

Abstract
Precision medicine is defined by the administration of drugs based on the tumor’s particular genetic characteristics. It is developing quickly in the field of cancer therapy. For example, KRAS, NRAS and BRAF genetic testing demonstrates its efficiency for precision medicine in colorectal cancer (CRC). Besides for these well-known mutations, the purpose of performing larger genetic testing in this pathology is unknown. Recent reports have shown that using the poly ADP ribose polymerase (PARP) inhibitor olaparib in patients with homologous repair enzyme deficiency gave positive clinical results in breast, ovarian and prostate cancers. We have reported here the cases of 2 patients with multi-treated metastatic CRC who underwent somatic and constitutional exome analyses. The analyses revealed a loss of function mutation in a homologous repair enzyme resulting in the loss of heterozygosity for both patients (Check2 for the first patient and RAD51C for the second one). Both patients were treated with off-label usage of olaparib. While the first patient showed clinical benefit, reduction of carcinoembryonic antigen tumor marker and radiologic response, the second patient quickly presented a progression of the tumor. Additional genetic analyses revealed a frameshift truncating mutation of the TP53BP1 gene in the patient who progressed. Interestingly, deficiency in TP53BP1 was previously described to confer resistance to olaparib in mice breast cancer models. Our findings suggest that exome analysis may be a helpful tool to highlight targetable mutations in CRC and that olaparib may be efficient in patients with a homologous repair deficiency.
Keywords: Colorectal cancer, Exome analysis, Genetic aberrations, Homologous repair deficiency, Precision medicine
Core tip: The role of genetic profiling in metastatic colorectal cancer for precision medicine is currently under investigation. This case report underlines for the first time, that olaparib may have some clinical efficiency in patients with homologous repair deficiency in colorectal tumor.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide with about 1 million new cases and over 500000 deaths every year[1]. Approximately 30% of patients with CRC develop an overt metastatic disease. Meanwhile, 40% to 50% of newly diagnosed patients already present a metastatic disease. For patients with non-operable metastatic CRC (mCRC), there are no curative options, but the use of palliative systemic chemotherapy dramatically enhances response rates, progression-free survival (PFS) and overall survival (OS)[2-4]. Currently, few drugs have demonstrated effectiveness in the treatment of metastatic colorectal disease and patients are treated based on the use of three cytotoxic chemotherapies: fluoropyrimidine, oxaliplatin and irinotecan, associated with targeted therapies [anti-EGFR (panitumumab and cetuximab) or anti-VEGF (bevacizumab) monoclonal antibodies].
In consequence, at the end of their treatment sequence, many patients still have a good performance status and would still be able to undergo treatment; unfortunately, we do not have any approved and efficient therapies to offer. Cancer cells are characterized by multiple genomic alterations. This finding has led to the development of molecularly targeted agents that inhibit mutated proteins. Such drugs are theoretically more specific for cancer cells bearing specific mutations. While such targeted agents have followed clinical development based on tumor location, most genetic molecular alterations exist across tumor types and histologies. This observation raises the possibility of shifting towards the administration of drugs based on molecular pattern rather than on histological status. Improvements in genetic sequencing have made possible the identification of multiple genomic molecular alterations in a timeframe compatible with clinical practice. Many retrospective and prospective trials have shown the usage and efficiency of off-label molecularly targeting agents. We report here the usage of exome testing in 2 patients with metastatic CRC and show for the first time that homologous repair deficiency could be targeted by poly ADP ribose polymerase (PARP) inhibitor in this pathology.
Sample preparation
Formalin-fixed paraffin-embedded (FFPE) tumors were characterized by a pathologist to determine the tumor cell content and sent to the molecular biology platform for DNA extraction. Tumor cell content was 85% for Case 1 and 60% for Case 2. Seven 15-μm slices were extracted using the Maxwell 16 FFPE Plus LEV DNA purification kit (Promega) according to the manufacturer’s protocol. Corresponding normal DNA was extracted from 500 μL ethylenediaminetetraacetic acid blood samples with the Maxwell 16 Blood DNA Purification System (Promega) according to the manufacturer’s instructions. DNA quality was assessed by spectrophotometry with absorbance at 230, 260 and 280 nm. DNA was quantified using a fluorimetric assay with a Qubit device.
Two-hundred nanogram of genomic DNA from normal and cancer cells were fragmented with a Covaris device to obtain fragments around 180-200 bp. Then, libraries were constructed and captured by using SureSelect Human All Exon v5 kit (Agilent), following the manufacturer’s protocol. Paired-end (2 × 151 bases) sequencing was performed on a NextSeq500 device (Illumina). Obtained sequences were aligned and annotated with the human Hg19 genome based on the SureSelect Human All Exon v5 manifest by using Burrows-Wheeler Aligne (BWA) and Genome Analysis toolkit (GATK) algorithms. Only sequences with a read depth of 10 × and a mutation allele frequency superior to 5% were conserved for the analysis. The analysis is fostered on 137 clinically relevant genes related to biological pathways linked to cancer predisposition or available targeted therapies (Supplementary Table 1). Copy number variations were studied by using Control-FREEC software as described[5,6].
CASE REPORT
Patient 1
A 58-year-old Caucasian male initially presented with abdominal pain. A CT scan, carried out in June 2011, revealed a metastatic sigmoid cancer with the following metastatic locations: mediastinal and lomboartic lymph nodes, lungs. A lymph node biopsy was performed and the diagnosis of wild-type KRAS, NRAS and BRAF, moderately differentiated Lieberkühn adenocarcinoma was made. From 2011 to 2015, the patient received different chemotherapeutic regimens. In July 2015, the patient’s lung metastases progressed and he started to show symptoms such as breathlessness and a permanent dry cough. As no approved chemotherapy or targeted therapy could be proposed, we performed somatic and constitutional exome analyses. We observed 479 somatic mutations. For clinical use, we analyzed a short list of 137 genes. It was interesting to see that among the 8 altered genes (Table 1), we observed a SMAD4 stop mutation, which is frequently found in CRC. An activating mutation of AKT1 was observed (Q79K). It could be targetable by protein kinase B (AKT)/mTor inhibitors. Surprisingly, we observed a constitutive Chek2 mutation (R117G), a gene involved in the homologous repair process. This mutation is cited in the public database for conferring a predisposition to cancer. Moreover, the analysis of copy number variation showed a loss of heterozygosity in chromosome 22 (from 29091114 to 29130709) (Figure 1A). This analysis suggested a complete deletion of Check2 function in tumor cells. After discussion of the case at the molecular tumor board, the patient was proposed to receive off-label PARP inhibitor olaparib which previously showed efficiency in patients with Chek2 mutation in metastatic prostate cancer[7]. One mo after beginning the therapy by PARP inhibitor, the patient declared reduction of cough and disappearance of breathlessness. After 3 mo we observed a reduction in carcinoembryonic antigen serum level (57 ng/mL to 25 ng/mL) and a tumor size reduction upon CT scan (Figure 2). No hematological toxicity was mentioned. Patient weight increased from 62 kg to 68 kg. However, despite this response, the patient died suddenly at home 4 mo after introduction of the therapy.
Table 1.
Alterations observed for Case 1 in the short list of clinically relevant genes
| Gene | Nucleotide variation | Protein variation | Origin | Impact |
| AKT1 | c.235C>A | Gln79Lys | Somatic | Activating |
| CCNE1 | c.1022C>T | Thr341Met | Somatic | Unknown |
| CHEK2 | c.478A>G | Arg117Gly | Constitutive | Likely pathogenic |
| CUL2 | c.70C>T | Pro24Ser | Somatic | Unknown |
| ERCC6 | c.4179G>A | Met1393Ile | Somatic | Unknown |
| PMS2 | c.1531A>G | Thr511Ala | Constitutive | Benign |
| SMAD4 | c.1091T>A | Leu364X | Somatic | Loss of function |
| SUFU | c.1211T>C | Met404Thr | Constitutive | Unknown |
Figure 1.
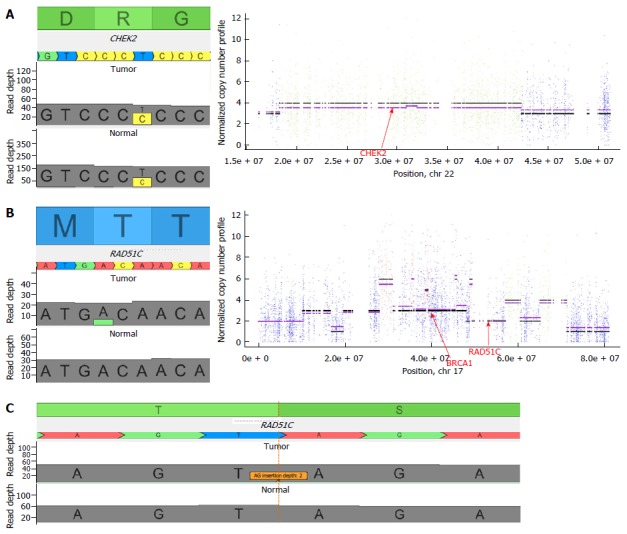
Genetic data of targeted alterations. A: Patient 1 presented a T>C variation in the CHEK2 gene inducing a constitutive deleterious R117G mutation (left panel). At the chromosomal level, it appeared that chromosome 22 harbored a loss of heterozygosity inducing (right panel) a complete inactivation of CHEK2; B: Patient 2 presented a A>G variation in the RAD51C gene inducing a somatic loss of function T287A mutation (left panel). At the chromosomal level, it appeared that chromosome 17 harbored an important loss of heterozygosity at the RAD51C locus inducing a complete inactivation of RAD51C; C: Patient 2 also harbored an AG insertion resulting in a frameshift truncating mutation in the TP53BP1 gene.
Figure 2.
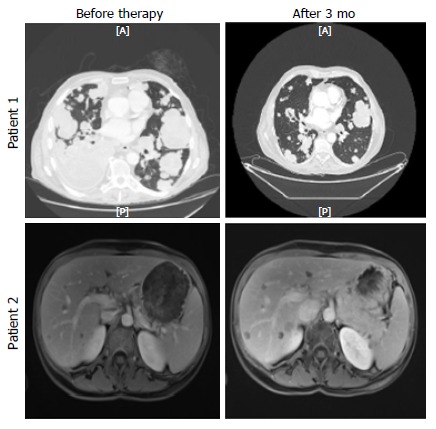
Computed tomography scan of patient 1 before and after 3 mo of olaparib (upper panel), and magnetic resonance imaging of liver metastasis of patient 2 before and after 3 mo of olaparib (lower panel).
Patient 2
A 49-year-old woman presented with rectal bleeding and diarrhea in March 2015. She underwent a colonoscopy with biopsies that revealed a KRAS mutated, well differentiated Lieberkühn rectal adenocarcinoma. The patient did not present any family history of CRC. The genetic testing did not show microsatellite instability. CT scan showed multiple, bilobar liver metastases. The patient was treated with FOLFIRINOX combination plus bevacizumab for 12 cycles. The CT scan showed major response in the liver and the rectum but no curative option could be proposed for the liver disease. Despite efficacy, the therapy had major toxicity with grade 3 peripheral neuropathy which precluded further usage of oxaliplatin. A therapeutic pause was proposed. The patient refused and asked for additional therapy. We performed somatic and constitutional exome analyses. We observed 905 somatic mutations. For clinical use, we analyzed a short list of 137 genes and interestingly, among the 13 altered genes (Table 2), we observed a potentially pathogenic APC mutation, which could predispose to CRC. Moreover, a KRAS activating mutation (G12D), commonly found in CRC, was observed. What was even more interesting is that we observed a somatic mutation of the homologous repair in RAD51C (T287A) previously reported to be associated with a loss of function[8]. Analysis of copy number variable showed a loss of heterozygosity in chromosome 17 from 56770004 to 56801461 (Figure 1B). This analysis suggested a complete deletion of RAD51C function in tumor cells. After discussion of the case at the molecular tumor board, the patient was proposed to receive off-label PARP inhibitor olaparib which previously showed efficacy in patients with RAD51 mutation in metastatic prostate cancer[7]. The patient received 3 mo of olaparib therapy without toxicity. Despite the absence of toxicity, the magnetic resonance imaging (Figure 2) showed tumor progression. Olaparib was stopped and the patient was included in a phase I clinical trial.
Table 2.
Alterations observed for Case 2 in the short list of clinically relevant genes
| Gene | Nucleotide variation | Protein variation | Origin | Impact |
| APC | c.3949G>C | Glu1317Gln | Constitutive | Potentially Pathogenic |
| BRCA1 | c.5180G>C | Gly1727Ala | Somatic | Unknown |
| BRCA1 | c.3119G>A | Ser1040Asn | Constitutive | SNP |
| BRCA1 | c.2521C>T | Arg841Trp | Somatic | Neutral |
| FGFR4 | c.1676G>A | Arg559Gln | Somatic | Unknown |
| GLI2 | c.1418G>A | Arg473His | Constitutive | Unknown |
| JAK2 | c.195A>C | Glu65Asp | Constitutive | Unknown |
| JAK3 | c.2164G>A | Val722Ile | Constitutive | Activating |
| KDR | c.2159G>A | Arg720Gln | Somatic | Unknown |
| KRAS | c.35G>A | Gly12Asp | Somatic | Activating |
| MET | c.2572G>A | Val858Met | Constitutive | Unknown |
| RAD51C | c.859A>G | Thr287Ala | Somatic | Loss of function |
| SLC28A1 | c.568G>T | Ala190Ser | Constitutive | SNP |
| SLC28A1 | c.1152delG | Val385Ser fsX16 | Constitutive | Loss of function |
| THRA | c.454C>T | Arg152X | Somatic | Loss of function |
| UIMC1 | c.43C>T | Arg15Trp | Constitutive | Unknown |
SNP: Single nucleotide polymorphism.
As loss of TP53BP1 was previously described to be involved in PARP inhibitor resistance in homologous repair deficient breast cancer models in mice[9], we searched for the mutation of this gene in both patients. While patient 1 had a wild-type TP53PB1 gene, patient 2 had a frameshift truncating insertion in TP53BP1 (AG insertion at chromosomal position 17:43766919) (Figure 1C), thus suggesting a loss of function of the protein.
DISCUSSION
The use of exome sequencing in cancer has largely taken place in the setting of research studies, such as The Cancer Genome Atlas. However, integration of exome sequencing into precision cancer care remains a challenging issue because samples issued from formalin-fixed tissues (i.e., FFPE) are difficult to analyze (due to small size and poor quality). Furthermore, bioinformatics approaches require detection of the wide-spectrum of mutations with the capacity to identify actionable mutations at an acceptable sensitivity and it remains an issue. This case report is the first to suggest efficacy of olaparib in CRC with homologous repair deficiency and to demonstrate that exome sequencing could be used to help drug repositioning. Accordingly, single-agent olaparib as well as combinations with irinotecan were previously tested in unselected CRC that is resistant to standard therapy without any clinical efficacy[10,11]. Such data raises the hypothesis that olaparib activity also requires molecular testing of homologous repair deficiency for CRC as requested for ovarian cancer.
Homologous repair is an enzymatic complex aimed at repairing DNA double-strand breaks. CHEK2 and RAD51 play a central role in the repair of DNA double-strand breaks performed by homologous repair[12]. After the detection of DNA double-strand breaks by ATM and CHEK2, BRCA1 is phosphorylated by these proteins. Rad52 binds recombinase polymerase amplification and displaces it to allow Rad51 binding. BRCA2 binds to Rad51 until BRCA2 becomes phosphorylated, releasing Rad51 and allowing it to localize to the double strand break with Rad52. Homologous repair-mediated repair goes on; Rad51 then forms a nucleoprotein filament that invades a homologous sequence and activates strand exchange to generate a crossover between the juxtaposed DNA[13]. Several studies have shown that the level of RAD51 protein expression is elevated in immortalized cells as well as in a wide variety of human cancer cell lines[14,15]. It is generally suggested that RAD51 overexpression results in an increased cellular resistance to radiation and some chemotherapeutic drugs, such as topoisomerase inhibitors or crosslinking agents[15-17]. In CRC, high expression of RAD51 is associated with poor prognosis[18]. Several in vitro studies have shown that an increase in RAD51 expression stimulates homologous repair, resulting in greater cellular resistance to treatment with crosslinking agents, etoposide or irradiation[15-17]. High numbers of RAD51 foci in tumor biopsies were also positively associated with greater chemoresistance in breast cancer patients[19]. Chek2 is also involved in the colorectal oncogenesis and protein truncating mutations in CHEK2 have been reported to confer higher risks of cancer of the breast and the prostate but also CRC[20].
Biallelic invalidation of homologous repair enzymes are still described to be a predictive marker of response to PARP inhibitor like olaparib in different diseases such as breast, ovarian and prostate cancer[7,21-23]. Our report is the first one that researched biallelic deficiency of homologous repair enzyme in metastatic CRC patients and underlines that such events could also occur in this pathology and could be targeted.
Resistance to PARP inhibitors is a key question and determining a predictive biomarker is important for the future design of clinical trials. Three mechanisms of resistance have been described: restoration of BRCA function by additional mutations[24-26], increased Mdr1 gene expression[27] or loss of TP53BP1[9]. Interestingly, TP53BP1 loss also induces resistance to the topoisomerase I inhibitor while tumor cells remain sensitive to DNA crosslinking agents like platinum, may thus explain the good response to FOLFIRINOX.
To conclude, we believe that this case report supports that large genetic characterization of metastatic CRC patients could be useful to find molecular hits that could be targeted by off-label targeted therapy. PARP inhibitors could be particularly useful is this context. Nevertheless, biallelic deficiency of homologous repair enzyme is a prerequisite to benefit from a PARP inhibitor therapy, but is not always associated to a PARP inhibitor response. Additional resistance pathways like TP53BP1 loss must be determined before prescribing PARP inhibitors.
COMMENTS
Case characteristics
Two case of multitreated metastatic colorectal cancer patients that benefit from genetic testing.
Clinical diagnosis
metastatic colorectal cancer.
Laboratory diagnosis
Whole exome sequencing revealed inactivating mutation in the homologous repair gene with genetic deficiency in RAD51C or Check2.
Treatment
Patients were treated with the poly ADP ribose polymerase inhibitor olaparib.
Experiences and lessons
One out the 2 patients gained clinical benefit from olaparib usage, thus suggesting that genetic testing could also be used in colorectal cancer to predict response to olaparib.
Peer-review
This case report highlighting the importance of the exome sequencing analysis before administering targeted therapy.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: France
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C, C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was approved by the local institutional review board.
Informed consent statement: Patients gave their written consent to authorize genetic analyses.
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Peer-review started: July 11, 2016
First decision: August 22, 2016
Article in press: September 28, 2016
P- Reviewer: Lakatos PL, Matsuda A, Meshikhes AW S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Liu WX
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, Bechstein WO, Primrose JN, Walpole ET, Finch-Jones M, Jaeck D, Mirza D, Parks RW, Collette L, Praet M, Bethe U, Van Cutsem E, Scheithauer W, Gruenberger T, EORTC Gastro-Intestinal Tract Cancer Group, Cancer Research UK, Arbeitsgruppe Lebermetastasen und-tumoren in der Chirurgischen Arbeitsgemeinschaft Onkologie (ALM-CAO), Australasian Gastro-Intestinal Trials Group (AGITG), Fédération Francophone de Cancérologie Digestive (FFCD) Perioperative chemotherapy with FOLFOX4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC Intergroup trial 40983): a randomised controlled trial. Lancet. 2008;371:1007–1016. doi: 10.1016/S0140-6736(08)60455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van den Eynde M, Hendlisz A. Treatment of colorectal liver metastases: a review. Rev Recent Clin Trials. 2009;4:56–62. doi: 10.2174/157488709787047558. [DOI] [PubMed] [Google Scholar]
- 4.Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–1047. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 5.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix-Lerosey I, Delattre O, Barillot E. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics. 2011;27:268–269. doi: 10.1093/bioinformatics/btq635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 9.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leichman L, Groshen S, O’Neil BH, Messersmith W, Berlin J, Chan E, Leichman CG, Cohen SJ, Cohen D, Lenz HJ, et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist. 2016;21:172–177. doi: 10.1634/theoncologist.2015-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen EX, Jonker DJ, Siu LL, McKeever K, Keller D, Wells J, Hagerman L, Seymour L. A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Invest New Drugs. 2016;34:450–457. doi: 10.1007/s10637-016-0351-x. [DOI] [PubMed] [Google Scholar]
- 12.Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature. 1998;391:401–404. doi: 10.1038/34937. [DOI] [PubMed] [Google Scholar]
- 13.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 14.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 15.Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int J Cancer. 2003;105:472–479. doi: 10.1002/ijc.11106. [DOI] [PubMed] [Google Scholar]
- 16.Slupianek A, Hoser G, Majsterek I, Bronisz A, Malecki M, Blasiak J, Fishel R, Skorski T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–4201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vispé S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998;26:2859–2864. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, Petersen C, Sauter G, Dikomey E, Borgmann K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer. 2013;132:2118–2126. doi: 10.1002/ijc.27907. [DOI] [PubMed] [Google Scholar]
- 19.Asakawa H, Koizumi H, Koike A, Takahashi M, Wu W, Iwase H, Fukuda M, Ohta T. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010;12:R17. doi: 10.1186/bcr2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cybulski C, Wokołorczyk D, Kładny J, Kurzawski G, Suchy J, Grabowska E, Gronwald J, Huzarski T, Byrski T, Górski B, et al. Germline CHEK2 mutations and colorectal cancer risk: different effects of a missense and truncating mutations? Eur J Hum Genet. 2007;15:237–241. doi: 10.1038/sj.ejhg.5201734. [DOI] [PubMed] [Google Scholar]
- 21.Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106:dju089. doi: 10.1093/jnci/dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16:87–97. doi: 10.1016/S1470-2045(14)71135-0. [DOI] [PubMed] [Google Scholar]
- 23.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 25.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]