is a paucity of information regarding the distribution of efavirenz in brain tissue. Due to impracticalities in obtaining brain tissue from patients, some groups have used concentrations in cerebrospinal fluid (CSF) as a surrogate for brain concentrations. The majority of pharmacokinetic (PK) studies have focused on describing efavirenz plasma concentrations and elucidating genetic factors that contribute to the variability in efavirenz PK or genetic associations to predict patients at risk of developing CNS toxicity (1, 7, 8). However, a few small studies investigated efavirenz PK in both plasma and CSF. CSF concentrations have been shown to be much lower (around 0.5%) than plasma concentrations. However, even at 0.5% of the plasma concentration, efavirenz concentrations in the CSF exceed the 50% inhibitory concentration (IC50) of efavirenz for wild-type HIV (9).
The appropriateness of CSF concentrations as a surrogate for brain concentrations is currently the subject of debate (10–12). It has been demonstrated in guinea pigs that brain tissue concentrations of nevirapine (NVP) not only differ from those in the CSF but also vary between brain regions (10). NVP uptake was shown to be 0.32 ml g−1 in the CSF, whereas NVP uptake was lower in the choroid plexus (0.25 ml g−1) and higher in the pituitary (1.61 ml g−1) than in the CSF (10). Indeed, concentrations of other antiretroviral drugs within CSF have been shown to vary depending on where the sample is taken. Lamivudine has been shown to be 5-fold higher in CSF sampled from the lumbar region than in ventricular CSF from rhesus monkeys (11). Although there are no comparable data for efavirenz in the literature, these data exemplify the challenges associated with predicting brain tissue concentrations based on CSF concentrations.
Physiologically based pharmacokinetic (PBPK) modeling is a bottom-up approach to simulate drug distributions in virtual patients. The approach mathematically describes physiological and molecular processes defining PK, integrating drug-specific properties (such as log P, Caco-2 cell apparent permeability, and affinities for transporters and metabolic enzymes) and patient-specific factors (such as height, weight, sex, organ volumes, and blood flow) (13). The model presented here is based on a full-body PBPK model supplemented with a 6-compartment model of the CNS and CSF, as previously described (14).
The aim of this investigation was to evaluate the efavirenz distribution in the CSF and the brain by using PBPK modeling. Simulated efavirenz PK data were then compared to available experimental data from rodents and clinical data from humans.
RESULTS
The protein binding of efavirenz in brain tissue was determined using rapid equilibrium dialysis (RED). The mean (± standard deviation) concentrations of efavirenz detected in the receiver chamber were 209.7 ± 33.4 ng ml−1 and 165 ± 22.0 ng ml−1 for 10% and 20% brain homogenates, respectively. The unbound fractions in brain tissue (fuBr) were calculated to be 0.00181 and 0.00212 for 10% and 20% brain homogenates, respectively. The average fuBr was 0.00197.
Following 5 weeks of oral dosing of efavirenz (10 mg kg of body weight−1), the median plasma concentration of efavirenz in rats was 69.7 ng ml−1 (interquartile range [IQR], 44.9 to 130.6 ng ml−1). The median efavirenz concentration in brain tissue was 702.9 ng ml−1 (IQR, 475.5 to 1,018.0 ng ml−1). The median tissue-to-plasma ratio was 9.5 (IQR, 7.0 to 10.9).
Simulation.
A standard dosing schedule of efavirenz (600 mg once daily) was simulated in 100 patients for the duration of 5 weeks. The results for efavirenz concentrations in plasma (Fig. 1A), CSF (Fig. 1B), and brain tissue (Fig. 1C) were all taken from the final 24 h of the simulation.
FIG 1.

Median (solid lines) simulated plasma (A), CSF (B), and brain tissue (C) concentrations of efavirenz during the final 24 h following 5 weeks of once-daily efavirenz (600 mg). Interquartile ranges are also shown (dotted lines).
The maximum concentration (Cmax), minimum concentration (Cmin), and area under the curve from 0 to 24 h (AUC0–24) for efavirenz in plasma were 3,916 ng ml−1 (IQR, 3,155 to 5,153 ng ml−1), 2,537 ng ml−1 (IQR, 1,942 to 3,779 ng ml−1), and 76,991 ng · h ml−1 (IQR, 62,170 to 107,560 ng · h ml−1). The CSF was predicted to have lower values for the efavirenz Cmax (50.96 ng ml−1 [IQR, 38.23 to 69.09 ng ml−1]), Cmin (47.8 ng ml−1 [IQR, 36.1 to 66.7 ng ml−1]), and AUC0–24 (1,193 ng · h ml−1 [IQR, 898 to 1,649 ng · h ml−1]). At 24 h, the efavirenz concentration in the CSF was 1.6% of the plasma concentration. The simulation predicted efavirenz concentrations in the brain to exceed those in the CSF and plasma, with a Cmax of 50,973 ng ml−1 (IQR, 39,122 to 66,177 ng ml−1), a Cmin of 49,566 ng ml−1 (IQR, 38,044 to 64,374 ng ml−1), and an AUC0–24 of 1,207,542 ng · h ml−1 (IQR, 926,900 to 1,567,974 ng · h ml−1). The brain tissue-to-plasma partition ratio at 24 h was 15.8.
The absorption constant (Ka) was predicted to be 0.19 h−1 (IQR, 0.18 to 0.21 h−1). The volume of distribution at steady state (VSS) and the elimination clearance (CL) were predicted to be 2.15 liters kg−1 (IQR, 2.06 to 2.31 liters kg−1) and 4.56 liters h−1 (IQR, 3.52 to 5.33 liters h−1), respectively. The absorbed fraction (fa) of efavirenz was predicted to have a median value of 0.46 (IQR, 0.44 to 0.49) and was used to calculate apparent VSS and apparent CL values of 323.31 liters kg−1 (IQR, 308.31 to 346.28 liters kg−1) and 9.79 liters h−1 (IQR, 7.54 to 11.41 liters h−1), respectively.
Comparison with clinical data.
The simulated PK parameters in plasma produced by the model were in agreement with data published from human trials and population PK (popPK) studies. Table 1 shows the results from the simulation and a number of clinical studies and popPK studies. The mean/median observed plasma concentrations of EFV ranged from 1,973 ng ml−1 to 3,180 ng ml−1 (9, 15–18). Simulated CL, VSS, and Ka values were 1.04-fold, 1.28-fold, and 0.6-fold different, respectively, from observed data (18). The average simulated concentration in CSF was 49.9 ng ml−1 (IQR, 36.6 to 69.7 ng ml−1), compared to a range of 11.1 ng ml−1 to 16.3 ng ml−1 observed in previously published clinical studies (9, 15).
TABLE 1.
Results from the simulation and a number of human trials and popPK studiesa
| Parameter | Simulated data |
Data from reference: |
|||||
|---|---|---|---|---|---|---|---|
| 15b | 9 | 16 | 17 | 18 | |||
| Mean (SD [or SE]) | Median (IQR) | Median (range) | Median (IQR) | Mean (range or SD) | Mean (SD or SE) | Mean (SE) | |
| Plasma concn (ng ml−1) | 3,183 (447) | 3,184 (2,219–4,851) | 3,718 (2,439–4,952) | 2,145 (1,384–4,423) | 1,973.8 (792.2–2,950.9 [range]) | 3,180 (1,610 [SD]) | |
| Plasma AUC (ng · h ml−1) | 91,924 (51,619) | 76,991 (62,170–107,560) | 86,280 | ||||
| Apparent CL (liters h−1) | 9.29 (0.26 [SE]) | 9.79 (7.54–11.44) | 9.61 (0.38 [SE]) | 9.4 (0.36) | |||
| Apparent VSS (liters kg−1) | 329.43 (2.38 [SE]) | 323.31 (308.31–346.28) | 291 (44.81 [SE]) | 252 (35.28) | |||
| Ka (h−1) | 0.20 (0.02) | 0.19 (0.18–0.21) | 0.3 (0.09) | ||||
| CSF concn (ng ml−1) | 49.9 (1.2) | 49.9 (36.6–69.7) | 16.3 (7.3–22.3) | 13.9 (4.1–21.2) | 11.1 (2.1–18.6 [SD]) | ||
| CSF AUC (ng · h ml−1) | 1,401 (809) | 1,193 (898–1,649) | 380 | ||||
| Brain tissue concn (ng ml−1) | 50,312.5 (438) | 50,343 (38,351–65,799) | |||||
| Brain tissue AUC (ng · h ml−1) | 1,397,820 (815,657) | 1,207,542 (926,900–1,567,974) | |||||
Means and medians are presented to allow comparisons of simulated and clinical data.
All samples in this study were obtained from a single patient over 24 h.
DISCUSSION
The presented data show that the PBPK model predicts efavirenz to accumulate in the brain at concentrations that far exceed those in the CSF. Human CSF concentrations were gathered from relatively small cohorts (n = 80 [9], n = 1 [15], and n = 10 [16]) and may not fully represent CSF concentrations in larger populations. Indeed, concentrations of efavirenz in the brain were predicted to exceed even plasma concentrations, with a brain-to-plasma ratio of 15.8. The rodent data presented here support the model prediction of a higher concentration of efavirenz in brain tissue, with a median tissue-to-plasma ratio of 9.5. Recently, efavirenz was demonstrated to accumulate in the brain tissue of a macaque. Following 8 days of orally administered efavirenz (60 mg kg−1), the concentrations in plasma and CSF were 541 and 3.30 ng ml−1, respectively. Concentrations of efavirenz in the cerebellum and basal ganglia were 6.86 μg g−1 (tissue-to-plasma ratio of 12.7) and 2.01 μg g−1 (tissue-to-plasma ratio of 3.7), respectively (19).
At present, only one study has examined efavirenz concentrations in human brain tissue (20). This study showed brain concentrations similar to historical CSF values and in disagreement with the data presented here. While participants in this analysis had detectable efavirenz in intracardiac serum by use of a qualitative assay, reliable dosing information was not routinely available, since the final care setting (home, hospice, or hospital) varied among individuals. Given this uncertainty regarding the final dosing interval, no precise information on the time of the last dose was available, which complicates interpretation of the reported brain concentrations. If the last efavirenz dose was administered, for example, 3 days prior to death, then the brain tissue concentrations may not accurately reflect those that occur in living, adherent patients. However, efavirenz has been shown to display a long plasma half-life (40 to 52 h) (21). This indicates that patients would have ceased receiving efavirenz for many days or had poor adherence in order to explain the very low concentrations observed. Despite this, the data predicted by the model are supported by robust data generated from the brain tissue concentrations for rats and monkeys (19).
Accumulation of efavirenz in brain tissue may be driven by physicochemical properties of efavirenz, in particular by lipophilicity. Since efavirenz is highly lipophilic (log P = 4.6) and has high accumulation levels in multiple cell types, it shows high cellular permeation (22). The brain has a high fat content, with approximately 60% of the brain consisting of fat (23). An additional factor that favors distribution is the high degree of protein binding of efavirenz. In plasma, efavirenz is highly protein bound (fu = 0.01) (24). Protein binding in the CSF is much lower, leading to more free efavirenz (fu = 0.238) (21). The data presented here for rapid equilibrium dialysis show the efavirenz fu in rodent brain tissue to be 0.00197. Taken collectively, the combination of a low fu and an affinity for the lipophilic environment of the brain favor accumulation of efavirenz in the CNS. Lipophilicity has been shown to be a significant factor in uptake of drugs into the brain (25). Lipophilicity, but not plasma protein binding, was shown to correlate with uptake of benzodiazepines, for example, into the brain. However, this study did not consider the fu in the brain, and the plasma fu may not be a good indicator of the brain fu. Kalvass et al. examined the fu in plasma and brain tissue for 34 drugs covering multiple drug classes. The data presented showed that plasma fu both under- and overestimated brain fu, depending on the drug (26).
Although this is the first study to employ PBPK modeling to investigate the efavirenz distribution in the CNS, PBPK modeling has been used previously to investigate efavirenz dose optimization, drug-drug interactions, and PK in special populations (22, 27).
Limitations of this work include the following: the presented model does not take genetic variability (i.e., CYP2B6 variants) into account, the brain fu values were generated for rodent brains rather than human brains, the current model is not able to estimate local concentrations in individual brain regions, and the permeation of efavirenz was calculated using a quantitative structure-activity relationship (QSAR) model of passive permeability, which often relies on extrapolated data obtained from animals with important differences from humans (28, 29). The CSF concentrations predicted by the model were approximately 3-fold greater than those observed in human patients. This indicates that the interactions between efavirenz and the blood-CSF barrier may not have been represented accurately. The permeation of efavirenz at the blood-CSF barrier was adjusted for the decreased surface area of the blood-CSF barrier, which is 1,000 times lower than that of the blood-brain barrier (BBB) (30). The assumption that the permeabilities of the two barriers are equal may be incorrect. However, these aspects can be expanded in future modeling strategies as the necessary input data emerge.
The BBB is highly effective at excluding xenobiotics from the CNS. Tight cellular junctions prevent paracellular transport of drugs, and the metabolizing enzymes and transport proteins remove drugs from the CNS. As such, another potential limitation of the model that warrants further elaboration is that distribution of efavirenz across the BBB may not be governed purely by passive permeability. The potential influence of influx and efflux transporters was not considered because efavirenz is not classified as the substrate of any transporter and effects of transporters on efavirenz PK have not been described. The model presented here potentially may be improved upon in the future if efavirenz is demonstrated to be a substrate for such transporters.
Numerous studies have linked efavirenz plasma concentrations to clinical evidence of CNS toxicity. Other studies have shown that efavirenz readily passes the BBB and is present in CSF. The simulations presented here indicate that plasma and CSF measurements may underestimate efavirenz exposure within the brain. The limitations associated with obtaining tissue biopsy specimens and paired plasma and CSF samples from patients make PBPK modeling an attractive tool for estimating such drug distribution.
MATERIALS AND METHODS
Animals and treatment.
Male Wistar rats (Charles River UK) weighing 180 to 220 g on arrival were used for PK analysis of efavirenz. Food and water were provided ad libitum. Following completion of dosing, all animals were sacrificed using an appropriate schedule 1 method (via exposure to CO2 at rising concentrations). All animal work was conducted in accordance with the Animals (Scientific Procedures) Act 1986 (ASPA), implemented by the United Kingdom Home Office.
Drug treatment.
Eight male Wistar rats were dosed with efavirenz (10 mg kg−1, with 2 ml kg−1 0.5% methylcellulose in distilled H2O) based on individual weights taken prior to dosing. The selected dose was based on scaling down the dose administered to adult humans (600 mg once daily given to an adult weighing 60/70 kg). The dose was also selected because it was previously administered to rats in a study examining anxiogenic effects, in which it was shown to induce anxiety in Wistar rats (31). Dosing was performed once daily via oral gavage over 5 weeks. The animals were terminated (via exposure to CO2 at rising concentrations) 2 h after the final dose, and blood was collected via cardiac puncture. Blood samples were centrifuged at 2,000 × g for 10 min at 4°C to separate plasma. Plasma was immediately frozen at −80°C and stored for later analysis. Brain tissue was also collected, and following washing in phosphate-buffered saline (PBS) for 30 s 3 times, it was immediately stored at −30°C for analysis.
RED.
The protein binding of efavirenz in brain tissue was determined using rapid equilibrium dialysis (RED) as described by Liu et al. (32). Untreated rat brain tissue was homogenized in 2 volumes (wt/vol) of 1% saline solution. Since efavirenz is highly protein bound, dilutions of brain tissue (10% and 20% brain tissue homogenates were prepared with 1% PBS) were used. Two hundred microliters of brain homogenate was spiked with 5,000 ng ml−1 efavirenz and added to the donor chamber. The receiver chamber contained 350 μl of Sorenson's buffer. The RED plate (Thermo, United Kingdom) was then placed in a shaking incubator for 4 h at 37°C and 100 rpm. A total of 250 μl was removed from the receiver chamber and frozen at −80°C for analysis. The fraction of unbound drug (fu) in brain tissue was then calculated from the diluted brain tissue by using the following formula (33):
where fu is the unbound fraction and D is the dilution factor.
Sample preparation for bioanalysis.
Efavirenz was extracted by protein precipitation. Twenty microliters of an internal standard (1,000 ng ml−1 lopinavir) was added to 100 μl of sample, standard, or quality control (QC), which was then treated with 400 μl of acetonitrile (ACN). Samples were then centrifuged at 4,000 × g for 10 min at 4°C. The supernatant fraction was transferred to a fresh glass vial and evaporated, and samples were placed in a rotary vacuum centrifuge at 30°C and then reconstituted in 140 μl of H2O-ACN (60:40). One hundred microliters of the sample was then transferred to a 200-μl chromatography vial. Five microliters of each sample was injected for analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Rat brain tissue was homogenized in 3 volumes (wt/vol) of plasma for 1 min at maximum power, using a Minilys homogenizer (Bertin Technologies, France). Extraction was performed using protein precipitation as detailed in the previous section. Recovery was tested at 3 levels (400 ng ml−1, 100 ng ml−1, and 20 ng ml−1). Mean recoveries were 95% (standard deviation, 8.9%) and 91% (standard deviation, 7.8%) for plasma and the brain, respectively. Samples generated from the RED experiment were pretreated with 20% ACN (PBS and Sorenson's buffer were spiked with 20% ACN in order to aid efavirenz solubility in these matrices), and the mean recovery was 84% (standard deviation, 11.6%).
Quantification of efavirenz.
Quantification was achieved via LC-MS/MS (TSQ Endura; Thermo Scientific) with an instrument operating in negative mode. The following ions were monitored for quantification in selected reaction monitoring scans: for efavirenz, m/z 315, >242.1, 244.0, and 250.0; and for the internal standard, lopinavir, m/z 627, >121.2, 178.1, and 198.1. A stock solution of 1 mg ml−1 efavirenz was prepared in methanol and stored at 4°C until use. A standard curve was prepared in plasma by serial dilution from 500 ng ml−1 to 1.9 ng ml−1, and an additional blank solution was also used.
Chromatographic separation was achieved using a multistep gradient with a Hypersil Gold C18 column (Thermo Scientific), using mobile phases A (100% H2O, 5 mM NH4HCO2) and B (100% ACN, 5 mM NH4HCO2). Chromatography was conducted over 8.55 min at a flow rate of 300 μl min−1. At the start of each run, mobile phase A was 90% until 0.1 min, and mobile phase B was then increased to 86% at 0.5 min. Mobile phase B was then gradually increased to 92% over 4.5 min, increased to 97% at 5.1 min, and held until 6 min. Mobile phase A was increased to 90% and held until the termination of the run at 8 min. Inter- and intra-assay variances in accuracy and precision were <15%.
PBPK parameters.
The full-body PBPK model used here was published previously, using equations from the physB model (Fig. 2) (13, 34). The model generates virtual patients based on a statistical description of human anatomy. The model simulates flow rates, organ volumes, and other tissue volumes based on anthropometric measures and allometric scaling.
FIG 2.
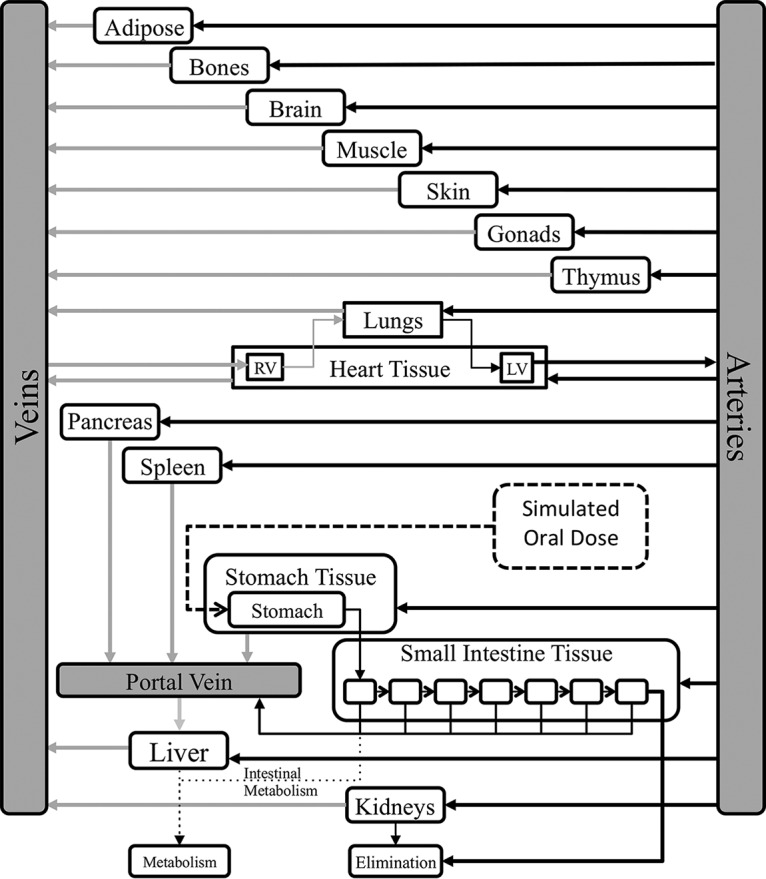
Full-body PBPK model. (Figure adapted from reference 34 with permission.)
Briefly, the equations required to simulate factors such as volume of distribution were previously published. Physicochemical properties from efavirenz data (including log P, molecular weight, and pKa) and in vitro data (permeation across Caco-2 cells and protein binding) were gathered from the literature and incorporated into the full-body model (22). The volume of distribution was simulated using the Poulin and Theil equation (35). This method describes the tissue-to-plasma ratio based on the individual organ volumes generated from the physB equations. Elimination clearance was calculated (using equation 1) by using allometric scaling of the metabolism of efavirenz in microsomes and accounting for the activities and abundances of cytochrome P450 2B6 (CYP2B6), CYP2A6, CYP1A2, CYP3A4, CYP3A5, and UGT2B7.
| (1) |
where “abundance” is the amount of enzyme expressed per microgram of microsomal protein and MPPGL is the amount of microsomal protein per gram of liver. Apparent clearance was calculated as the product of the TCLint values of all the enzymes contributing to the metabolism of efavirenz. Systemic clearance was calculated using equation 2, in which Qhv is the hepatic flow rate and fu is the unbound fraction in plasma (34).
| (2) |
The CNS portion of the model was based on validated parameters describing CNS and CSF physiology and anatomy (14). A schematic of this model is shown in Fig. 3. Physiological and physicochemical properties used are displayed in Table 2. The equations used in the model presented here are as follows.
| (3) |
Equation 3 shows a 3-descriptor QSAR model of the permeability surface area product (log PS) of the BBB as developed by Liu et al. (28). The three descriptors are log D (octanol/water partition coefficient at pH 7.4), vasbase (van der Waals surface area of the basic atoms), and TPSA (van der Waals polar surface area). The permeability surface area product of the blood-CSF barrier was calculated by dividing the permeability surface area product of the BBB by 1,000 to reflect the smaller surface area of the blood-CSF barrier (30).
| (4) |
Equation 4 describes the movement of efavirenz from arterial plasma to the brain, where EFVAr is the concentration of arterial efavirenz, fu is the unbound fraction in plasma, R is the blood-to-plasma ratio, EFVBr is the concentration of efavirenz in the brain, Qecf is the flow of brain extracellular fluid, fuBr is the unbound fraction in the brain, and PSB is permeability surface area product of the BBB.
| (5) |
| (6) |
| (7) |
| (8) |
Equations 5 to 8 describe the movement of efavirenz from the brain to the CSF, including movement across the blood-CSF barrier. The CSF is subdivided into 4 compartments: the left ventricle (LV), the third and fourth ventricles (TFV), the cisterna magna (CM), and the subarachnoid space (SAS). In the equations, EFVVe is the concentration of efavirenz in veins, fu is the unbound fraction in plasma, R is the blood-to-plasma ratio, EFVBr is the concentration of efavirenz in the brain, EFVCSF is the concentration of efavirenz in the CSF compartments, Qecf is the flow of brain extracellular fluid, Qcsf is the flow of CSF, fuCSF is the unbound fraction in CSF, fuBr is the unbound fraction in the brain, and PSE is the permeability surface area of the blood-CSF barrier.
FIG 3.
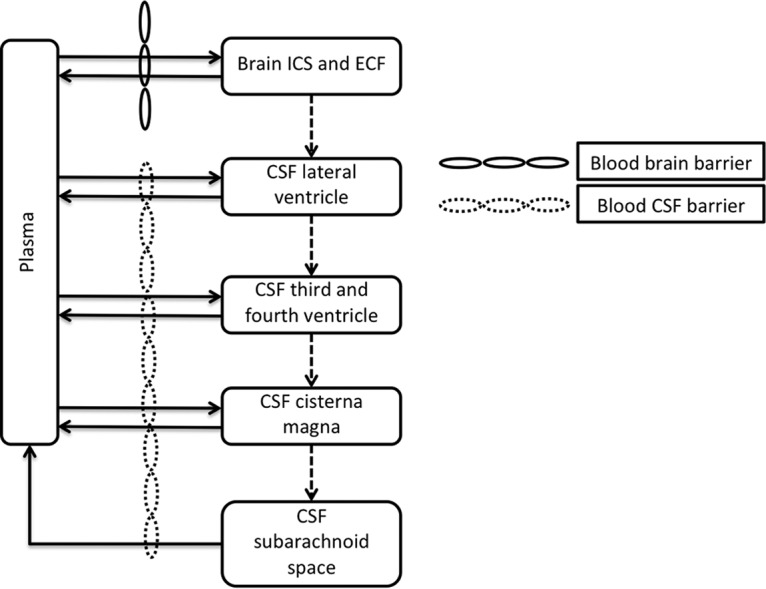
Diagram of the CNS component of the PBPK model to describe efavirenz movement within the CNS. The brain compartment is comprised of the total volume of extracellular fluid (ECF) and intracellular space (ICS).
TABLE 2.
Physiological and physicochemical variables used to generate the PBPK modela
| Model parameter | Value | Reference |
|---|---|---|
| Molecular weight | 315.7 | 22 |
| Log P | 4.6 | 22 |
| pKa | 10.2 | 22 |
| Caco-2 cell permeability (10−6 cm/s) | 2.5 | 22 |
| Unbound fraction | ||
| Plasma | 0.01 | 24 |
| CSF | 0.238 | 21 |
| Brain tissue | 0.00197 | |
| PSB | 2.47 | |
| PSE | 0.00247 | |
| Qcsf (ml/min) | 0.175 | 14 |
| Qecf (ml/min) | 0.4 | 14 |
| Brain ICS (ml) | 960 | 14 |
| Brain ECF (ml) | 240 | 14 |
| CSF vol (ml) | ||
| LV | 22.5 | 14 |
| TFV | 22.5 | 14 |
| CM | 7.5 | 14 |
| SAS | 90 | 14 |
ICS, intracellular space; ECF, extracellular fluid; LV, left ventricle; TFV, third and fourth ventricles; CM, cisterna magna; SAS, subarachnoid space.
Simulation design.
A virtual cohort of 100 patients was generated, and a once-daily dose of efavirenz (600 mg) was simulated over 5 weeks. Patient age (minimum of 18 years and maximum of 60 years), weight (minimum of 40 kg and maximum of 100 kg), height (minimum of 1.5 m and maximum of 2.1 m), and body mass index (minimum of 18 and maximum of 30) were generated from random normally distributed values. The PK in plasma, CSF, and brain tissue were recorded during the final 24 h at steady state. Plasma and CSF PK simulations were compared to previous data generated from clinical trials. Brain tissue-to-plasma ratios were also calculated and compared to data generated from rodents.
Materials.
Male Wistar rats were purchased from Charles River (Oxford, United Kingdom). Efavirenz powder (>98% pure) was purchased from LGM Pharma Inc. (Boca Raton, FL). All other consumables were purchased from Sigma-Aldrich (Dorset, United Kingdom).
ACKNOWLEDGMENTS
P.C., R.K.R.R., D.M.M., N.J.L., S.L., A.O., and M.S. wrote the manuscript, P.C. and M.S. designed research, P.C., R.K.R.R., D.M.M., and N.J.L. performed research, and P.C., R.K.R.R., and M.S. analyzed data.
S.L. is funded by NIH research awards, including grants HHSN271201000036C, R01 MH58076, R01 MH92225, P50 DA26306, and P30 MH62512. He has received support for research projects from Abbott, Merck, Tibotec, and GlaxoSmithKline. He has consulted for Gilead Sciences, GlaxoSmithKline, Merck, and Tibotec and has received lecture honoraria from Abbott and Boehringer-Ingelheim. A.O. has received research funding from Merck, Pfizer, and AstraZeneca and consultancies from Merck and Norgine and is a coinventor on patents relating to HIV nanomedicines. M.S. has received research funding from ViiV and Janssen.
REFERENCES
- 1.Sanchez Martin A, Cabrera Figueroa S, Cruz Guerrero R, Hurtado LP, Hurle AD, Carracedo Alvarez A. 2013. Impact of pharmacogenetics on CNS side effects related to efavirenz. Pharmacogenomics 14:1–10. doi: 10.2217/pgs.13.111. [DOI] [PubMed] [Google Scholar]
- 2.Vrouenraets SM, Wit FW, van Tongeren J, Lange JM. 2007. Efavirenz: a review. Expert Opin Pharmacother 8:851–871. doi: 10.1517/14656566.8.6.851. [DOI] [PubMed] [Google Scholar]
- 3.Leutscher PD, Stecher C, Storgaard M, Larsen CS. 2013. Discontinuation of efavirenz therapy in HIV patients due to neuropsychiatric adverse effects. Scand J Infect Dis 45:645–651. doi: 10.3109/00365548.2013.773067. [DOI] [PubMed] [Google Scholar]
- 4.Theys K, Camacho RJ, Gomes P, Vandamme AM, Rhee SY, Portuguese HIV-1 Resistance Study Group. 2015. Predicted residual activity of rilpivirine in HIV-1 infected patients failing therapy including NNRTIs efavirenz or nevirapine. Clin Microbiol Infect 21:607.e1–607.e8. doi: 10.1016/j.cmi.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Fumaz CR, Tuldra A, Ferrer MJ, Paredes R, Bonjoch A, Jou T, Negredo E, Romeu J, Sirera G, Tural C, Clotet B. 2002. Quality of life, emotional status, and adherence of HIV-1-infected patients treated with efavirenz versus protease inhibitor-containing regimens. J Acquir Immune Defic Syndr 29:244–253. doi: 10.1097/00042560-200203010-00004. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Molina JA. 2002. Safety and tolerance of efavirenz in different antiretroviral regimens: results from a national multicenter prospective study in 1,033 HIV-infected patients. HIV Clin Trials 3:279–286. doi: 10.1310/3Q91-YT2D-BUT4-8HN6. [DOI] [PubMed] [Google Scholar]
- 7.Wyen C, Hendra H, Siccardi M, Platten M, Jaeger H, Harrer T, Esser S, Bogner JR, Brockmeyer NH, Bieniek B, Rockstroh J, Hoffmann C, Stoehr A, Michalik C, Dlugay V, Jetter A, Knechten H, Klinker H, Skaletz-Rorowski A, Fatkenheuer G, Egan D, Back DJ, Owen A, German Competence Network for HIV/ AIDS Coordinators. 2011. Cytochrome P450 2B6 (CYP2B6) and constitutive androstane receptor (CAR) polymorphisms are associated with early discontinuation of efavirenz-containing regimens. J Antimicrob Chemother 66:2092–2098. doi: 10.1093/jac/dkr272. [DOI] [PubMed] [Google Scholar]
- 8.Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. 2001. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 15:71–75. doi: 10.1097/00002030-200101050-00011. [DOI] [PubMed] [Google Scholar]
- 9.Best BM, Koopmans PP, Letendre SL, Capparelli EV, Rossi SS, Clifford DB, Collier AC, Gelman BB, Mbeo G, McCutchan JA, Simpson DM, Haubrich R, Ellis R, Grant I, CHARTER Group. 2011. Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J Antimicrob Chemother 66:354–357. doi: 10.1093/jac/dkq434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibbs JE, Gaffen Z, Thomas SA. 2006. Nevirapine uptake into the central nervous system of the guinea pig: an in situ brain perfusion study. J Pharmacol Exp Ther 317:746–751. doi: 10.1124/jpet.105.098459. [DOI] [PubMed] [Google Scholar]
- 11.Blaney SM, Daniel MJ, Harker AJ, Godwin K, Balis FM. 1995. Pharmacokinetics of lamivudine and BCH-189 in plasma and cerebrospinal fluid of nonhuman primates. Antimicrob Agents Chemother 39:2779–2782. doi: 10.1128/AAC.39.12.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen DD, Artru AA, Adkison KK. 2004. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev 56:1825–1857. doi: 10.1016/j.addr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Bosgra S, van Eijkeren J, Bos P, Zeilmaker M, Slob W. 2012. An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit Rev Toxicol 42:751–767. doi: 10.3109/10408444.2012.709225. [DOI] [PubMed] [Google Scholar]
- 14.Westerhout J, Ploeger B, Smeets J, Danhof M, de Lange EC. 2012. Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J 14:543–553. doi: 10.1208/s12248-012-9366-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yilmaz A, Watson V, Dickinson L, Back D. 2012. Efavirenz pharmacokinetics in cerebrospinal fluid and plasma over a 24-hour dosing interval. Antimicrob Agents Chemother 56:4583–4585. doi: 10.1128/AAC.06311-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tashima KT, Caliendo AM, Ahmad M, Gormley JM, Fiske WD, Brennan JM, Flanigan TP. 1999. Cerebrospinal fluid human immunodeficiency virus type 1 (HIV-1) suppression and efavirenz drug concentrations in HIV-1-infected patients receiving combination therapy. J Infect Dis 180:862–864. doi: 10.1086/314945. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez A, Cabrera S, Santos D, Valverde MP, Fuertes A, Dominguez-Gil A, Garcia MJ, Tormes G. 2011. Population pharmacokinetic/pharmacogenetic model for optimization of efavirenz therapy in Caucasian HIV-infected patients. Antimicrob Agents Chemother 55:5314–5324. doi: 10.1128/AAC.00194-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csajka C, Marzolini C, Fattinger K, Decosterd LA, Fellay J, Telenti A, Biollaz J, Buclin T. 2003. Population pharmacokinetics and effects of efavirenz in patients with human immunodeficiency virus infection. Clin Pharmacol Ther 73:20–30. doi: 10.1067/mcp.2003.22. [DOI] [PubMed] [Google Scholar]
- 19.Thompson CG, Bokhart MT, Sykes C, Adamson L, Fedoriw Y, Luciw PA, Muddiman DC, Kashuba AD, Rosen EP. 2015. Mass spectrometry imaging reveals heterogeneous efavirenz distribution within putative HIV reservoirs. Antimicrob Agents Chemother 59:2944–2948. doi: 10.1128/AAC.04952-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bumpus N, Ma Q, Best B, Moore D, Ellis RJ, Crescini M, Achim C, Fletcher C, Masliah E, Grant I, Letendre S, CNTN Group. Antiretroviral concentrations in brain tissue are similar to or exceed those in CSF, poster 436. 22nd Conf Retroviruses Opportunistic Infect, Seattle, WA, 23 to 26 February 2015. [Google Scholar]
- 21.Avery LB, Sacktor N, McArthur JC, Hendrix CW. 2013. Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: applying the law of mass action to predict protein-free drug concentration. Antimicrob Agents Chemother 57:1409–1414. doi: 10.1128/AAC.02329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siccardi M, Almond L, Schipani A, Csajka C, Marzolini C, Wyen C, Brockmeyer NH, Boffito M, Owen A, Back D. 2012. Pharmacokinetic and pharmacodynamic analysis of efavirenz dose reduction using an in vitro-in vivo extrapolation model. Clin Pharmacol Ther 92:494–502. doi: 10.1038/clpt.2012.61. [DOI] [PubMed] [Google Scholar]
- 23.Chang CY, Ke DS, Chen JY. 2009. Essential fatty acids and human brain. Acta Neurol Taiwan 18:231–241. [PubMed] [Google Scholar]
- 24.Almond LM, Hoggard PG, Edirisinghe D, Khoo SH, Back DJ. 2005. Intracellular and plasma pharmacokinetics of efavirenz in HIV-infected individuals. J Antimicrob Chemother 56:738–744. doi: 10.1093/jac/dki308. [DOI] [PubMed] [Google Scholar]
- 25.Pajouhesh H, Lenz GR. 2005. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalvass JC, Maurer TS, Pollack GM. 2007. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666. doi: 10.1124/dmd.106.012294. [DOI] [PubMed] [Google Scholar]
- 27.Siccardi M, Olagunju A, Seden K, Ebrahimjee F, Rannard S, Back D, Owen A. 2013. Use of a physiologically-based pharmacokinetic model to simulate artemether dose adjustment for overcoming the drug-drug interaction with efavirenz. In Silico Pharmacol 1:4. doi: 10.1186/2193-9616-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Tu M, Kelly RS, Chen C, Smith BJ. 2004. Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos 32:132–139. doi: 10.1124/dmd.32.1.132. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann C, van de Wetering K, van de Steeg E, Wagenaar E, Vens C, Schinkel AH. 2008. Species-dependent transport and modulation properties of human and mouse multidrug resistance protein 2 (MRP2/Mrp2, ABCC2/Abcc2). Drug Metab Dispos 36:631–640. doi: 10.1124/dmd.107.019620. [DOI] [PubMed] [Google Scholar]
- 30.Pardridge WM. 2002. Drug and gene delivery to the brain: the vascular route. Neuron 36:555–558. doi: 10.1016/S0896-6273(02)01054-1. [DOI] [PubMed] [Google Scholar]
- 31.O'Mahony SM, Myint AM, Steinbusch H, Leonard BE. 2005. Efavirenz induces depressive-like behaviour, increased stress response and changes in the immune response in rats. Neuroimmunomodulation 12:293–298. doi: 10.1159/000087107. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, Monshouwer M. 2009. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos 37:787–793. doi: 10.1124/dmd.108.024125. [DOI] [PubMed] [Google Scholar]
- 33.Kalvass JC, Maurer TS. 2002. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 34.Rajoli RK, Back DJ, Rannard S, Freel Meyers CL, Flexner C, Owen A, Siccardi M. 2015. Physiologically based pharmacokinetic modelling to inform development of intramuscular long-acting nanoformulations for HIV. Clin Pharmacokinet 54:639–650. doi: 10.1007/s40262-014-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poulin P, Theil FP. 2002. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156. [DOI] [PubMed] [Google Scholar]