

Abstract
Liver fibrosis is a reversible wound-healing process aimed at maintaining organ integrity, and presents as the critical pre-stage of liver cirrhosis, which will eventually progress to hepatocellular carcinoma in the absence of liver transplantation. Fibrosis generally results from chronic hepatic injury caused by various factors, mainly viral infection, schistosomiasis, and alcoholism; however, the exact pathological mechanisms are still unknown. Although numerous drugs have been shown to have antifibrotic activity in vitro and in animal models, none of these drugs have been shown to be efficacious in the clinic. Importantly, hepatic stellate cells (HSCs) play a key role in the initiation, progression, and regression of liver fibrosis by secreting fibrogenic factors that encourage portal fibrocytes, fibroblasts, and bone marrow-derived myofibroblasts to produce collagen and thereby propagate fibrosis. These cells are subject to intricate cross-talk with adjacent cells, resulting in scarring and subsequent liver damage. Thus, an understanding of the molecular mechanisms of liver fibrosis and their relationships with HSCs is essential for the discovery of new therapeutic targets. This comprehensive review outlines the role of HSCs in liver fibrosis and details novel strategies to suppress HSC activity, thereby providing new insights into potential treatments for liver fibrosis.
Keywords: Liver cirrhosis, Fibrosis, Hepatic stellate cells, Etiology, Pathology, Treatment
Core tip: This review discusses the molecular mechanisms of liver fibrosis with respect to hepatic stellate cells (HSCs). In particular, we describe the functional significance of HSCs with respect to major events triggering fibrosis and novel therapeutic strategies to suppress the activity of activated HSCs.
INTRODUCTION
Liver fibrosis is a complex fibrogenic and inflammatory process that results from chronic liver injury and represents an early step in the progression of liver cirrhosis. Cirrhosis is a major health problem worldwide, owing to the lack of effective treatment methods[1,2]. During hepatic fibrosis, continuous accumulation of extracellular matrix (ECM) extremely rich in collagen I and III leads to scar deposition and liver fibrosis[3,4]. When left untreated, this condition can develop into cirrhosis and subsequent portal hypertension, hepatic encephalopathy, and/or liver failure, and lead to an increased risk of hepatocellular carcinoma (HCC), which can ultimately cause organ failure and death[2,4]. Liver transplantation is currently regarded as the only treatment method for cirrhosis and is generally inadequate[3]. During chronic liver disease, ongoing liver injury results in excessive ECM deposition with limited remodeling, which inevitably leads to scarring and fibrosis[5]. In comparison, the liver can quickly re-establish its structural integrity in response to acute injury, even when a substantial portion of the organ is damaged[6].
Hepatic stellate cells (HSCs) localize to the perisinusoidal space between hepatocytes and sinusoidal endothelial cells and are the primary source of activated myofibroblasts and portal fibroblasts that drive the fibrogenic process[7]. Quiescent HSCs (qHSCs) mostly function as vitamin A reserves[8]. In response to liver injury, inflammatory mediators promote HSC activation and subsequent differentiation into myofibroblasts[9]. Activated HSCs (aHSCs) are a major source of collagen in the liver and can abundantly secrete ECM proteins, tissue inhibitors of metalloproteinases, and matrix metalloproteinases (MMPs) that elicit liver architecture remodeling[9,10]. Importantly, HSCs are responsible for as much as 80% of total fibrillar collagen I in the fibrotic liver[8-11]; thus, aHSC depletion is critical for the resolution of fibrosis.
Based on these findings, we provide a comprehensive review summarizing the etiology and pathological characteristics of hepatic fibrosis, and detail the potential therapeutic targets for suppression of aHSC function.
ETIOLOGY AND PATHOLOGICAL CHARACTERISTICS OF HEPATIC FIBROSIS
Liver fibrosis is a complex process that results from various forms of chronic hepatic disease and is associated with excess hepatocellular death[2,12,13]. The main etiologies of liver fibrosis are schistosome and chronic viral hepatitis infection, nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), and cholestatic and autoimmune liver disease[1,14-17]. Liver fibrosis, which is characterized by the excessive deposition ECM proteins[18], involves both parenchymal and nonparenchymal hepatic cells, as well as infiltrating immune cells[3,19]. Furthermore, different organs, such as the adipose tissue, bile duct, intestine, and muscle, can also affect the development of liver fibrosis. Moreover, several essential signaling pathways have important roles in fibrosis. The complex interactions among these signaling pathways, diverse cells, and different organs contribute to the progression of liver fibrosis[20]. Upon fibrogenic initiation, qHSCs differentiate into aHSCs, upon which they lose the intracellular lipid droplets and acquire a myofibroblastic phenotype characterized by marked upregulation of a-smooth muscle actin (a-SMA, ACTA2), desmin (DES), and type I collagen (COL1A1)[8-10]. The sustained buildup of collagens distorts the liver parenchyma and vascular architecture, resulting in impaired liver function, scar deposition, and liver fibrosis[1,2,12,14,17]. The initiation, progression, and resolution of liver fibrosis involving HSCs are present in Figure 1.
Figure 1.
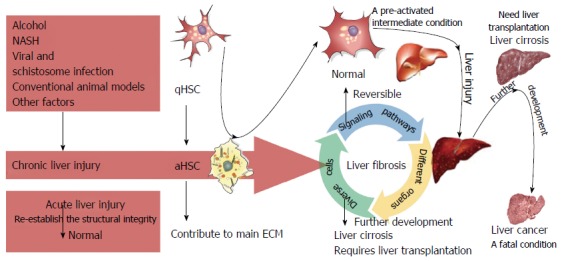
Initiation, progression, and resolution of liver fibrosis involving hepatic stellate cells. Upon various types of chronic injury - including that caused by alcohol, viral and schistosome infection, nonalcoholic steatohepatitis (NASH), and other factors - hepatic stellate cells (HSCs) transdifferentiate from quiescent HSCs to activated HSCs, the latter of which secrete abundant extracellular proteins that contribute to liver fibrosis. Liver fibrosis is thought to be a reversible condition owing to the elimination of causative agents and different strategies of limiting HSC activation; however, they cannot totally return to a quiescent status of the naive HSCs. Instead, they exhibit a pre-activated intermediate condition with an increased sensitivity to injury. Thus, preventing recurrent chronic liver injury is of great importance in patients undergoing treatment for liver fibrosis. Untreated or relapsed fibrosis progresses to liver cirrhosis, which often requires hepatic transplantation.
Viral and schistosome infection
Viral infections such as those caused by hepatitis B virus (HBV) and hepatitis C virus (HCV) induce hepatic inflammation and thereby contribute to the cyclical process of inflammation, necrosis, and regeneration[21]. Within this inflammatory microenvironment, continuous infiltration of immune cells and secreted inflammatory cytokines leads to liver injury, triggering a progressive cascade of hepatic lobule reconstruction that promotes liver fibrosis and cirrhosis[22].
Schistosomiasis is a major chronic disease that occurs in humans living in endemic regions, owing to substantial pathologic liver fibrosis caused by from an accumulation of parasitic eggs[23]. Ongoing antigenic stimulation from the trapped ova results in immune cell recruitment to the sites of infection, leading to the formation of periovular granulomas and eventual fibrosis[24]. Liver fibrosis often begins 6 wk after infection, when the Th2 immune response predominates and subsequently subsides at 12 wk postinfection. The Th17 response has also been associated with severe hepatic inflammation; however, the function of B cells in schistosome-induced pathology remains controversial. Because immune cell-derived chemokines play a vital role in schistosome-induced pathology[25,26], one method to hamper disease progression could be by modulating chemokine production to limit hepatic eosinophil recruitment[27]. Importantly, although praziquantel therapy effectively kills adult Schistosoma, it has diminutive effects on liver fibrogenesis or portal hypertension[28,29]; thus, new strategies to treat schistosomiasis are urgently needed.
Alcohol
Excessive alcohol abuse causes steatohepatitis that can progress to ALD. Most patients are generally asymptomatic, and ALD is easily reversible when patients abstain from alcohol consumption. Otherwise, they will develop into liver fibrosis. Acetaldehyde is regarded as a major intermediate in alcohol-induced fibrogenesis[30,31], and recent studies have delineated the mechanisms through which transforming growth factor (TGF)-β/small mother against decapentaplegic (SMAD) signaling is enhanced by acetaldehyde[32]. Additionally, acetaldehyde-induced fibrogenesis is also thought to involve members of the basic transcription element binding protein[33,34], CAAT/enhanced-binding protein[35,36], and acetaldehyde-responsive element[37]. Cytochrome P450 2E1 (CYP2E1) protein is a member of the microsomal ethanol oxidizing system responsible for ethanol metabolism and is crucial for alcohol-induced fibrogenesis[38]. This mechanism is readily observed in hepatocyte and HSC cocultures with enhanced collagen I protein synthesis resulting from CYP2E1-dependent reactive oxygen species generation[39]. Correspondingly, ethanol-mediated lipid peroxidation is effectively blocked in CYP2E1-/- mice[40], whereas oxidative stress and hepatic fibrogenesis is elevated in transgenic mice with CYP2E1 overexpression[41]. Moreover, the calcium regulatory protein osteopontin (OPN) has demonstrated protective effects in early alcohol-induced liver injury by binding lipopolysaccharide and blocking tumor necrosis factor-alpha (TNF-α) function in the liver[42]. OPN is also positively correlated with fibrosis in patients with ALD[43].
Nonalcoholic steatohepatitis
Nonalcoholic steatohepatitis (NASH) is a relatively common chronic liver disease with histological characteristics similar to that of ALD[44]. NASH presents as balloon-like hepatocellular injury with or without hepatic fibrosis in liver biopsies[45] and is the intermediate between NAFLD and cirrhosis[46]. NASH occurs when sustained oxidative stress prevents the proliferation of mature liver cells, resulting in excess necrosis and an overgrowth of liver progenitor cells (oval cells)[47]. In addition, the inflammatory response to cellular necrosis induces the progressive release of platelet-derived growth factor, TGF-β, TNF-α, and other inflammatory factors, such as interleukin (IL)-1, by resident immune cells[48]. These inflammatory signals result in the activation and proliferation of HSCs and induce differentiation of HSCs into myofibroblasts, further driving ECM synthesis and ultimately liver fibrosis[49].
Animal models of liver fibrogenesis
Liver fibrosis takes years to develop in most patients and results from an interplay of several risk factors, including HBV and HCV infection, alcohol abuse, and metabolic syndromes attributed to obesity, insulin resistance, and diabetes[50]. Accordingly, animal models used to study the pathophysiology of liver fibrosis, cirrhosis, and HCC should mimic the general disease patterns found in human counterparts.
Currently, in vivo models of liver fibrosis can be divided into five categories based on etiology: chemical, dietary, surgical, genetically modified, and infection[51]. The chemicals commonly used to cause hepatic lesions and induce liver fibrosis include ethanol, carbon tetrachloride (CCl4)[52], thioacetamide[53], dimethylnitrosamine[54], and diethylnitrosamine[55]. A number of specific diets, such as the methionine- and choline-deficient diet[56], high-fat diet[57], and choline-deficient L-amino acid-defined diet[58], can be used to induce progression of NAFLD to hepatic fibrosis in experimental animals. Moreover, common bile duct ligation (BDL) can also lead to cholestatic injury and periportal biliary fibrosis[59]. In the past decade, multidrug resistance-associated protein 2-deficient (Mdr2-/-) mice[60] and Alms1foz/foz fat Aussie mice[61] have been used to study the functional relevance of specific signaling pathways in the formation of liver fibrosis and identify novel drug targets. Finally, infections with HBV[62] and Schistosoma parasites[63] are also popular models of liver fibrosis.
NOVEL THERAPEUTIC TARGETS IN LIVER FIBROSIS
Liver fibrosis was once deemed irreversible; however, early liver fibrosis is now managed by clinical treatment, and overwhelming evidence suggests that advanced fibrosis may likely be reversible once the injurious stimulus is removed[64]. Since aHSCs are the primary mediators of liver pathology in this process, several molecules required for HSC activation are considered potential therapeutic targets[9,64,65]. The following section details recent novel targets identified for the treatment of liver fibrosis through suppression of HSC activation.
Key molecules in liver fibrosis
Mitra and colleagues reported that IL-30 attenuates hepatic fibrosis by inducing natural killer group 2D (NKG2D)/ribonucleic acid export 1 crosstalk between aHSCs and natural killer T (NKT) cells and is therefore an ideal therapy for liver fibrosis. Mechanistically, IL-30 treatment promotes surface NKG2D expression on liver NKT cells to subsequently enhance their cytotoxic activity towards aHSCs, thereby inhibiting liver fibrosis[66]. Another molecule, hydrogen peroxide-inducible clone-5 (Hic-5) is a TGF-β1-inducible focal adhesion protein that facilitates cell proliferation and ECM expansion in various organs[67]. Previous studies have shown that Hic-5 contributes to vascular restoration and restructuring[67,68]; however, a recent study revealed that Hic-5 expression also plays a critical role in attenuating fibrosis by enhancing TGF-β-induced Smad2 phosphorylation via the downregulation of Smad7 in both human and mouse aHSCs[69]. Taken together, these data indicate that Hic-5 is a novel therapeutic target and a potential marker of activated HSCs. Additionally, acyl-coenzyme A: cholesterol acyltransferase (ACAT) is comprised of two isoenzymes-ACAT1 and ACAT2-and functions as a catalyst to convert free cholesterol (FC) to cholesteryl esters[70]. FC accumulation has been shown to regulate HSC activation and the development of liver fibrosis by promoting Toll-like receptor 4 signal transduction. Because ACAT1 plays an essential role in regulating FC accumulation in HSCs[71], studies have focused on developing new ACAT1-directed therapeutic interventions for the treatment of liver fibrosis. The roles of IL-30, Hic-5, and ACAT1 in liver fibrosis are presented in Figure 2.
Figure 2.
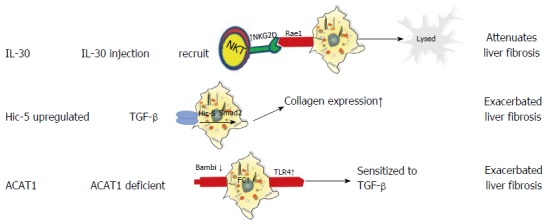
Roles of interleukin-30, hydrogen peroxide inducible clone 5, and cholesterol acyltransferase 1 in liver fibrosis.
Regulatory CD4+ T cells
Regulatory T (Treg) cells function to modulate HCV-dependent liver fibrosis by regulating the interaction between NK cells and aHSCs[72,73]. Specifically, Treg cells act in a cell-contact-dependent manner to reduce NK cell activity against HSCs and downregulate vital NKT-activating ligands on HSCs by secreting soluble IL-8 and/or TGF-β1[73]. This mechanism may also be present in fibrosis, resulting from other etiologies; however, further studies are needed to confirm this hypothesis.
Macrophages
Macrophages, which can be classified as M1 (classically activated) macrophages and M2 (alternatively activated) macrophages, play dual roles in the progression and resolution of liver fibrosis. Typically, M1 macrophages produce inflammatory cytokines, whereas M2 macrophages regulate inflammatory responses and tissue repair. The imbalance of M1 and M2 macrophages mediates the progression and resolution of liver fibrosis[74]. During the early stages of liver injury, bone marrow-derived monocytes are extensively recruited to the liver and then differentiate into inflammatory macrophages (mostly M1 macrophages) to produce pro-inflammatory and profibrotic cytokines, thereby promoting inflammatory responses and HSC activation. Afterwards, recruited macrophages switch their phenotypic (mostly M2 macrophages) to secrete MMPs, the main enzymes degrading ECM, to facilitate fibrosis resolution[20,75,76].
Role of signal transduction in the progression of liver fibrosis
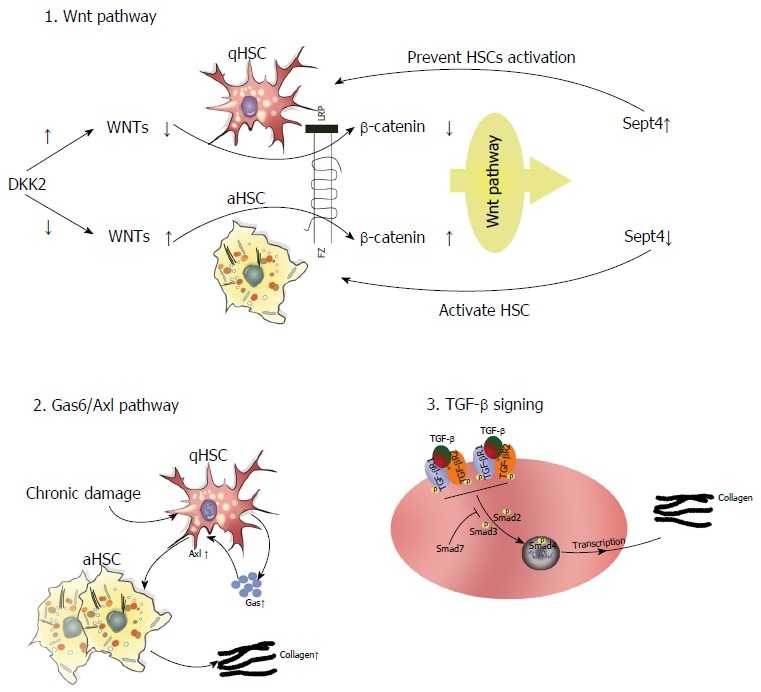
Several intracellular signaling pathways are involved in the pathophysiology of liver fibrosis. In this section, we detail the functional significance of three key signaling axes in this process: Gas6/Axl, TGF-β/Smad, and target of Wnt signaling pathway (Figure 3).
Figure 3.
Roles of the Wnt, TGF-β/Smad, and Gas6/Axl signaling pathways in the progression of liver fibrosis.
Gas6/Axl pathway: The TAM (Tyro3, Axl, Mer) receptor ligand Gas6 is a vitamin K-dependent protein with an extremely high affinity for the Axl receptor. Gas6 is primarily expressed by Kupffer cells, whereas Axl is found in both macrophages and qHSCs in the normal liver[77,78]. Study has demonstrated that CCl4-induced liver fibrosis elicits Gas6/Axl pathway activation to promote HSC activation. Notably, Axl knockout disrupts this pathway, thereby attenuating hepatic fibrosis[78]. Clinical trials have also shown increased Gas6 and Axl serum levels in patients with HCV infection and ALD[78]. As such, targeting Axl may be a potential method to remediate liver fibrosis.
TGF-β/Smad signaling: TGF-β regulates ECM metabolism and tissue fibrosis through the overproduction of type I collagen in both mice and humans. Recent studies have demonstrated that TGF-β/Smad signaling plays a crucial role in the progression of hepatic fibrosis caused by parasitic infection, including Schistosoma, Clonorchis sinensis, and Echinococcus multilocularis, as well as other etiological factors[79,80]. More specifically, TGF-β1 ligation to TGF-β type I (TGFβRI) and type II receptors induces Smad2/3 phosphorylation and its subsequent interaction with Smad4. The Smad2/3/4 complex can then translocate to the nucleus and induce the expression of profibrotic genes, namely collagen type I. Strikingly, Smad7 can block TGF-β signaling through various means[80-82], such as binding TGFβRI to inhibit the interaction-dependent activation of Smad2, collaborating with other effectors to induce TGFβRI degradation, and regulating the Wnt/β-catenin pathway to influence TGF-β-induced apoptosis[83]. Similarly, targeting of Smad7 enhances TGF-β pathway activation[84].
Wnt pathway: Several studies have demonstrated that aberrant Wnt/β-catenin signaling affects the progression of fibrotic disorders. Wnt comprises an evolutionarily conserved family of excreted lipid-modified glycoproteins that can be classified into at least three signaling pathways: Necdin-Wnt, noncanonical (β-catenin-independent), and canonical (β-catenin-dependent). In the Necdin-Wnt pathway, HSC activation and differentiation require the downregulation of peroxisome proliferator-activated receptor γ (PPARγ). Necdin is a melanoma antigen family protein preferentially expressed in aHSCs that promotes myogenic and neuronal differentiation while suppressing adipogenesis. Notably, Necdin silencing restores PPARγ-mediated Wnt pathway inhibition to effectively reverse HSC activation[85,86]. In the canonical pathway, Wnt ligation to cell surface receptors elicits downstream signaling that stabilizes β-catenin, which can then translocate into the nucleus, bind T cell factor/lymphoid enhancer-binding factor (TCF/LEF) promoter, and induce gene expression to exert biological effects[87,88]. Alternatively, noncanonical Wnt signaling occurs via the β-catenin-independent planar cell polarity and noncanonical Wnt/Ca2+ pathways. Thus, a collective understanding of Wnt signaling mechanisms may provide novel insights into the pathophysiology of liver fibrosis. A recent study also showed that DKK2 (a Wnt antagonist and target of the Wnt pathway) connects Sept4 (a subunit of the septin cytoskeleton expressed in qHSCs) and the activation of HSCs, thereby mediating the progression of liver fibrosis. The expression of DKK2 is high in primary cultured HSCs. However, DKK2 expression is reduced when Sept is not expressed in a mice model of CCL4-induced fibrosis. The high expression of DKK2 in qHSCs inhibits Wnts and thereby affects downstream β-catenin signaling. This results in suppression of the Wnt signaling pathway, leading to increased expression of Sept4 and preventing HSC activation[87].
HAb18G/CD147: HAb18G/CD147 is induced by TGF-β1 stimulation and is highly expressed on sinusoidal aHSCs, where it colocalizes with a-SMA. Transient transfection of CD147 in LX-2 cells results in increased expression of mRNAs encoding α-SMA, TIMP-1, α1(I) collagen, and TGF-β1. In contrast, MMP-13 and MMP-2 levels are markedly reduced, suggesting that HAb18G/CD147 promotes HSC activation. Consistent with this, HAb18G/CD147-targeting antibodies block HSC activation, thereby inhibiting liver fibrogenesis[89]. These data support the potential role for HAb18G/CD147 in liver fibrosis; however, further studies are needed to confirm these findings.
microRNAs and HSCs in liver fibrosis
Recently, microRNAs (miRNAs) have also been found to play multifaceted roles in hepatic fibrosis, including those in HSC activation and proliferation and production of ECM proteins[3,11]. Previous studies have indicated that human and murine miRNAs participate in liver fibrosis. For example, miR-199a, antisense miR-199a*, miR-200a, and miR-200b are dramatically upregulated in a mouse model of liver fibrosis[90]. Conversely, the miR-29 family is downregulated in aHSCs when compared with that in qHSCs, both in vivo and in vitro[91].
miR-133a is specifically downregulated in HSCs during fibrogenesis, but is overexpressed in primary murine HSC, resulting in attenuation of collagen expression[91]. Similarly, CCL4-induced miR-122 expression is markedly lower in aHSCs and fibrotic liver tissue. Cell experiments have also shown that miR-133a overexpression inhibits both LX2 and primary murine HSC proliferation and prevents the progression of liver fibrosis[92-94]. Furthermore, both miR-15b and miR-16 facilitate qHSC apoptosis by targeting Bcl-2 and the caspase signaling cascade[95].
Promising therapies for liver fibrosis
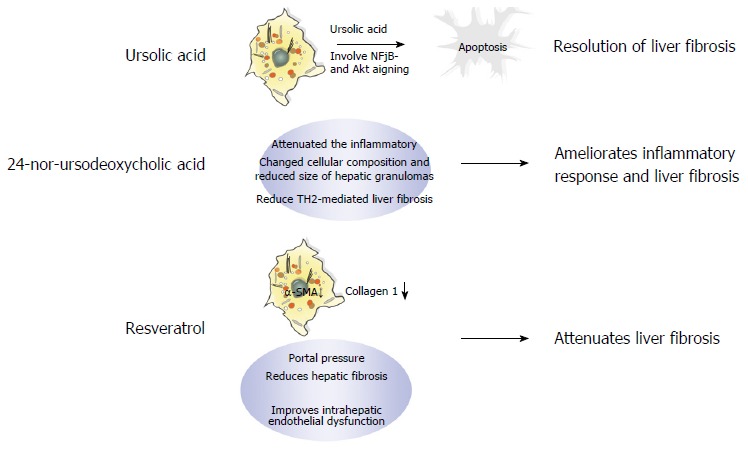
Although several antifibrotic drug candidates have recently been evaluated, these drugs have failed to show increased therapeutic efficacy over those drugs currently used in the clinic, e.g., ursolic acid (UA), 24-nor-ursodeoxycholic acid (norUDCA), and resveratrol. UA is a pentacyclic triterpenoid compound with a wide spectrum of pharmacological activities found in various edible fruits and medicinal plants. Studies have demonstrated that UA induces apoptotic culture-activated HSC death due to inhibition of nuclear factor kappa B and AKT in HSCs, but not in isolated qHSCs in vitro. In addition, UA alleviates liver fibrosis induced by both BDL and chronic thioacetamide administration in vivo. As shown in Figure 4, the mechanism of UA-induced apoptosis may be attributed to its suppression of cell survival pathways and the activation of downstream caspases via the mitochondrial permeability transition[96].
Figure 4.
Mechanism of action of three potential therapeutic drugs-ursolic acid, 24-nor-ursodeoxycholic acid, and resveratrol-for treating fibrosis.
The bile acid derivative norUDCA is a promising new treatment option for liver fibrosis that significantly reduces liver fibrosis in chronically infected Schistosoma mansoni mice by limiting T-cell proliferation and IL-13 and IL-4 serum levels (Figure 4). Moreover, norUDCA has anti-inflammatory properties demonstrated by the low expression of MHC class II on dendritic cells and macrophages after norUDCA treatment[28].
Finally, the natural polyphenol flavonoid resveratrol has a broad range of beneficial biological functions, including anti-inflammatory[97] and antioxidant[98] properties[99]. In addition, resveratrol is believed to ameliorate obesity-related complications by mimicking caloric restriction[100] through activation of key metabolic regulators, including NAD+-dependent deacetylase (SIRT1)[101], AMP-activated protein kinase[102], and nuclear factor erythroid-2 related factor 2[103]. Furthermore, oxidative damage and inflammation are closely related to the HSC activation process. For example, SIRT1 activation inhibits the expression of muscle-related genes, such as MyoD[104]. Moreover, studies have demonstrated the beneficial effects of resveratrol in different models of liver steatosis[105-108]. Superoxide dismutase activity is necessary for the reduction of oxygen free radicals and protects against lipid peroxidation, thereby inhibiting HSC activation and limiting the progression of liver fibrosis[109]. The mechanisms through which resveratrol alleviates fibrosis are shown in Figure 4. Although resveratrol has been shown to have beneficial biological functions in the antifibrotic response, its efficacy in NAFLD is insignificant; indeed, a meta-analysis conducted by Zhang et al[110] indicated that resveratrol can only improve LDL and total cholesterol levels in patients with NAFLD.
CONCLUSION
In this review, we outlined some major etiological and pathological characteristics of hepatic fibrosis and described several promising approaches for liver fibrosis therapy. We strongly believe that liver fibrosis will be cured through the combined application of these therapeutics; however, further studies are necessary to support this hypothesis.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Peer-review started: August 26, 2016
First decision: September 29, 2016
Article in press: November 16, 2016
P- Reviewer: Chow WK S- Editor: Qi Y L- Editor: A E- Editor: Zhang FF
References
- 1.Weiskirchen R, Tacke F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig Dis. 2016;34:410–422. doi: 10.1159/000444556. [DOI] [PubMed] [Google Scholar]
- 2.Pinzani M. Pathophysiology of Liver Fibrosis. Dig Dis. 2015;33:492–497. doi: 10.1159/000374096. [DOI] [PubMed] [Google Scholar]
- 3.van Dijk F, Olinga P, Poelstra K, Beljaars L. Targeted Therapies in Liver Fibrosis: Combining the Best Parts of Platelet-Derived Growth Factor BB and Interferon Gamma. Front Med (Lausanne) 2015;2:72. doi: 10.3389/fmed.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63:1038–1039. doi: 10.1016/j.jhep.2015.03.039. [DOI] [PubMed] [Google Scholar]
- 5.Schuppan D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin Res Hepatol Gastroenterol. 2015;39 Suppl 1:S51–S59. doi: 10.1016/j.clinre.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Nicolas CT, Wang Y, Nyberg SL. Cell therapy in chronic liver disease. Curr Opin Gastroenterol. 2016;32:189–194. doi: 10.1097/MOG.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josan S, Billingsley K, Orduna J, Park JM, Luong R, Yu L, Hurd R, Pfefferbaum A, Spielman D, Mayer D. Assessing inflammatory liver injury in an acute CCl4 model using dynamic 3D metabolic imaging of hyperpolarized [1-(13)C]pyruvate. NMR Biomed. 2015;28:1671–1677. doi: 10.1002/nbm.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lepreux S, Desmoulière A. Human liver myofibroblasts during development and diseases with a focus on portal (myo)fibroblasts. Front Physiol. 2015;6:173. doi: 10.3389/fphys.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D, He L, Guo H, Chen H, Shan H. Targeting activated hepatic stellate cells (aHSCs) for liver fibrosis imaging. EJNMMI Res. 2015;5:71. doi: 10.1186/s13550-015-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–1492. doi: 10.1002/cphy.c120035. [DOI] [PubMed] [Google Scholar]
- 11.Rao HY, Wei L, Li J, Zhang LF, Chen HY, Zhu LM, Liu F, Sun Y, Wang H. Liver fibrosis and hepatic stellate cells improvement of chronic hepatitis C patients by interferon-beta-1a with or without sustained viral response. Hepatogastroenterology. 2009;56:328–334. [PubMed] [Google Scholar]
- 12.Kitano M, Bloomston PM. Hepatic Stellate Cells and microRNAs in Pathogenesis of Liver Fibrosis. J Clin Med. 2016;5:pii. E38. doi: 10.3390/jcm5030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elpek GÖ. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol. 2014;20:7260–7276. doi: 10.3748/wjg.v20.i23.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poelstra K. Liver fibrosis in 2015: Crucial steps towards an effective treatment. Nat Rev Gastroenterol Hepatol. 2016;13:67–68. doi: 10.1038/nrgastro.2015.224. [DOI] [PubMed] [Google Scholar]
- 15.Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. 2015;22:512–518. doi: 10.1002/jhbp.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid P, Bregenzer A, Huber M, Rauch A, Jochum W, Müllhaupt B, Vernazza P, Opravil M, Weber R. Progression of Liver Fibrosis in HIV/HCV Co-Infection: A Comparison between Non-Invasive Assessment Methods and Liver Biopsy. PLoS One. 2015;10:e0138838. doi: 10.1371/journal.pone.0138838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacchi P, Cima S, Corbella M, Comolli G, Chiesa A, Baldanti F, Klersy C, Novati S, Mulatto P, Mariconti M, et al. Liver fibrosis, microbial translocation and immune activation markers in HIV and HCV infections and in HIV/HCV co-infection. Dig Liver Dis. 2015;47:218–225. doi: 10.1016/j.dld.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Baiocchini A, Montaldo C, Conigliaro A, Grimaldi A, Correani V, Mura F, Ciccosanti F, Rotiroti N, Brenna A, Montalbano M, et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS One. 2016;11:e0151736. doi: 10.1371/journal.pone.0151736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toosi AE. Liver Fibrosis: Causes and Methods of Assessment, A Review. Rom J Intern Med. 2015;53:304–314. doi: 10.1515/rjim-2015-0039. [DOI] [PubMed] [Google Scholar]
- 20.Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gastroenterol. 2015;39 Suppl 1:S60–S63. doi: 10.1016/j.clinre.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagao Y, Kawahigashi Y, Sata M. Association of Periodontal Diseases and Liver Fibrosis in Patients With HCV and/or HBV infection. Hepat Mon. 2014;14:e23264. doi: 10.5812/hepatmon.23264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin H, Ha NB, Ahmed A, Ayoub W, Daugherty TJ, Lutchman GA, Garcia G, Nguyen MH. Both HCV and HBV are major causes of liver cancer in Southeast Asians. J Immigr Minor Health. 2013;15:1023–1029. doi: 10.1007/s10903-013-9871-z. [DOI] [PubMed] [Google Scholar]
- 23.Zoni AC, Catalá L, Ault SK. Schistosomiasis Prevalence and Intensity of Infection in Latin America and the Caribbean Countries, 1942-2014: A Systematic Review in the Context of a Regional Elimination Goal. PLoS Negl Trop Dis. 2016;10:e0004493. doi: 10.1371/journal.pntd.0004493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Immunity to schistosomiasis. Lancet. 1987;1:1015–1016. [PubMed] [Google Scholar]
- 25.al-Fawaz IM, al-Rasheed SA, al-Majed SA, Ashour M. Schistosomiasis associated with a mediastinal mass: case report and review of the literature. Ann Trop Paediatr. 1990;10:293–297. doi: 10.1080/02724936.1990.11747445. [DOI] [PubMed] [Google Scholar]
- 26.Immunopathology of schistosomiasis. Lancet. 1987;2:194–195. [PubMed] [Google Scholar]
- 27.Chuah C, Jones MK, Burke ML, McManus DP, Gobert GN. Cellular and chemokine-mediated regulation in schistosome-induced hepatic pathology. Trends Parasitol. 2014;30:141–150. doi: 10.1016/j.pt.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Sombetzki M, Fuchs CD, Fickert P, Österreicher CH, Mueller M, Claudel T, Loebermann M, Engelmann R, Langner C, Sahin E, et al. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J Hepatol. 2015;62:871–878. doi: 10.1016/j.jhep.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Praticò L, Mariani B, Brunetti E, Maserati R, Bruno A, Novati S, Chichino G. Failure of repeated treatment with praziquantel and arthemeter in four patients with acute schistosomiasis. J Travel Med. 2014;21:133–136. doi: 10.1111/jtm.12098. [DOI] [PubMed] [Google Scholar]
- 30.Moshage H, Casini A, Lieber CS. Acetaldehyde selectively stimulates collagen production in cultured rat liver fat-storing cells but not in hepatocytes. Hepatology. 1990;12:511–518. doi: 10.1002/hep.1840120311. [DOI] [PubMed] [Google Scholar]
- 31.Casini A, Cunningham M, Rojkind M, Lieber CS. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology. 1991;13:758–765. [PubMed] [Google Scholar]
- 32.Svegliati-Baroni G, Inagaki Y, Rincon-Sanchez AR, Else C, Saccomanno S, Benedetti A, Ramirez F, Rojkind M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42:343–352. doi: 10.1002/hep.20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anania FA, Potter JJ, Rennie-Tankersley L, Mezey E. Effects of acetaldehyde on nuclear protein binding to the nuclear factor I consensus sequence in the alpha 2(I) collagen promoter. Hepatology. 1995;21:1640–1648. [PubMed] [Google Scholar]
- 34.Attard FA, Wang L, Potter JJ, Rennie-Tankersley L, Mezey E. CCAAT/enhancer binding protein beta mediates the activation of the murine alpha1(I) collagen promoter by acetaldehyde. Arch Biochem Biophys. 2000;378:57–64. doi: 10.1006/abbi.2000.1803. [DOI] [PubMed] [Google Scholar]
- 35.García-Trevijano ER, Iraburu MJ, Fontana L, Domínguez-Rosales JA, Auster A, Covarrubias-Pinedo A, Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 36.Greenwel P, Domínguez-Rosales JA, Mavi G, Rivas-Estilla AM, Rojkind M. Hydrogen peroxide: a link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology. 2000;31:109–116. doi: 10.1002/hep.510310118. [DOI] [PubMed] [Google Scholar]
- 37.Svegliati-Baroni G, Ridolfi F, Di Sario A, Saccomanno S, Bendia E, Benedetti A, Greenwel P. Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology. 2001;33:1130–1140. doi: 10.1053/jhep.2001.23788. [DOI] [PubMed] [Google Scholar]
- 38.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bell LN, Temm CJ, Saxena R, Vuppalanchi R, Schauer P, Rabinovitz M, Krasinskas A, Chalasani N, Mattar SG. Bariatric surgery-induced weight loss reduces hepatic lipid peroxidation levels and affects hepatic cytochrome P-450 protein content. Ann Surg. 2010;251:1041–1048. doi: 10.1097/SLA.0b013e3181dbb572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butura A, Nilsson K, Morgan K, Morgan TR, French SW, Johansson I, Schuppe-Koistinen I, Ingelman-Sundberg M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J Hepatol. 2009;50:572–583. doi: 10.1016/j.jhep.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 41.Patouraux S, Bonnafous S, Voican CS, Anty R, Saint-Paul MC, Rosenthal-Allieri MA, Agostini H, Njike M, Barri-Ova N, Naveau S, et al. The osteopontin level in liver, adipose tissue and serum is correlated with fibrosis in patients with alcoholic liver disease. PLoS One. 2012;7:e35612. doi: 10.1371/journal.pone.0035612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagoshi S. Osteopontin: Versatile modulator of liver diseases. Hepatol Res. 2014;44:22–30. doi: 10.1111/hepr.12166. [DOI] [PubMed] [Google Scholar]
- 43.Morales-Ibanez O, Domínguez M, Ki SH, Marcos M, Chaves JF, Nguyen-Khac E, Houchi H, Affò S, Sancho-Bru P, Altamirano J, et al. Human and experimental evidence supporting a role for osteopontin in alcoholic hepatitis. Hepatology. 2013;58:1742–1756. doi: 10.1002/hep.26521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–1719. [PubMed] [Google Scholar]
- 45.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 46.de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48 Suppl 1:S104–S112. doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 47.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 48.Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Devière J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 49.Fujii H, Kawada N. Inflammation and fibrogenesis in steatohepatitis. J Gastroenterol. 2012;47:215–225. doi: 10.1007/s00535-012-0527-x. [DOI] [PubMed] [Google Scholar]
- 50.Blachier M, Leleu H, Peck-Radosavljevic M, Valla DC, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58:593–608. doi: 10.1016/j.jhep.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 51.Crespo Yanguas S, Cogliati B, Willebrords J, Maes M, Colle I, van den Bossche B, de Oliveira CP, Andraus W, Alves VA, Leclercq I, et al. Experimental models of liver fibrosis. Arch Toxicol. 2016;90:1025–1048. doi: 10.1007/s00204-015-1543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan RA, Khan MR, Sahreen S, Ahmed M, Shah NA. Carbon tetrachloride-induced lipid peroxidation and hyperglycemia in rat: a novel study. Toxicol Ind Health. 2015;31:546–553. doi: 10.1177/0748233713475503. [DOI] [PubMed] [Google Scholar]
- 53.Wallace MC, Hamesch K, Lunova M, Kim Y, Weiskirchen R, Strnad P, Friedman SL. Standard operating procedures in experimental liver research: thioacetamide model in mice and rats. Lab Anim. 2015;49:21–29. doi: 10.1177/0023677215573040. [DOI] [PubMed] [Google Scholar]
- 54.Oh SW, Kim DH, Ha JR, Kim DY. Anti-fibrotic effects of a methylenedioxybenzene compound, CW209292 on dimethylnitrosamine-induced hepatic fibrosis in rats. Biol Pharm Bull. 2009;32:1364–1370. doi: 10.1248/bpb.32.1364. [DOI] [PubMed] [Google Scholar]
- 55.Jin N, Deng J, Chadashvili T, Zhang Y, Guo Y, Zhang Z, Yang GY, Omary RA, Larson AC. Carbogen gas-challenge BOLD MR imaging in a rat model of diethylnitrosamine-induced liver fibrosis. Radiology. 2010;254:129–137. doi: 10.1148/radiol.09090410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jha P, Knopf A, Koefeler H, Mueller M, Lackner C, Hoefler G, Claudel T, Trauner M. Role of adipose tissue in methionine-choline-deficient model of non-alcoholic steatohepatitis (NASH) Biochim Biophys Acta. 2014;1842:959–970. doi: 10.1016/j.bbadis.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, Hirose H, Ito M, Ishihara A, Iwaasa H, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37:50–57. doi: 10.1111/j.1872-034X.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 58.Denda A, Kitayama W, Kishida H, Murata N, Tsutsumi M, Tsujiuchi T, Nakae D, Konishi Y. Development of hepatocellular adenomas and carcinomas associated with fibrosis in C57BL/6J male mice given a choline-deficient, L-amino acid-defined diet. Jpn J Cancer Res. 2002;93:125–132. doi: 10.1111/j.1349-7006.2002.tb01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park KC, Park JH, Jeon JY, Kim SY, Kim JM, Lim CY, Lee TH, Kim HK, Lee HG, Kim SM, et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br J Pharmacol. 2014;171:4820–4830. doi: 10.1111/bph.12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 61.Arsov T, Larter CZ, Nolan CJ, Petrovsky N, Goodnow CC, Teoh NC, Yeh MM, Farrell GC. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun. 2006;342:1152–1159. doi: 10.1016/j.bbrc.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 62.Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, Esposito A, Ruggeri ZM, Chisari FV, Iannacone M, et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci USA. 2012;109:E2165–E2172. doi: 10.1073/pnas.1209182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Huang D, Gao W, Yan J, Zhou W, Hou X, Liu M, Ren C, Wang S, Shen J. Lack of IL-17 signaling decreases liver fibrosis in murine schistosomiasis japonica. Int Immunol. 2015;27:317–325. doi: 10.1093/intimm/dxv017. [DOI] [PubMed] [Google Scholar]
- 64.Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol. 2015;62:S15–S24. doi: 10.1016/j.jhep.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 65.Mallat A, Lotersztajn S. Reversion of hepatic stellate cell to a quiescent phenotype: From myth to reality? J Hepatol. 2013;59:383–386. doi: 10.1016/j.jhep.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 66.Mitra A, Satelli A, Yan J, Xueqing X, Gagea M, Hunter CA, Mishra L, Li S. IL-30 (IL27p28) attenuates liver fibrosis through inducing NKG2D-rae1 interaction between NKT and activated hepatic stellate cells in mice. Hepatology. 2014;60:2027–2039. doi: 10.1002/hep.27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim-Kaneyama JR, Lei XF, Arita S, Miyauchi A, Miyazaki T, Miyazaki A. Hydrogen peroxide-inducible clone 5 (Hic-5) as a potential therapeutic target for vascular and other disorders. J Atheroscler Thromb. 2012;19:601–607. doi: 10.5551/jat.10736. [DOI] [PubMed] [Google Scholar]
- 68.Wu JR, Hu CT, You RI, Pan SM, Cheng CC, Lee MC, Wu CC, Chang YJ, Lin SC, Chen CS, et al. Hydrogen peroxide inducible clone-5 mediates reactive oxygen species signaling for hepatocellular carcinoma progression. Oncotarget. 2015;6:32526–32544. doi: 10.18632/oncotarget.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lei XF, Fu W, Kim-Kaneyama JR, Omoto T, Miyazaki T, Li B, Miyazaki A. Hic-5 deficiency attenuates the activation of hepatic stellate cells and liver fibrosis through upregulation of Smad7 in mice. J Hepatol. 2016;64:110–117. doi: 10.1016/j.jhep.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 70.Billheimer JT, Cromley DA, Kempner ES. The functional size of acyl-coenzyme A (CoA): cholesterol acyltransferase and acyl-CoA hydrolase as determined by radiation inactivation. J Biol Chem. 1990;265:8632–8635. [PubMed] [Google Scholar]
- 71.Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Usui S, Furuhashi H, Kimura A, Nishiyama K, et al. Acyl-CoA: cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. 2014;61:98–106. doi: 10.1016/j.jhep.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 72.Xue Y, Michalopoulos G. Tregs: A Therapeutic Target for the Treatment of Portal Fibrosis? Dig Dis Sci. 2015;60:1878–1880. doi: 10.1007/s10620-015-3687-8. [DOI] [PubMed] [Google Scholar]
- 73.Glässner A, Eisenhardt M, Kokordelis P, Krämer B, Wolter F, Nischalke HD, Boesecke C, Sauerbruch T, Rockstroh JK, Spengler U, et al. Impaired CD4+ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J Hepatol. 2013;59:427–433. doi: 10.1016/j.jhep.2013.04.029. [DOI] [PubMed] [Google Scholar]
- 74.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–1473. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, Stavitsky AB, Bucala R, Nagy LE. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J Leukoc Biol. 2015;97:161–169. doi: 10.1189/jlb.3A0614-280R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D, Baeck C, Hittatiya K, Eulberg D, Luedde T, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 2014;63:1960–1971. doi: 10.1136/gutjnl-2013-306294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsai WB, Long Y, Kuo MT. Gas6/Axl in arginine-starvation therapy. Oncoscience. 2015;2:659–660. doi: 10.18632/oncoscience.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bárcena C, Stefanovic M, Tutusaus A, Joannas L, Menéndez A, García-Ruiz C, Sancho-Bru P, Marí M, Caballeria J, Rothlin CV, et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J Hepatol. 2015;63:670–678. doi: 10.1016/j.jhep.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan C, Wang L, Li B, Zhang BB, Zhang B, Wang YH, Li XY, Chen JX, Tang RX, Zheng KY. The expression dynamics of transforming growth factor-β/Smad signaling in the liver fibrosis experimentally caused by Clonorchis sinensis. Parasit Vectors. 2015;8:70. doi: 10.1186/s13071-015-0675-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Calabrese F, Valente M, Giacometti C, Pettenazzo E, Benvegnu L, Alberti A, Gatta A, Pontisso P. Parenchymal transforming growth factor beta-1: its type II receptor and Smad signaling pathway correlate with inflammation and fibrosis in chronic liver disease of viral etiology. J Gastroenterol Hepatol. 2003;18:1302–1308. doi: 10.1046/j.1440-1746.2003.03162.x. [DOI] [PubMed] [Google Scholar]
- 81.Qu Y, Zong L, Xu M, Dong Y, Lu L. Effects of 18α-glycyrrhizin on TGF-β1/Smad signaling pathway in rats with carbon tetrachloride-induced liver fibrosis. Int J Clin Exp Pathol. 2015;8:1292–1301. [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JH, Jang EJ, Seo HL, Ku SK, Lee JR, Shin SS, Park SD, Kim SC, Kim YW. Sauchinone attenuates liver fibrosis and hepatic stellate cell activation through TGF-β/Smad signaling pathway. Chem Biol Interact. 2014;224:58–67. doi: 10.1016/j.cbi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 83.Luo L, Li N, Lv N, Huang D. SMAD7: a timer of tumor progression targeting TGF-β signaling. Tumour Biol. 2014;35:8379–8385. doi: 10.1007/s13277-014-2203-7. [DOI] [PubMed] [Google Scholar]
- 84.Wang ZH, Zhang QS, Duan YL, Zhang JL, Li GF, Zheng DL. TGF-β induced miR-132 enhances the activation of TGF-β signaling through inhibiting SMAD7 expression in glioma cells. Biochem Biophys Res Commun. 2015;463:187–192. doi: 10.1016/j.bbrc.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 85.Yu F, Lu Z, Huang K, Wang X, Xu Z, Chen B, Dong P, Zheng J. MicroRNA-17-5p-activated Wnt/β-catenin pathway contributes to the progression of liver fibrosis. Oncotarget. 2016;7:81–93. doi: 10.18632/oncotarget.6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma ZG, Lv XD, Zhan LL, Chen L, Zou QY, Xiang JQ, Qin JL, Zhang WW, Zeng ZJ, Jin H, et al. Human urokinase-type plasminogen activator gene-modified bone marrow-derived mesenchymal stem cells attenuate liver fibrosis in rats by down-regulating the Wnt signaling pathway. World J Gastroenterol. 2016;22:2092–2103. doi: 10.3748/wjg.v22.i6.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, Lv XW, Jin Y, Li J. Wnt signaling in liver fibrosis: progress, challenges and potential directions. Biochimie. 2013;95:2326–2335. doi: 10.1016/j.biochi.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 88.Li W, Zhu C, Li Y, Wu Q, Gao R. Mest attenuates CCl4-induced liver fibrosis in rats by inhibiting the Wnt/β-catenin signaling pathway. Gut Liver. 2014;8:282–291. doi: 10.5009/gnl.2014.8.3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang DW, Zhao YX, Wei D, Li YL, Zhang Y, Wu J, Xu J, Chen C, Tang H, Zhang W, et al. HAb18G/CD147 promotes activation of hepatic stellate cells and is a target for antibody therapy of liver fibrosis. J Hepatol. 2012;57:1283–1291. doi: 10.1016/j.jhep.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 90.Murakami Y, Toyoda H, Tanaka M, Kuroda M, Harada Y, Matsuda F, Tajima A, Kosaka N, Ochiya T, Shimotohno K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One. 2011;6:e16081. doi: 10.1371/journal.pone.0016081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He Y, Huang C, Lin X, Li J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie. 2013;95:1355–1359. doi: 10.1016/j.biochi.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 92.Roderburg C, Luedde M, Vargas Cardenas D, Vucur M, Mollnow T, Zimmermann HW, Koch A, Hellerbrand C, Weiskirchen R, Frey N, et al. miR-133a mediates TGF-β-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J Hepatol. 2013;58:736–742. doi: 10.1016/j.jhep.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 93.Halász T, Horváth G, Pár G, Werling K, Kiss A, Schaff Z, Lendvai G. miR-122 negatively correlates with liver fibrosis as detected by histology and FibroScan. World J Gastroenterol. 2015;21:7814–7823. doi: 10.3748/wjg.v21.i25.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen WJ, Dong R, Chen G, Zheng S. microRNA-222 modulates liver fibrosis in a murine model of biliary atresia. Biochem Biophys Res Commun. 2014;446:155–159. doi: 10.1016/j.bbrc.2014.02.065. [DOI] [PubMed] [Google Scholar]
- 95.Guo CJ, Pan Q, Li DG, Sun H, Liu BW. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J Hepatol. 2009;50:766–778. doi: 10.1016/j.jhep.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 96.Wang X, Ikejima K, Kon K, Arai K, Aoyama T, Okumura K, Abe W, Sato N, Watanabe S. Ursolic acid ameliorates hepatic fibrosis in the rat by specific induction of apoptosis in hepatic stellate cells. J Hepatol. 2011;55:379–387. doi: 10.1016/j.jhep.2010.10.040. [DOI] [PubMed] [Google Scholar]
- 97.Das S, Das DK. Anti-inflammatory responses of resveratrol. Inflamm Allergy Drug Targets. 2007;6:168–173. doi: 10.2174/187152807781696464. [DOI] [PubMed] [Google Scholar]
- 98.Schmatz R, Perreira LB, Stefanello N, Mazzanti C, Spanevello R, Gutierres J, Bagatini M, Martins CC, Abdalla FH, Daci da Silva Serres J, et al. Effects of resveratrol on biomarkers of oxidative stress and on the activity of delta aminolevulinic acid dehydratase in liver and kidney of streptozotocin-induced diabetic rats. Biochimie. 2012;94:374–383. doi: 10.1016/j.biochi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 99.Di Pascoli M, Diví M, Rodríguez-Vilarrupla A, Rosado E, Gracia-Sancho J, Vilaseca M, Bosch J, García-Pagán JC. Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. J Hepatol. 2013;58:904–910. doi: 10.1016/j.jhep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 100.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 101.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 102.Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Haskó G, Pacher P. Endothelial Nrf2 activation: a new target for resveratrol? Am J Physiol Heart Circ Physiol. 2010;299:H10–H12. doi: 10.1152/ajpheart.00436.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J Biol Chem. 2009;284:21872–21880. doi: 10.1074/jbc.M109.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ahn J, Cho I, Kim S, Kwon D, Ha T. Dietary resveratrol alters lipid metabolism-related gene expression of mice on an atherogenic diet. J Hepatol. 2008;49:1019–1028. doi: 10.1016/j.jhep.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 107.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–G842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho SJ, Jung UJ, Choi MS. Differential effects of low-dose resveratrol on adiposity and hepatic steatosis in diet-induced obese mice. Br J Nutr. 2012;108:2166–2175. doi: 10.1017/S0007114512000347. [DOI] [PubMed] [Google Scholar]
- 109.Tian XP, Yin YY, Li X. Effects and mechanisms of Acremoniumterricola milleretal mycelium on liver fibrosis induced by carbon tetrachloride in rats. Am J Chin Med. 2011;39:537–550. doi: 10.1142/S0192415X11009019. [DOI] [PubMed] [Google Scholar]
- 110.Zhang C, Yuan W, Fang J, Wang W, He P, Lei J, Wang C. Efficacy of Resveratrol Supplementation against Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Placebo-Controlled Clinical Trials. PLoS One. 2016;11:e0161792. doi: 10.1371/journal.pone.0161792. [DOI] [PMC free article] [PubMed] [Google Scholar]