

Abstract
AIM
To evaluate the effects of asymmetric dimethylarginine (ADMA) in renal arteries from portal hypertensive and cirrhotic rats.
METHODS
Rat renal arteries from Sham (n = 15), pre-hepatic portal hypertension (PPVL; n = 15) and bile duct ligation and excision-induced cirrhosis (BDL; n = 15) were precontracted with norepinephrine, and additional contractions were induced with ADMA (10-6-10-3 mol/L), an endogenous inhibitor of nitric oxide (NO) synthase. Concentration-response curves to acetylcholine (1 × 10-9-3 × 10-6 mol/L) were determined in precontracted renal artery segments with norepinephrine in the absence and in the presence of ADMA. Kidneys were collected to determine the protein expression and activity of dimethylarginine dimethylaminohydrolase (DDAH), an enzyme that catabolizes ADMA.
RESULTS
In renal arteries precontracted with norepinephrine, ADMA caused endothelium-dependent contractions. The pD2 values to ADMA were similar in the Sham and PPVL groups (4.20 ± 0.08 and 4.11 ± 0.09, P > 0.05, respectively), but were lower than those of the BDL group (4.79 ± 0.16, P < 0.05). Acetylcholine-induced endothelium-dependent relaxation that did not differ, in terms of pD2 and maximal relaxation, among the 3 groups studied. Treatment with ADMA (3 × 10-4 mol/L) inhibited acetylcholine-induced relaxation in the 3 groups, but the inhibition was higher (P < 0.05) in the BDL group compared with that for the Sham and PPVL groups. The mRNA and protein expression of DDAH-1 were similar in kidneys from the three groups. Conversely, DDAH-2 expression was increased (P < 0.05) in PPVL and further enhanced (P < 0.05) in the BDL group. However, renal DDAH activity was significantly decreased in the BDL group.
CONCLUSION
Cirrhosis increased the inhibitory effect of ADMA on basal- and induced-release of NO in renal arteries, and decreased DDAH activity in the kidney.
Keywords: Portal hypertension, Cirrhosis, Nitric oxide, Asymmetric dimethylarginine, Nitric oxide inhibitors, Dimethylarginine dimethylaminohydrolase
Core tip: Cirrhosis is associated with renal dysfunction and renal vasoconstriction. This constriction leads to decreased renal blood flow and glomerular filtration. Decreased nitric oxide (NO) bioavailability is involved in these effects. Although plasma levels of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NO synthase, are increased in cirrhosis, the effects of ADMA on renal arteries under this pathological condition are unknown. Therefore, the present work studied the effects of ADMA on basal- and stimulated-NO release in renal arteries from portal hypertensive and cirrhotic rats and the renal expression and activity of dimethylarginine dimethylaminohydrolase, an enzyme that catabolizes ADMA.
INTRODUCTION
The progression of cirrhosis is frequently associated with an impairment in renal function manifested by the appearance of sodium and water retention and the accumulation of fluid within the interstitial tissue and peritoneal cavity[1]. As the disease develops, vasoconstriction of the renal vascular bed commonly results in a reduced glomerular filtration rate and eventually in renal failure[2]. The mechanisms leading to renal dysfunction in cirrhosis involve the activation of the vasoconstrictor and sodium-retaining systems in an attempt to preserve the tubular function[1].
In the kidney, nitric oxide (NO) has numerous physiological roles including the regulation of renal hemodynamics[3,4]. Studies using NO synthase (NOS) inhibitors have demonstrated that NO plays a significant role in maintaining normal vascular tone in the renal vascular bed[4,5]. Basal release of NO from the vessel wall has been described in humans[6,7] and in human renal artery[8]. In the kidney, the basal release of NO induces a substantially lower vascular resistance compared to other organs[4,5].
The plasma levels of NG,NG-asymmetric dimethylarginine (ADMA), an endogenous NOS inhibitor[9], are significantly increased in various pathological conditions, including end-stage chronic renal failure[10,11], cirrhosis[12-14] and hepatorenal syndrome[15]. In human renal artery, ADMA induces a progressive inhibition of NO synthesis and a diminished response to endothelium-mediated relaxation[8]. In spite of the increased ADMA plasma levels in patients with cirrhosis and hepatorenal syndrome, the role of ADMA in renal dysfunction associated to cirrhosis has almost been overlooked, and no attempt has been made to determine the effects of ADMA on the vascular tone of renal arteries during portal hypertension and cirrhosis.
Dimethylarginine dimethylaminohydrolase (DDAHs) degrade ADMA to citrulline and dimethylamine, whereas NG-nitro-L-arginine methyl ester (L-NAME), another inhibitor of NOS, is not degraded by DDAHs[16,17]. DDAHs are expressed as type 1 and 2 isoforms[18] and are widely distributed in various organs and tissues, including the kidney and renal vascular bed[18-20].
The present study hypothesized that one mechanism involved in the renal vasoconstriction associated with cirrhosis could be the elevated levels of ADMA in cirrhosis that may decrease basal- and induced-release of NO by the endothelium of renal vessels. To verify this, the present study investigated the effects of ADMA and L-NAME on both the basal, as well as the stimulated release of NO in the renal arteries of rats with pre-hepatic portal hypertension without cirrhosis and in rats with portal hypertension and secondary biliary cirrhosis induced by ligation and excision of the bile duct. Furthermore, it assessed the effects of portal hypertension and cirrhosis on the renal expression of DDAH-1, DDAH-2 and renal DDAH activity.
MATERIALS AND METHODS
Male Sprague-Dawley rats (200-250 g) were acquired from Charles River, and housed according to institutional guidelines (constant room temperature 22 °C, 12 h light/dark cycle, 60% humidity, standard rat chow and water ad libitum). All protocols were approved by the Institutional Ethics Committee at the University of Valencia (No. UV20121124), and conformed to the Guide for the Care and Use of Laboratory Animals published in Directive 2010/63/EU of the European Parliament.
Rats were assigned to a sham-operated (Sham) group (n = 15), partial portal vein ligation (PPVL) group (n = 15) or bile duct ligation and excision (BDL) group (n = 15) in a random way. After induction of anesthesia by isoflurane (5%, by induction chamber), rats received isoflurane 2%-3% by mask. To assess the adequacy of anesthesia during the surgery, parameters such as responsiveness (e.g., no response to toe pinching), respiratory rate, and heart rate were monitored. Analgesia with Butorphanol was used pre-operatively for preemptive analgesia and post-operatively every 4-12 h during the day of the surgery.
Surgical procedures
Surgical procedures were performed as described previously[21]. Briefly, pre-hepatic portal hypertension induced by partial portal vein ligation was performed by placing a 20-gauge needle on the portal vein. A non-absorbable surgical thread ligature was placed around the needle and portal vein, and the needle was then withdrawn. The studies were performed 14-16 d after PPVL, when the hyperdynamic circulation accompanying portal hypertension was fully established. Secondary biliary cirrhosis was induced by bile duct ligation and excision. The bile duct was cut between a ligature close to the hilum of the liver and another one close to the duodenum. The studies were performed 28 d after BDL when secondary biliary cirrhosis had developed. For the sham operation, the duodenum, portal vein, and bile duct were exposed during laparotomy, and the abdomen was closed 15 min later.
On the day of the experiment, mean arterial pressure (MAP) and portal pressure (PP) measurements were performed while the rats were kept under isoflurane anesthesia, as previously described[21]. Briefly, MAP and PP were measured by catheterization of the right carotid artery and ileocolic vein, respectively. Pressure was transmitted through a Statham pressure transducer and recorded continuously. The external zero reference was placed at the midportion of the rat.
Biochemical analysis
Blood drawn from the carotid artery in the anesthetized rat was collected after hemodynamic assay. The plasma was separated and stored at -20 °C until total bilirubin and creatinine levels were assayed in an autoanalyzer, according to the manufacturer’s instructions.
Isolated rat renal artery preparation
The renal arteries were isolated and cleaned of connective tissue under a dissecting microscope. Segments (4 mm in length) of renal artery were cut for isometric recording of tension. Outside diameter of the rings was measured using an ocular micrometer within a Wild M8 zoom microscope (Heerbrugg, Switzerland) and ranged from 0.8 to 1.4 mm. In some experiments the endothelium was removed mechanically by inserting a roughened stainless-steel wire into the lumen and gently rolling the vessel ring on wet filter paper.
Two stainless-steel holders (100 μm in diameter) were introduced through the arterial lumen and placed in a 5 mL tissue bath containing modified Krebs-Henseleit solution of the following mmol/L composition: NaCl 115; KCl 4.6; KH2PO4 1.2; MgCl2 1.2; CaCl2 2.5; NaHCO3 25; glucose 11.1; EDTA 0.01, pH 7.3-7.4. Indomethacin (10-5 mol/L) was added to the Krebs-Henseleit solution in order to block the cyclooxygenase-derived substances that could interfere with the effects of the NOS inhibitors. The solution was continuously gassed with 95% O2-5% CO2 while the temperature was maintained at 37 °C with a circulating water jacket and a heat pump. One holder was fixed to the organ bath wall and the other was connected to a strain gauge (model FT03; Grass Instruments Division of Astro-Med Inc, United States). Changes in isometric force were recorded by use of Chart v. 4.2.3 software and a MacLab/8e data acquisition system (ADInstruments, Australia). Once the optimal resting tension was reached (1 g), each ring was allowed to attain this steady level of tension during a 1-h accommodation period before testing. Following this, smooth muscle function was assessed by exposing the arterial rings to receptor-independent depolarizing agent KCl (60 mmol/L) until the contraction reached a stable plateau. After washout and return to stable baseline, functional integrity of the endothelium was confirmed routinely by the presence of relaxation induced by acetylcholine (10-6 mol/L) during contraction obtained with norepinephrine (3 × 10-7 to 1 × 10-6 mol/L). Arteries in which acetylcholine reversed the norepinephrine-induced tone by more than 70% were designated as endothelium intact and arteries in which acetylcholine caused less than 15% relaxation were designated as without endothelium.
To assess the effects of portal hypertension and cirrhosis on renal artery contractility, we performed in artery rings from each group cumulative concentration-response curves to KCl (10-120 mmol/L), an agent that induces contraction by facilitating Ca2+ entry through voltage-dependent Ca2+ channels.
The basal release of NO is revealed when endothelium-intact artery rings are precontracted and an additional contraction is induced by NOS inhibitor. This additional contraction provides a functional indication of NO release. Therefore, the ability of ADMA (10-6-10-3 mol/L) or L-NAME (10-6-10-3 mol/L) to inhibit basal activity of NO was assessed from its enhancement of low-levels of contraction (approximately 200-300 mg) induced by norepinephrine (1 × 10-7-3 × 10-7 mol/L) in endothelium-containing renal artery rings. The ability of L-arginine (10-3 mol/L) to either protect against or reverse the enhancement by ADMA or L-NAME was also assessed. Additionally, the effects of both ADMA and L-NAME were examined on norepinephrine-induced tone in endothelium-denuded rings.
Concentration-response curves to acetylcholine (1 × 10-9-3 × 10-6 mol/L), an endothelium-dependent vasorelaxant, were determined in precontracted segments with norepinephrine (3 × 106 mol/L), in the absence and in the presence of ADMA (3 × 10-4 mol/L) or L-NAME (3 × 10-4 mol/L) that were added to the organ bath 20 min before starting the concentration-response curve.
All substances and drugs were purchased from Sigma-Aldrich Chemical Co. (United States). Drugs were prepared and diluted in distilled water except for indomethacin, which was dissolved in absolute ethanol. Stock solutions of the drugs were freshly prepared every day.
Real Time PCR analyses
Samples of cortical tissue from the kidney of each rat were immediately collected into RNAlater RNA stabilization reagent (Thermo Fisher Scientific, United States) following the manufacturer's instructions. Total RNA was isolated and reverse transcribed as previously described[21]. Ready-to-use primers and probes from the Assay-on-demand service of Applied Biosystems were used for the quantification of DDAH-1 and DDAH-2 (Rn00574200_m1 and Rn01525775_g1, respectively) and endogenous reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 4352338E). The qRT-PCR was carried out using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, United States). Samples were run in triplicate and fold changes were generated for each sample by calculating 2-ΔΔCT[22].
Western blotting
Equal amounts of protein from renal cortical homogenates (100 μg total protein) were resolved in SDS-PAGE on 12% gels and electroblotted onto polyvinylidene difluoride membranes. After 1 h blocking with 5% milk in phosphate-buffered saline with 0.1% (v/v) Tween 20 (PBST), membranes were incubated in PBST containing 0.1% milk with a specific primary antibody: monoclonal goat anti-rat DDAH1 antibody (Santa Cruz Biotechnology, United States; 1:500 dilution, overnight incubation at 4 °C) or goat anti-rat DDAH2 antibody (Santa Cruz Biotechnology, United States, 1:200 dilution, overnight incubation at 4 °C). After 4 washes with PBST, membranes were incubated for 1 h with a horseradish peroxidase-labeled antibody at a 1:2000 dilution in PBST containing 1% milk. After 4 additional washes, the membranes were incubated with chemiluminescent reagent according to the manufacturer’s protocols (Inmuno-StarTM HRP Substrate Kit, Bio-Rad, United States), and the chemiluminescent signal was visualized by the LAS-1000 imaging system (Fujifilm, Japan). Densitometric analyses of Western blots were performed using Image Reader LAS-1000 Pro v2.3 software. All membranes were reblogged using a monoclonal antibody anti β-actin (1:2500, Sigma-Aldrich, United States) as a loading control. Data were normalized to corresponding values of β-actin densitometry.
DDAH activity
DDAH activity was measured as described previously by a colorimetric assay[23]. Kidney cortex was homogenized in 5 volumes of 0.1 mol/L sodium phosphate buffer (pH 6.5). Protein concentration of the homogenate was determined using the BCA protein assay (Thermo Fisher, United States) according to the manufacturer’s instructions. The final protein concentration of the homogenate was adjusted to 20 mg/mL with sodium phosphate buffer. Then, 100 μL of homogenate were preincubated with urease (100 U/mL) for 15 min at 37 °C, and then incubated with 1 mmol/L ADMA for 60 min at 37 °C. After deproteinization with 0.5 mL of 4% sulfosalicylic acid, 100 μL of supernatant was incubated with 100 μL of a mixture composed by one part of diacetyl monoxime (0.8% wt/v in 5% acetic acid) and two parts of antipyrine (0.5% wt/v in 50% sulfuric acid) at 90 °C for 1 h. Each sample was analyzed with a paired blank, in which ADMA was omitted. The amounts of L-citrulline formed were determined by spectrophotometry at 466 nm. The DDAH activity was represented as μmol L-citrulline formatted/g protein/min at 37 °C.
Statistical analysis
All values are expressed as mean ± SEM. The contractile effects were expressed as absolute tension (milligrams-force). Relaxation was expressed as a percentage of the norepinephrine-induced contraction. The pD2 (negative logarithm of the molar concentration at which half-maximum response occurs) was determined from individual concentration-response curves by non-linear regression analysis. Area under the concentration-response curve (AUC) was calculated from each individual concentration-response curve to acetylcholine and was expressed as arbitrary units. The contribution of NO to the vascular relaxation induced by acetylcholine was calculated by subtracting from the AUC for acetylcholine the AUC for acetylcholine in the presence of L-NAME or ADMA. All n values are presented as the number of rats. One- or two-way analyses of variance (ANOVA) were performed followed by Bonferroni’s post-test. The level of statistical significance was P < 0.05. The statistical analysis was carried out using Prism 4 software (GraphPad Software Inc., United States).
RESULTS
Morphological features, hemodynamic and biochemical parameters
Morphological characteristics, hemodynamic, and biochemical parameters of the Sham, PPVL, and BDL groups are summarized in Table 1. Both the PPVL and BDL groups led to the characteristic hemodynamic changes found in portal hypertension, with higher values in PP and lower MAP compared to the Sham rats, suggesting the presence of a hyperdynamic state. As expected, the PPVL and BDL groups exhibited higher spleen weights than did Sham rats. In the BDL group, the rats became visibly icteric by the 3rd wk following surgery, weight gain was decreased, and they had higher total bilirubin values than the Sham or PPVL rats. Creatinine concentrations were within the normal range in the three groups. The Sham rats displayed normal post-operative recovery.
Table 1.
Morphological characteristics, hemodynamic and biochemical parameters of the Sham, partial portal vein ligation, and bile duct ligation groups
| Sham | PPVL | BDL | |
| Body weight gain (g) | 39 ± 6 | 35 ± 6 | 10 ± 8ac |
| Spleen weight (g) | 0.7± 0.1 | 1.3 ± 0.1a | 1.4 ± 0.2a |
| Liver weight (g) | 11.6 ± 0.6 | 10.9 ± 0.7 | 16.9 ± 0.7ac |
| Mean arterial pressure (mmHg) | 116 ± 9 | 95 ± 5a | 92 ± 6a |
| Portal pressure (mmHg) | 7 ± 1 | 17 ± 2a | 19 ± 3a |
| Bilirubin (mg/dL) | 0.12 ± 0.03 | 0.15 ± 0.03 | 9.91 ± 0.07ac |
| Creatinine (mg/dL) | 0.75 ± 0.07 | 0.73 ± 0.06 | 0.78 ± 0.06 |
P < 0.05 vs Sham group and
P < 0.05 vs PPVL group. PPVL: Partial portal vein ligation; BDL: Bile duct ligation.
Effects of KCl
In the Sham group, KCl caused concentration-dependent contractions with a pD2 of 1.49 ± 0.01 and a maximal contraction of 1018 ± 83 mg (Figure 1 and Table 2). In the PPVL group, neither maximal contraction nor pD2 values to KCl were affected (Figure 1 and Table 2). In the renal artery rings of the BDL group, maximal contraction to KCl was decreased (P < 0.05) compared to the Sham and PPVL groups (Figure 1 and Table 2). There were no differences among groups in the sensitivity to KCl as demonstrated by similar pD2 values (Table 2).
Figure 1.
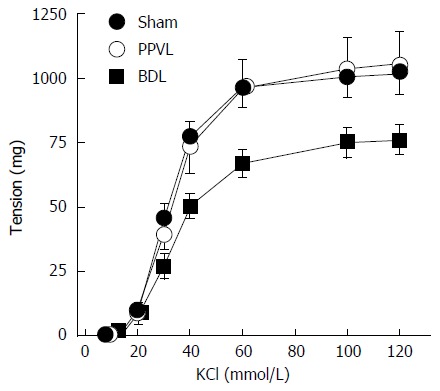
Effects of portal hypertension and cirrhosis on contractile effects induced by high extracellular concentrations of KCl in rat renal arteries. PPVL: Pre-hepatic portal hypertension; BDL: Bile duct ligation.
Table 2.
pD2 values and maximal responses of the concentration-response curves to KCl (10-120 mmol/L) in renal arteries from Sham, partial portal vein ligation and bile duct ligation groups
| n | pD2 | Emax (mg) | |
| Sham | 8 | 1.49 ± 0.01 | 1018 ± 83 |
| PPVL | 8 | 1.46 ± 0.01 | 1050 ± 131 |
| BDL | 8 | 1.46 ± 0.01 | 762 ± 59ac |
pD2, - log M of KCl causing 50% of the maximal contraction; Emax, maximal contraction; n = number of rats;
P < 0.05 vs Sham group and
P < 0.05 vs PPVL group.
Effects of NOS inhibitors on basal NO
At resting tension, the addition of L-NAME (10-6-10-3 mol/L) or ADMA (10-6-10-3 mol/L) did not show significant changes in tension (results not shown). Following the induction of a low level of contraction (210 ± 50 mg) with norepinephrine (1 × 10-7-3 × 10-7 mol/L), the addition of L-NAME (10-6-10-3 mol/L) or ADMA (10-6-10-3 mol/L) led to concentration-dependent increases in tension (Figure 2). The pD2 values for the concentration-response curves to L-NAME were similar in the Sham, PPVL and BDL groups (Table 3). The pD2 values for the ADMA curves were similar in Sham and PPVL, but were lower (P < 0.05) than those for the BDL group, suggesting an increased sensitivity to ADMA in renal arteries from cirrhotic rats. In the Sham, PPVL and BDL groups, pD2 values of the ADMA curves were lower (P < 0.05) than those for L-NAME, suggesting a decreased sensitivity to ADMA in all groups. The maximal responses to ADMA and L-NAME were similar in the Sham and PPVL groups (Figure 2 and Table 3). Conversely, in the BDL group the maximal responses to both ADMA and L-NAME were reduced (P < 0.05) compared with those for Sham and PPVL rats. The contractile effect induced by ADMA and L-NAME was prevented or reverted by L-arginine 10-3 mol/L, a precursor of NO synthesis, in all the groups studied (Figure 2).
Figure 2.
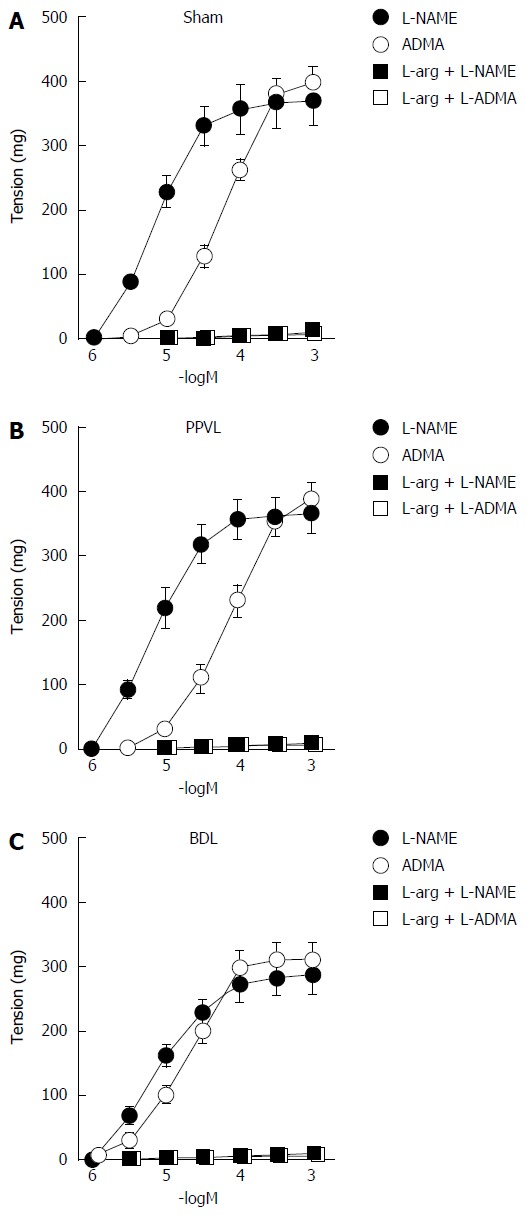
Effects of nitric oxide synthase inhibitors on basal nitric oxide release in renal artery. Contractions induced by L-NAME (n = 8) and ADMA (n = 8) on rings of rat renal artery with endothelium from Sham, PPVL, and BDL groups in the absence and in the presence of L-arginine (L-arg, 10-3 mol/L, n = 6). Contractions were determined after evoking submaximal tone with 10-7-3 × 10-7 mol/L norepinephrine. PPVL: Pre-hepatic portal hypertension; BDL: Bile duct ligation; ADMA: Asymmetric dimethylarginine; L-NAME: NG-nitro-L-arginine methyl ester.
Table 3.
pD2 values and maximal responses of the concentration-response curves to NG-nitro-L-arginine methyl ester and asymmetric dimethylarginine in renal arteries from Sham, partial portal vein ligation, and bile duct ligation groups, after precontraction with norepinephrine
| n | pD2 | Emax (mg) | |
| Sham | |||
| L-NAME | 8 | 5.35 ± 0.17 | 370 ± 36 |
| ADMA | 8 | 4.20 ± 0.08e | 400 ± 26 |
| PPVL | |||
| L-NAME | 8 | 5.30 ± 0.16 | 365 ± 30 |
| ADMA | 8 | 4.11 ± 0.09e | 388 ± 25 |
| BDL | |||
| L-NAME | 8 | 5.24 ± 0.16 | 285 ± 29ac |
| ADMA | 8 | 4.79 ± 0.16ace | 310 ± 27ac |
pD2, - log M of substance causing 50% of the maximal contraction; Emax, maximal contraction; n = number of rats;
P < 0.05 vs Sham group with the same treatment,
P < 0.05 vs PPVL group with the same treatment and
P < 0.05 vs L-NAME treatment in the same group. ADMA: Asymmetric dimethylarginine; L-NAME: NG-nitro-L-arginine methyl ester.
Effects of NOS inhibitors on acetylcholine-induced relaxation
In renal arteries from the Sham group, acetylcholine (1 × 10-9-3 × 10-6 mol/L) caused endothelium-dependent relaxation (pD2 = 7.95 ± 0.08 and Emax = 93% ± 3%) in rings precontracted with norepinephrine (Figure 3A). The relaxation induced by acetylcholine did not differ, in terms of pD2 and maximal relaxation, among the 3 groups studied (Figure 3A and Table 4). No relaxation was observed in response to acetylcholine in renal arteries without endothelium (Figure 3A).
Figure 3.
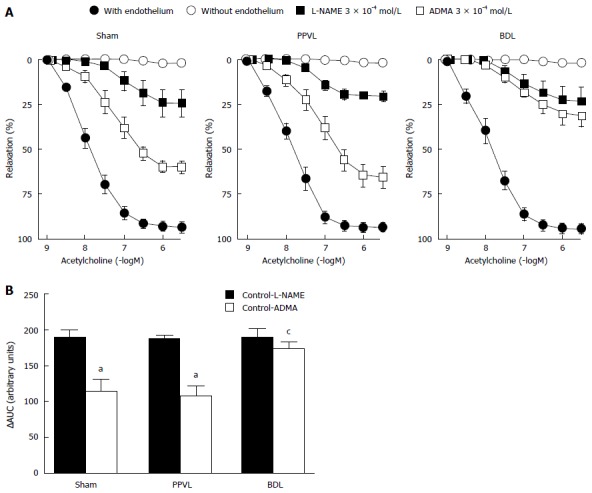
Effects of nitric oxide synthase inhibitors on acetylcholine-induced relaxation in renal artery. A: Concentration-response curves to acetylcholine on rings of rat renal artery from Sham, PPVL, and BDL groups in artery rings with endothelium (n = 8) and without endothelium (n = 6) and in artery rings with endothelium in the presence of L-NAME (3 × 10-4 mol/L; n = 8) or ADMA (3 × 10-4 mol/L; n = 8). Relaxation is expressed as a percentage of the contraction in response to norepinephrine; B: Difference between the areas under curves (AUCs) from artery rings with endothelium (Control) and treated with L-NAME or ADMA. aP < 0.05 vs L-NAME treated group and cP < 0.05 vs Sham and PPVL groups treated with ADMA. PPVL: Pre-hepatic portal hypertension; BDL: Bile duct ligation.
Table 4.
pD2 and maximal response values for the concentration-response curves to acetylcholine in renal arteries from Sham, partial portal vein ligation, and bile duct ligation groups in the absence (Control) and in the presence of NG-nitro-L-arginine methyl ester or asymmetric dimethylarginine
| Acetylcholine | n | pD2 | Emax (%) |
| Sham | |||
| Control | 8 | 7.95 ± 0.08 | 93 ± 3 |
| L-NAME (3 × 10-4 mol/L) | 8 | 7.13 ± 0.28a | 24 ± 7a |
| ADMA (3 × 10-4 mol/L) | 8 | 7.27 ± 0.13ac | 60 ± 3ac |
| PPVL | |||
| Control | 8 | 7.92 ± 0.08 | 94 ± 2 |
| L-NAME (3 × 10-4 mol/L) | 8 | 7.24 ± 0.11a | 21 ± 3a |
| ADMA (3 × 10-4 mol/L) | 8 | 7.18 ± 0.15a | 65 ± 6ac |
| BDL | |||
| Control | 8 | 7.91 ± 0.10 | 95 ± 3 |
| L-NAME (3 × 10-4 mol/L) | 8 | 7.13 ± 0.35a | 24 ± 8a |
| ADMA (3 × 10-4 mol/L) | 8 | 7.17 ± 0.22a | 32 ± 6ace |
pD2, - log M of acetylcholine causing 50% of the maximal relaxation; Emax, maximal relaxation expressed as a percentage of the contraction in response to 3 × 10-6 mol/L norepinephrine; n = number of rats.
P < 0.05 vs control group,
P < 0.05 vs L-NAME treated group and
P < 0.05 vs Sham and PPVL groups with the same treatment. ADMA: Asymmetric dimethylarginine; L-NAME: NG-nitro-L-arginine methyl ester.
The relaxation induced by acetylcholine was inhibited by the treatment with L-NAME (3 × 10-4 mol/L) in renal arteries from the three groups. In the Sham, PPVL and BDL groups, the inhibitions of maximal relaxations induced by acetylcholine in the presence of L-NAME were similar (P > 0.05) 69% ± 4%, 73% ± 3%, and 71% ± 5%, respectively (Figure 3A and Table 4). The pD2 values decreased (P < 0.05) likewise in the presence of L-NAME compared to those for the untreated segments, providing evidence that NO induced by acetylcholine is similar in the three groups. Treatment with ADMA (3 ×10-4 mol/L) inhibited acetylcholine-induced relaxation in the 3 groups, but the inhibition was higher (P < 0.05) in the BDL group compared with the Sham and PPVL groups (Figure 3A and Table 4). In renal arteries from the BDL group, ADMA induced a greater (P < 0.05) inhibition of maximal relaxation than it did in the Sham and PPVL groups, but sensitivity (evidenced by pD2 values) was unchanged (Figure 3A and Table 4). When areas under the curve (AUC) were analyzed, L-NAME inhibited the NO-mediated relaxation similarly in the 3 groups (Figure 3B). Likewise, ADMA inhibited NO release in the Sham and PPVL groups; in BDL group the inhibitory effects of ADMA were increased (Figure 3B).
Expression of DDAHs
We performed real-time RT-PCR on kidneys from the Sham, PPVL, and BDL groups (n = 6 per group). The DDAH-1 mRNA expression was similar in kidneys from the three groups (Figure 4A). In contrast, the DDAH-2 mRNA expression was increased (P < 0.05) in the PPVL compared to that for the Sham group, and in the BDL group, it was further enhanced (Figure 4B). The level of DDAH-2 mRNA expression in PPVL and BDL rats increased 1.33- and 1.64-fold, respectively. Densitometry analysis of Western blot confirmed that DDAH 1 was equally expressed in the kidney of the three groups (Figure 4C and E). Conversely, DDAH-2 expression in kidney was increased in the PPVL group and further increased in the BDL group (Figure 4D and F).
Figure 4.
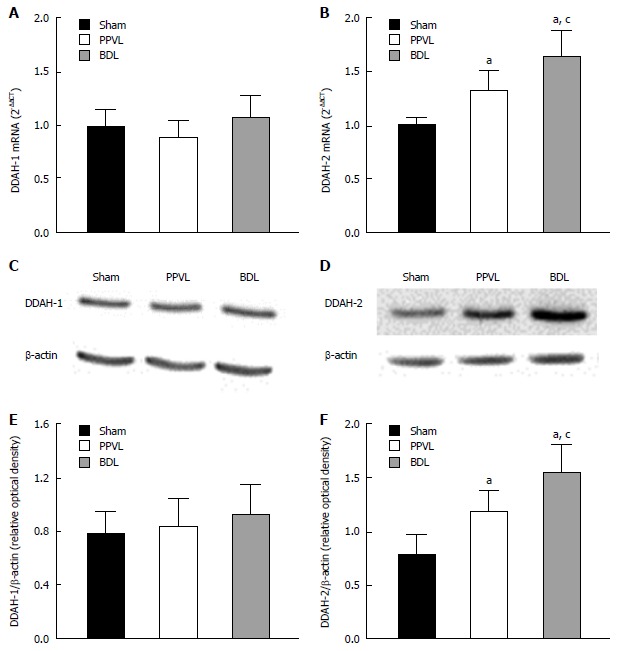
DDAH1 and DDAH2 expression in kidneys from portal hypertensive and cirrhotic rats. A and B: DDAH1 and DDAH2 mRNA expression in kidney from Sham, PPVL, and BDL groups normalized to the expression of GAPDH, which was used as an endogenous reference gene; C and D: Immunoblot analysis in single kidney probed with antibodies against DDAH1, DDAH2 or β-actin, as indicated; E and F: Graphs show the results of densitometric analyses from pooled data, plotted as optical densitometry relative to the signal obtained by β-actin. Each data set represents the mean ± SEM derived from 6 independent experiments. aP < 0.05 vs Sham group and cP < 0.05 vs PPVL group. DDAH: Dimethylarginine dimethylaminohydrolase.
DDAH activity
We determined the effects of portal hypertension and cirrhosis on renal DDAH activity in crude tissue lysates. Renal DDAH activity was increased, but not significantly (P > 0.05), in kidneys from the PPVL group. The DDAH activity in the BDL group, however, was significantly reduced (61% ± 7%, P < 0.05) compared to that for the Sham and PPVL groups (Figure 5).
Figure 5.
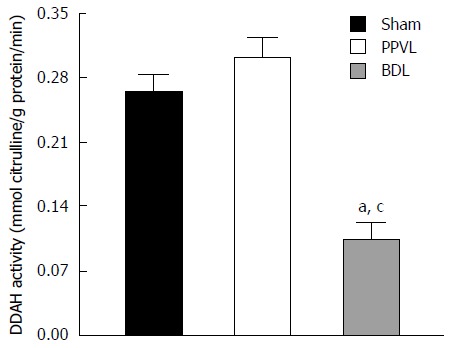
Effects of portal hypertension and cirrhosis on renal dimethylarginine dimethylaminohydrolase activity. Bar graphs represent DDAH activity in kidney from Sham, PPVL, and BDL groups. Each data set represents the mean ± SEM derived from 6 independent experiments. aP < 0.05 vs Sham group and cP < 0.05 vs PPVL group. DDAH: Dimethylarginine dimethylaminohydrolase.
DISCUSSION
The results of the present study demonstrate that both the basal- and induced-release of NO are inhibited by ADMA, with a higher effect in renal arteries from rats with secondary biliary cirrhosis. The increased effect of ADMA inhibiting NO synthesis together with decreased renal DDAH activity indicates that the accumulation of ADMA during cirrhosis could make the renal artery prone to vasoconstriction.
One finding of the present report demonstrates a decreased contraction in renal arteries from cirrhotic rats in response to a high extracellular K+ concentration, which causes the depolarization and subsequent opening of voltage-dependent Ca2+ channels. Vascular hypocontractility in cirrhosis is a multifactorial phenomenon where several mechanisms have been identified and contribute to impaired vasoconstriction. These include the overproduction of vasodilators and decreased responsiveness to vasoconstrictors. Although NO overproduction is widely accepted as main culprit of vasodilation in cirrhosis[24-26], several studies have shown that other factors besides NO are involved in the pathogenesis of vascular hypocontractility. It is known that in cirrhosis, a component of hypocontractility is found in isolated vessels, even though the endothelium is removed and NOS is pharmacologically inhibited[27-29]. It is noteworthy that the cirrhosis-impaired Rho kinase pathway results in decreased phosphorylation of Ca2+ sensitizing proteins, increased myosin light chain phosphatase activity and decreased Ca2+ sensitivity[30]. Although our results in vitro show a reduced contraction of renal arteries during cirrhosis, the in vivo activation of vasoactive systems on renal circulation during hyperdynamic circulation associated to portal hypertension and cirrhosis[1] could develop into an excessive contraction of renal artery.
The basal release of NO was determined indirectly by measuring the effects of ADMA and L-NAME in precontracted artery rings. We found that in renal arteries both NOS inhibitors markedly increased the vascular tone, suggesting an important basal NO synthesis. The contractile effects induced by NOS inhibitors were endothelium-dependent and reversed by L-arginine, the substrate for NO synthesis, thus demonstrating that ADMA and L-NAME increase arterial tone by inhibiting the basal release of endothelial NO.
The maximal contraction induced by NOS inhibitors in renal arteries from cirrhotic rats was lower than those in control or portal hypertensive rats. The hypocontractility cannot be attributed to a lower basal release of NO, since the smooth muscle of renal arteries from cirrhotic rats showed hypocontractility in response to KCl. Therefore, differences in the level of contraction in response to NOS inhibitors would not reflect changes in basal NO generation.
That notwithstanding, we found a similar sensitivity to L-NAME in the three groups studied. Furthermore, when comparing the sensitivity of the two inhibitors, a decreased sensitivity to ADMA as compared with that for L-NAME was observed in the three groups. The concentration-response curve to ADMA was significantly displaced to the left for BDL, as compared to those for Sham and PPVL rats; this represents indirect evidence of a decreased ability of DDAH to catabolize ADMA. Interestingly, no significant differences in the contractile response to ADMA were observed between Sham and PPVL rats. Since BDL and PPVL rats had similar increases in portal pressure, and liver damage was only present in the BDL group, it is conceivable that in renal arteries the liver dysfunction is the main factor causing the altered responses to the ADMA.
Since DDAH is highly specific for the degradation of ADMA, but not L-NAME[16], these changes in ADMA sensitivity could be related to changes in DDAH activity. Another study addressing the localization of DDAH and NOS in the rat kidney has shown co-localization of the two enzymes[20]. Therefore, the close relationship between DDAH and NOS in the kidney supports the idea that DDAH regulates ADMA levels and NOS activity[19,31]. The increased sensitivity to ADMA in renal arteries from the BDL group offers a reasonable indication that decreased DDAH activity and the accumulation of ADMA occur in the vessel wall, enhancing the inhibitory effect on NO biosynthesis.
Both ADMA and L-NAME inhibited acetylcholine-induced relaxation in renal arteries indicating that NO pathways contribute to this effect. As expected, L-NAME markedly inhibited the relaxation induced by acetylcholine. Although it has been demonstrated that ADMA preferentially blocks basal NO release, but it has little effect on acetylcholine-induced relaxation[7,21,32], ADMA markedly inhibited the acetylcholine-induced relaxation in renal arteries from cirrhotic rats. It has been demonstrated that ADMA inhibits basal- and stimulated-release of NO in human renal arteries[8] and other arterial beds where acetylcholine-induced relaxation is mainly dependent on endothelial NO, such as the human middle cerebral artery[33] and internal mammary artery[34].
The present functional analyses in renal arteries from cirrhotic rats demonstrates for the first time that the increased ability of ADMA to inhibit NOS could be related, at least in part, to a lower activity of DDAH and a lesser degradation of ADMA. This reinforces the role of DDAH in controlling the NO bioavailability, and its impairment during cirrhosis might be a mechanism involved in the increased renal artery contraction during cirrhosis.
The study shows that the mRNA and protein expressions of DDAH-1 were unchanged in kidneys from the PPVL and BDL groups, thus suggesting that portal hypertension and cirrhosis do not control renal DDAH-1 expression. The present results confirm previous studies demonstrating unchanged levels of DDAH-1 expression in kidneys from young cirrhotic rats[35]. In contrast, it has been demonstrated that there is a decreased hepatic DDAH-1 expression[36] and increased DDAH-1 expression in mesenteric arteries[21] from BDL rats. Although the mechanisms involved in this different regulation of DDAH-1 expression are not apparent, they could be related to the organ involved.
The results of the present work suggest an association between portal hypertension, cirrhosis and DDAH-2 expression. The kidneys of rats with portal hypertension exhibited a higher expression of DDAH-2 than the kidneys of control rats, and those of the BDL group exhibited a further increase of DDAH-2. In this case, a similar pattern of expression has been shown in mesenteric arteries[21] and liver[36] from BDL rats. Surprisingly, DDAH-2 protein expression was unaltered in kidneys from young BDL rats[35], suggesting an age-dependent regulation of the expression of DDAH-2 induced by cirrhosis.
To determine whether the different patterns in DDAH protein expression were correlated with enzymatic activity, in vitro ADMA degradation by DDAH was measured. Renal DDAH activity was unchanged in the PPVL group, but was significantly reduced in the BDL group, pointing out the liver dysfunction as a main factor responsible for the decreased DDAH activity as opposed to the portal hypertension and hyperdynamic circulation. These data confirm previous findings that demonstrate the inhibitory effect of cirrhosis in the renal DDAH activity in young rats[35].
There is a growing body of evidence that DDAH activity is inhibited by superoxide[37,38]. It has been demonstrated that ADMA can uncouple endothelial NOS and initiate superoxide generation by NOS[39]. This finding suggests that an increased concentration of ADMA during liver dysfunction[12,13,15] could be an initial point for further NOS uncoupling, increased superoxide and DDAH inhibition, therefore a further increase of ADMA, thereby initiating a feed-forward reaction. Accordingly, it has been hypothesized that there is a possible role for ADMA in the development of hepatorenal syndrome[40], a pathology characterized by an excessive vasoconstriction of renal circulation[1].
In cholestatic patients, a correlation between oxidative stress during obstructive jaundice and renal dysfunction has recently been established[41]. The levels of bilirubin were progressively increased from benign to malignant evolution[41] which is in concordance with pro-oxidant capacity of toxic bile acids[42]. Therefore, it is possible that the hyperbilirubinemia associated with the BDL model of cirrhosis could increase the oxidative stress in the kidney and inhibit renal DDAH. Renal dysfunction in cirrhosis is a common complication, characterized by marked renal artery contraction as a consequence of the activation of several vasoactive pathways[1]. Therefore, the increased inhibitory effects of ADMA on NO synthesis in renal arteries from BDL rats could be another factor contributing to the vasoconstriction associated with cirrhosis. DDAH activators or ADMA-reducing agents may be a potential therapeutic approach to managing the vascular renal dysfunction associated with cirrhosis.
In conclusion, both basal- and induced-NO release are inhibited in renal arteries by ADMA, an effect that is increased in cirrhotic rats. The results of the present study confirm that liver dysfunction is the main factor in the decreased renal DDAH activity and supports the notion that the vascular renal system is highly exposed to ADMA during cirrhosis. Furthermore, our data show an increased DDAH-2 expression, but a reduced DDAH activity in the kidney, associated with cirrhosis.
COMMENTS
Background
Increased renal vascular contraction is a major cause for the development of renal dysfunction in cirrhosis. Several observations have shown that nitric oxide (NO) inhibition is associated with decreased renal plasma flow and increased renal vascular resistance, suggesting that NO exerts a tonic relaxing effect on the renal circulation. Therefore, the kidney is highly vulnerable to the accumulation of asymmetric dimethylarginine (ADMA), an endogenous NO synthase inhibitor. The plasma levels of ADMA are significantly increased in cirrhosis and hepatorenal syndrome. No attempts, however, have been made to determine the effects of ADMA on the vascular tone of renal arteries from portal hypertensive and cirrhotic rats.
Research frontiers
Evidence indicates that in the BDL group dimethylarginine dimethylaminohydrolase (DDAH) activity is reduced in kidneys and ADMA inhibits the basal and stimulated NO in renal arteries more efficiently. High levels of ADMA in the plasma of patients with cirrhosis and hepatorenal syndrome have been previously described and could be responsible, in part, for the contraction and decreased vasodilation of renal arteries during the development of cirrhosis.
Innovations and breakthroughs
This findings draw attention to the role of ADMA and DDAH in the renal vascular dysfunction associated with cirrhosis. Since the enhanced sensitivity to ADMA and inhibition of DDAH is observed in BDL rats but not in PPVL ones, these effects are related to liver dysfunction more than are the portal hypertension and hyperdynamic circulation.
Applications
DDAH emerges as an important regulator of NO bioavailability in the renal artery. The DDAH activators or ADMA-reducing agents may be a potential therapeutic approach to managing the vascular renal dysfunction associated with cirrhosis.
Terminology
Hepatorenal syndrome is defined as the development of renal failure in patients with severe liver disease, acute or chronic, in the absence of any other identifiable cause of renal pathology.
Peer-review
ADMA is a new molecule that its value as a marker is being tested for many diseases and situations; cardiovascular diseases, statin usage, etc. The study is a well designed and conducted one. It may contribute to the pathophysiology and to the development strategies to prevent/treat of hepatorenal syndrome.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Spain
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the University of Valencia Institutional Ethics Committee.
Institutional animal care and use committee statement: All protocols were approved by the Institutional Ethics Committee at the University of Valencia (No. UV20121124), and conformed to the Guide for the Care and Use of Laboratory Animals published in Directive 2010/63/EU of the European Parliament.
Conflict-of-interest statement: The authors declare no conflict of interests.
Data sharing statement: No additional data are available.
Peer-review started: July 30, 2016
First decision: August 29, 2016
Article in press: October 10, 2016
P- Reviewer: Bayraktar Y, Fernandez-Rodriguez CM, Fierbinteanu-Braticevici C, Ozen H, Thomopoulos KC S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Ginès P, Guevara M, Arroyo V, Rodés J. Hepatorenal syndrome. Lancet. 2003;362:1819–1827. doi: 10.1016/S0140-6736(03)14903-3. [DOI] [PubMed] [Google Scholar]
- 2.Ginès A, Escorsell A, Ginès P, Saló J, Jiménez W, Inglada L, Navasa M, Clària J, Rimola A, Arroyo V. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993;105:229–236. doi: 10.1016/0016-5085(93)90031-7. [DOI] [PubMed] [Google Scholar]
- 3.Bachmann S, Mundel P. Nitric oxide in the kidney: synthesis, localization, and function. Am J Kidney Dis. 1994;24:112–129. doi: 10.1016/s0272-6386(12)80170-3. [DOI] [PubMed] [Google Scholar]
- 4.Majid DS, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am J Hypertens. 2001;14:74S–82S. doi: 10.1016/s0895-7061(01)02073-8. [DOI] [PubMed] [Google Scholar]
- 5.Mount PF, Power DA. Nitric oxide in the kidney: functions and regulation of synthesis. Acta Physiol (Oxf) 2006;187:433–446. doi: 10.1111/j.1748-1716.2006.01582.x. [DOI] [PubMed] [Google Scholar]
- 6.Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- 7.Mittermayer F, Schaller G, Pleiner J, Vychytil A, Sunder-Plassmann G, Hörl WH, Wolzt M. Asymmetrical dimethylarginine plasma concentrations are related to basal nitric oxide release but not endothelium-dependent vasodilation of resistance arteries in peritoneal dialysis patients. J Am Soc Nephrol. 2005;16:1832–1838. doi: 10.1681/ASN.2004121109. [DOI] [PubMed] [Google Scholar]
- 8.Segarra G, Medina P, Vila JM, Chuan P, Domenech C, Torondel B, Lluch A. Inhibition of nitric oxide activity by arginine analogs in human renal arteries. Am J Hypertens. 2001;14:1142–1148. doi: 10.1016/s0895-7061(01)02208-7. [DOI] [PubMed] [Google Scholar]
- 9.Vallance P, Leone A, Calver A, Collier J, Moncada S. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J Cardiovasc Pharmacol. 1992;20 Suppl 12:S60–S62. doi: 10.1097/00005344-199204002-00018. [DOI] [PubMed] [Google Scholar]
- 10.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 11.Kielstein JT, Böger RH, Bode-Böger SM, Schäffer J, Barbey M, Koch KM, Frölich JC. Asymmetric dimethylarginine plasma concentrations differ in patients with end-stage renal disease: relationship to treatment method and atherosclerotic disease. J Am Soc Nephrol. 1999;10:594–600. doi: 10.1681/ASN.V103594. [DOI] [PubMed] [Google Scholar]
- 12.Lluch P, Torondel B, Medina P, Segarra G, Del Olmo JA, Serra MA, Rodrigo JM. Plasma concentrations of nitric oxide and asymmetric dimethylarginine in human alcoholic cirrhosis. J Hepatol. 2004;41:55–59. doi: 10.1016/j.jhep.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Lluch P, Segarra G, Medina P. Asymmetric dimethylarginine as a mediator of vascular dysfunction in cirrhosis. World J Gastroenterol. 2015;21:9466–9475. doi: 10.3748/wjg.v21.i32.9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrigno A, Di Pasqua LG, Berardo C, Richelmi P, Vairetti M. Liver plays a central role in asymmetric dimethylarginine-mediated organ injury. World J Gastroenterol. 2015;21:5131–5137. doi: 10.3748/wjg.v21.i17.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lluch P, Mauricio MD, Vila JM, Segarra G, Medina P, Del Olmo JA, Rodrigo JM, Serra MA. Accumulation of symmetric dimethylarginine in hepatorenal syndrome. Exp Biol Med (Maywood) 2006;231:70–75. doi: 10.1177/153537020623100108. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa T, Kimoto M, Sasaoka K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J Biol Chem. 1989;264:10205–10209. [PubMed] [Google Scholar]
- 17.Jacobi J, Sydow K, von Degenfeld G, Zhang Y, Dayoub H, Wang B, Patterson AJ, Kimoto M, Blau HM, Cooke JP. Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation. 2005;111:1431–1438. doi: 10.1161/01.CIR.0000158487.80483.09. [DOI] [PubMed] [Google Scholar]
- 18.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J. 1999;343 Pt 1:209–214. [PMC free article] [PubMed] [Google Scholar]
- 19.Kimoto M, Tsuji H, Ogawa T, Sasaoka K. Detection of NG,NG-dimethylarginine dimethylaminohydrolase in the nitric oxide-generating systems of rats using monoclonal antibody. Arch Biochem Biophys. 1993;300:657–662. doi: 10.1006/abbi.1993.1091. [DOI] [PubMed] [Google Scholar]
- 20.Tojo A, Welch WJ, Bremer V, Kimoto M, Kimura K, Omata M, Ogawa T, Vallance P, Wilcox CS. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney Int. 1997;52:1593–1601. doi: 10.1038/ki.1997.490. [DOI] [PubMed] [Google Scholar]
- 21.Serna E, Mauricio MD, Lluch P, Segarra G, Cortina B, Lluch S, Medina P. Basal release of nitric oxide in the mesenteric artery in portal hypertension and cirrhosis: role of dimethylarginine dimethylaminohydrolase. J Gastroenterol Hepatol. 2013;28:880–886. doi: 10.1111/jgh.12119. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Tain YL, Baylis C. Determination of dimethylarginine dimethylaminohydrolase activity in the kidney. Kidney Int. 2007;72:886–889. doi: 10.1038/sj.ki.5002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee FY, Colombato LA, Albillos A, Groszmann RJ. N omega-nitro-L-arginine administration corrects peripheral vasodilation and systemic capillary hypotension and ameliorates plasma volume expansion and sodium retention in portal hypertensive rats. Hepatology. 1993;17:84–90. [PubMed] [Google Scholar]
- 25.Sieber CC, Groszmann RJ. Nitric oxide mediates hyporeactivity to vasopressors in mesenteric vessels of portal hypertensive rats. Gastroenterology. 1992;103:235–239. doi: 10.1016/0016-5085(92)91118-n. [DOI] [PubMed] [Google Scholar]
- 26.Sieber CC, Lopez-Talavera JC, Groszmann RJ. Role of nitric oxide in the in vitro splanchnic vascular hyporeactivity in ascitic cirrhotic rats. Gastroenterology. 1993;104:1750–1754. doi: 10.1016/0016-5085(93)90655-v. [DOI] [PubMed] [Google Scholar]
- 27.Heller J, Trebicka J, Shiozawa T, Schepke M, Neef M, Hennenberg M, Sauerbruch T. Vascular, hemodynamic and renal effects of low-dose losartan in rats with secondary biliary cirrhosis. Liver Int. 2005;25:657–666. doi: 10.1111/j.1478-3231.2005.01053.x. [DOI] [PubMed] [Google Scholar]
- 28.Hennenberg M, Biecker E, Trebicka J, Jochem K, Zhou Q, Schmidt M, Jakobs KH, Sauerbruch T, Heller J. Defective RhoA/Rho-kinase signaling contributes to vascular hypocontractility and vasodilation in cirrhotic rats. Gastroenterology. 2006;130:838–854. doi: 10.1053/j.gastro.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 29.Lin HC, Yang YY, Huang YT, Lee TY, Hou MC, Lee FY, Lee SD. Vascular contractile response and signal transduction in endothelium-denuded aorta from cirrhotic rats. World J Gastroenterol. 2005;11:2306–2312. doi: 10.3748/wjg.v11.i15.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hennenberg M, Trebicka J, Biecker E, Schepke M, Sauerbruch T, Heller J. Vascular dysfunction in human and rat cirrhosis: role of receptor-desensitizing and calcium-sensitizing proteins. Hepatology. 2007;45:495–506. doi: 10.1002/hep.21502. [DOI] [PubMed] [Google Scholar]
- 31.MacAllister RJ, Parry H, Kimoto M, Ogawa T, Russell RJ, Hodson H, Whitley GS, Vallance P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br J Pharmacol. 1996;119:1533–1540. doi: 10.1111/j.1476-5381.1996.tb16069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Zobaidy MJ, Craig J, Martin W. Differential sensitivity of basal and acetylcholine-induced activity of nitric oxide to blockade by asymmetric dimethylarginine in the rat aorta. Br J Pharmacol. 2010;160:1476–1483. doi: 10.1111/j.1476-5381.2010.00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segarra G, Medina P, Ballester RM, Lluch P, Aldasoro M, Vila JM, Lluch S. Effects of some guanidino compounds on human cerebral arteries. Stroke. 1999;30:2206–2211. doi: 10.1161/01.str.30.10.2206. [DOI] [PubMed] [Google Scholar]
- 34.Segarra G, Medina P, Vila JM, Martínez-León JB, Ballester RM, Lluch P, Lluch S. Contractile effects of arginine analogues on human internal thoracic and radial arteries. J Thorac Cardiovasc Surg. 2000;120:729–736. doi: 10.1067/mtc.2000.109537. [DOI] [PubMed] [Google Scholar]
- 35.Tain YL, Hsieh CS, Chen CC, Sheen JM, Lee CT, Huang LT. Melatonin prevents increased asymmetric dimethylarginine in young rats with bile duct ligation. J Pineal Res. 2010;48:212–221. doi: 10.1111/j.1600-079X.2010.00745.x. [DOI] [PubMed] [Google Scholar]
- 36.Mookerjee RP, Mehta G, Balasubramaniyan V, Mohamed Fel Z, Davies N, Sharma V, Iwakiri Y, Jalan R. Hepatic dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis and is a target for therapy in portal hypertension. J Hepatol. 2015;62:325–331. doi: 10.1016/j.jhep.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sydow K, Munzel T. ADMA and oxidative stress. Atheroscler Suppl. 2003;4:41–51. doi: 10.1016/s1567-5688(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 38.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–H3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 39.Böger RH, Bode-Böger SM, Tsao PS, Lin PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J Am Coll Cardiol. 2000;36:2287–2295. doi: 10.1016/s0735-1097(00)01013-5. [DOI] [PubMed] [Google Scholar]
- 40.Nijveldt RJ, Teerlink T, van Leeuwen PA. The asymmetrical dimethylarginine (ADMA)-multiple organ failure hypothesis. Clin Nutr. 2003;22:99–104. doi: 10.1054/clnu.2002.0614. [DOI] [PubMed] [Google Scholar]
- 41.Martínez-Cecilia D, Reyes-Díaz M, Ruiz-Rabelo J, Gomez-Alvarez M, Villanueva CM, Álamo J, Muntané J, Padillo FJ. Oxidative stress influence on renal dysfunction in patients with obstructive jaundice: A case and control prospective study. Redox Biol. 2016;8:160–164. doi: 10.1016/j.redox.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan KP, Yang M, Ito S. Activation of nuclear factor (erythroid-2 like) factor 2 by toxic bile acids provokes adaptive defense responses to enhance cell survival at the emergence of oxidative stress. Mol Pharmacol. 2007;72:1380–1390. doi: 10.1124/mol.107.039370. [DOI] [PubMed] [Google Scholar]