

Summary
Objective
Co‐administration of amylin and leptin induces synergistic and clinically meaningful (>10%) weight loss that is attenuated as the degree of obesity increases. We explored whether calorie restriction (CR) could restore amylin/leptin synergy in very obese rats.
Methods
Sprague Dawley rats on high‐fat diet (696 ± 8 g, n = 72) were randomized to three cohorts (C1–C3). Rats in C1 were administered vehicle, rat amylin (50 µg kg−1 d−1), murine leptin (125 µg kg−1 d−1) or amylin and leptin for 28 days (n = 6 per group) via subcutaneous minipump. Simultaneously, C2 and C3 rats initiated CR. After moderate (12.4 ± 0.3%, 86.7 ± 2.8 g; C2) or severe (24.9 ± 0.3%, 172.7 ± 4.7 g; C3) weight loss, amylin and/or leptin was administered as described.
Results
In C1, leptin did not alter weight, and amylin induced 40.2 ± 6.1 g weight loss (−6.0 ± 0.9%), which was not enhanced by leptin (44.4 ± 4.9 g, −6.1 ± 0.8%). In C2, vehicle‐treated (75.1 ± 7.8 g weight change from start of treatment, 1.1 ± 0.8% difference from start of pre‐CR phase) and leptin‐treated rats (68.6 ± 9.2 g, −1.3 ± 1.0%) rebounded to pre‐restriction weight that was attenuated by amylin (29.2 ± 11.4 g, −6.2 ± 0.7%). Leptin did not enhance the effect of amylin (22.8 ± 11.7 g, −8.3 ± 1.5%). In C3, vehicle‐treated and leptin‐treated rats regained most of their weight (161.9 ± 11.8, −2.3 ± 0.8% and 144.6 ± 9.5 g, −2.3 ± 0.9%, respectively), which was attenuated by amylin (91.1 ± 16.8 g, −11.2 ± 0.7%), but not enhanced by leptin (83.0 ± 7.6 g, −10.7 ± 0.8%).
Conclusions
Extreme obesity associated with leptin resistance perturbs amylin/leptin weight loss synergy in rats, which cannot be restored by pre‐treatment weight loss.
Keywords: Diabetes, obesity, synergy
Introduction
Leptin, a hormone secreted by adipose tissue, has long been touted as a potential anti‐obesity molecule 1. The role of endogenous leptin in the regulation of energy balance is clear, yet exogenously administered leptin has remained intractable as a therapy for obesity 2. Leptin administration to leptin‐deficient rodents and humans dramatically improves obesity and metabolic disease 3, 4, and recombinant human leptin (metreleptin) was recently approved as an adjunct to diet for use in individuals with leptin‐insufficient congenital or acquired generalized lipodystrophy. In leptin‐deficient states like generalized lipodystrophy, metreleptin has been reported to improve the profound insulin resistance and other metabolic abnormalities present in that population 5. However, in states like obesity, the phenomenon of leptin resistance is uniformly observed and is felt to be a consequence in part of the endogenous hyperleptinemia associated with obesity 2, 6. While the mechanism or molecular foundation of leptin resistance remains unknown, the potential ability to restore leptin sensitivity in states of obesity‐associated leptin resistance remains an attractive area of research for anti‐obesity pharmacotherapies 7.
Amylin is a peptide hormone co‐secreted with insulin from pancreatic β‐cells, which has primary physiological roles in the regulation of glucose and energy balance via its activity at specific receptors in the area postrema of the hindbrain 8. In humans with obesity, the synthetic amylin analogue pramlintide inhibited food intake and induced significant weight loss 9, 10. Amylin has been demonstrated to be one of the few molecules that is able to restore leptin sensitivity in obesity. The co‐administration of amylin and leptin (in rodents) or pramlintide and metreleptin (in humans) elicited synergistic clinically meaningful effects on weight loss 11, 12, 13, 14. Leptin resistance, however, is not so easily, nor uniformly, abolished. In studies in extremely diet‐induced obese (DIO) rats (average body weight ~800 g), we reported that amylin/leptin synergy was less evident compared with more moderately DIO (average body weight ~500 g) rats 15. This was further reflected in the results of a 52‐week blinded, placebo‐controlled phase II study of pramlintide/metreleptin in which the most robust efficacy was seen in patients with a body mass index less than 35 kg m−2 16.
As a means to potentially understand whether pramlintide/metreleptin could have therapeutic utility in the most obese subjects where the incidence and impact of weight‐related comorbidities is most profound 17, we investigated whether the loss of amylin‐mediated leptin synergy in a state of extreme obesity could be rescued by weight loss induced via caloric restriction prior to pharmacotherapy.
Materials and methods
Animal experiments
All studies were approved by the Institutional Animal Care and Use Committee at Amylin Pharmaceuticals, LLC in accordance with Animal Welfare Act guidelines. Animals were housed individually in standard caging at 22 C in a 12‐h light : dark cycle. Male Sprague Dawley rats (n = 85; Charles River Laboratories, Wilmington, MA, USA) were pre‐fattened on a high‐fat diet (HFD; 32% kcal per fat; D12266B, Research Diets, Brunswick, NJ, USA) at Charles River from 6 weeks of age until 12 weeks of age. Upon arrival, the mean body weight of the animals was 441 ± 4 g, and rats were maintained on the same diet for a further 22 weeks (34 weeks of age). At this time, the final study population (n = 72) was selected, and mean body weight was 696 ± 8 g.
The final population was subjected to a body composition assessment (EchoMRI, Echo Medical Systems, Houston, TX, USA) and randomized to three cohorts (each n = 24) based on body weight. Rats in cohort 1 (C1) were subsequently randomized to four weight‐matched groups (n = 6 per group) for vehicle or drug treatment (described in the succeeding text). Remaining rats in cohorts 2 and 3 (C2 and C3) were subjected to 50% CR based on mean 24‐h food intake for the previous 5 days (average food intake for the whole group of rats was 17.6 ± 0.3 g; rats in C2 and C3 were allocated 8.8 g of diet per day, just prior to lights off). At this level of CR, body weight loss of the rats began to plateau by day 5, so on day 6, the degree of CR was increased to 75%, and rats were allocated 4.4 g of diet per day and throughout the remainder of the CR phase. On day 15 of CR when animals had reached 12.4% body weight loss, rats in C2 were subjected to a second body composition assessment (EchoMRI), and on day 16, rats were randomized to four weight‐matched groups for vehicle or drug administration. On day 34 of CR, the target weight loss of 25% was reached in the remaining C3 rats, and they were administered a body composition assessment (EchoMRI) and randomized to vehicle or drug treatment groups based on body weight.
Drug administration and plasma analysis
On day 0 (C1), day 16 (C2) or day 35 (C3) of the CR phase, rats were implanted subcutaneously with two osmotic minipumps (Alzet, Cupertino, CA model 2ML4), delivering either vehicle (50% dimethyl sulfoxide)/vehicle (sterile water), amylin (50 µg kg−1 d−1)/vehicle (sterile water), leptin (125 µg kg−1 d−1)/vehicle (50% dimethyl sulfoxide) or amylin (50 µg kg−1 d−1)/leptin (125 µg kg−1 d−1) for 28 days. These doses were selected from a previous study that mapped amylin + leptin weight loss synergy across a range of doses using response surface methodology 12. Minipumps were prepared the day before implantation and were incubated in sterile saline at 37 °C overnight. Minipumps were implanted in the subscapular space, one in either side of the spine, in sterile conditions with the rats under isoflurane anaesthesia. Following minipump implantation, all animals were allowed ad libitum access to HFD (D12266B, research diets).
Vehicle or drug was administered via minipump for 28 days. Body weight and food intake were measured weekly.On day 28 of drug treatment, rats were administered a final body composition assessment (EchoMRI), then euthanized via anaesthetic (isoflurane) overdose and cardiac blood collected via cardiac puncture. Blood was collected into a heparin sodium‐coated tube, and plasma glucose, triglycerides and total cholesterol were measured using an Olympus AU400e Bioanalyzer (Olympus America, Irving, TX, USA).
Statistical analyses
Single‐point data were analysed using one‐way analysis of variance, with Bonferroni post hoc test. For longitudinal group comparisons, data were analysed using two‐way analysis of variance with drug * time as variables and Bonferroni repeated measures post hoc test. Significance was assumed for p < 0.05. Graphs and statistical analyses were generated using Prism 6 for Windows (Graphpad Software, San Diego, CA, USA). All data are expressed as mean ± SEM.
Results
For the extremely obese rats in C1 (mean body weight 696 ± 8 g), sustained infusion of leptin had no effect on body weight, whereas amylin administration elicited the expected effect on body weight inducing 40.2 ± 6.1 g (6.0 ± 0.9%) body weight loss over the 4‐week infusion period (Figures 1 and 2A). Weight loss was associated with reductions in cumulative food intake (Table 1) and significantly reduced fat mass (Figure 3A) and sparing of fat‐free mass (Table 1). The addition of leptin to amylin exerted no additional benefit on body weight (44.4 ± 4.9 g; 6.1 ± 0.8% weight loss), fat mass or food intake (Figures 1 and 2A and Table 1), confirming that synergy between amylin and leptin is lost in states of extreme obesity.
Figure 1.
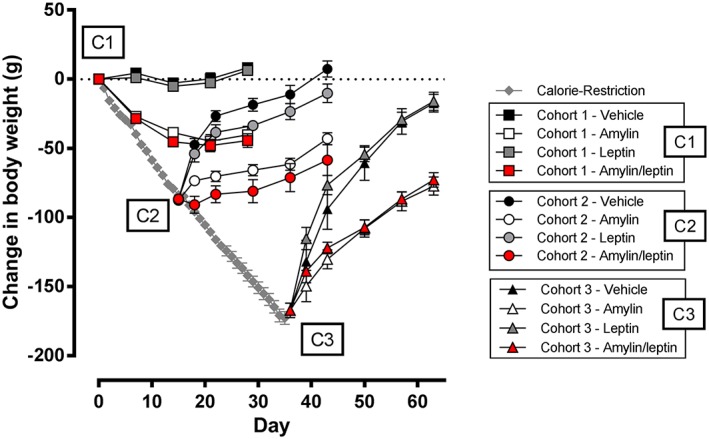
Change in weight of very DIO rats (starting body weight 696 g) subjected to amylin, leptin or amylin/leptin infusion. C1, cohort 1: no calorie restriction; C2, cohort 2: 12.1% weight loss via calorie restriction; C3, cohort 3: 25% weight loss via calorie restriction.
Figure 2.
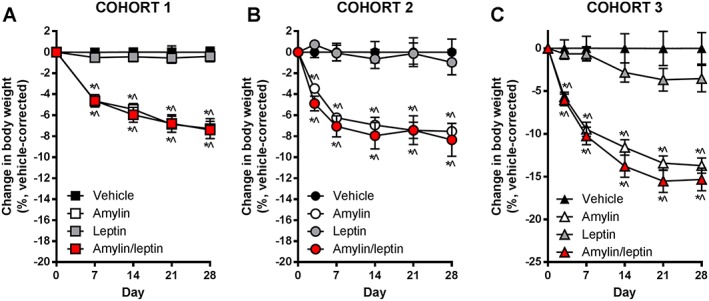
Change in body weight for (A) cohort 1, (B) cohort 2 and (C) cohort 3 rats from start of drug treatment, expressed as change in body weight in percent, corrected to vehicle. * p < 0.05 vs. vehicle; ^p < 0.05 vs. leptin monotherapy.
Table 1.
Fat‐free mass expressed as grams or percent of body weight of DIO rats at baseline (cohort 1; after prolonged high‐fat diet feeding), after CR to induce 11% (cohort 2) or 25% (cohort 3) body weight loss and at termination after 28 days of vehicle or drug treatment
| Cohort | Treatment | Baseline fat‐free mass (g) | Post‐CR fat‐free mass (g) | Terminal fat‐free mass (g) | Baseline fat‐free mass (%) | Post‐CR fat‐free mass (%) | Terminal fat‐free mass (%) | Total food intake (g) |
|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Vehicle | 441 ± 17 | — | 410 ± 13 | 64.1 ± 3.5 | — | 60.7 ± 3.2 | 464 ± 15 |
| Amylin | 423 ± 14 | — | 385 ± 11 | 63.7 ± 1.7 | — | 63.6 ± 1.6 | 336 ± 23‡, § | |
| Leptin | 437 ± 8 | — | 396 ± 8 | 63.0 ± 1.7 | — | 58.0 ± 1.9 | 466 ± 12 | |
| Amylin/leptin | 451 ± 16 | — | 403 ± 12 | 62.5 ± 2.0 | — | 61.5 ± 2.3 | 369 ± 20‡, § | |
| Cohort 2 | Vehicle | 443 ± 20 | 391 ± 15* | 416 ± 17*, † | 64.3 ± 2.6 | 64.0 ± 2.6 | 62.1 ± 2.2 | 538 ± 19 |
| Amylin | 455 ± 19 | 402 ± 16* | 408 ± 19* | 64.1 ± 0.6 | 64.3 ± 0.8 | 64.6 ± 0.4 | 442 ± 16‡, § | |
| Leptin | 458 ± 30 | 397 ± 19* | 416 ± 22* | 65.2 ± 1.9 | 65.0 ± 1.8 | 63.3 ± 0.9 | 550 ± 21‡ | |
| Amylin/leptin | 455 ± 13 | 395 ± 9* | 402 ± 13* | 65.1 ± 2.8 | 64.9 ± 2.6 | 66.0 ± 2.7 | 438 ± 23‡, § | |
| Cohort 3 | Vehicle | 459 ± 13 | 369 ± 15* | 418 ± 15*, † | 66.3 ± 3.6 | 72.0 ± 2.8* | 64.5 ± 3.1† | 692 ± 55 |
| Amylin | 443 ± 16 | 354 ± 12* | 396 ± 21*, † | 64.3 ± 2.4 | 68.7 ± 1.5* | 68.1 ± 1.0* | 486 ± 33‡, § | |
| Leptin | 452 ± 24 | 379 ± 16* | 416 ± 21*, † | 65.7 ± 1.8 | 73.1 ± 1.3* | 64.5 ± 1.8† | 645 ± 32 | |
| Amylin/leptin | 426 ± 6 | 352 ± 8* | 384 ± 8*, † | 62.1 ± 0.6 | 67.7 ± 0.6* | 66.3 ± 1.0* | 508 ± 23‡ |
Total cumulative food intake is also shown for the 28‐day treatment period.
p < 0.05 vs. baseline.
p < 0.05 vs. post‐CR within treatment group.
p < 0.05 vs. vehicle.
p < 0.05 vs. leptin within cohort.
CR, calorie restriction; DIO, diet‐induced obese.
Figure 3.
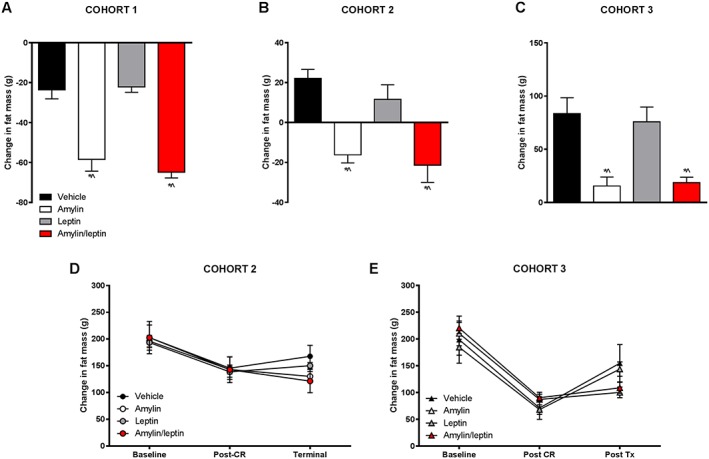
Change in fat mass for (A) cohort 1, (B) cohort 2 and (C) cohort 3 rats from start of drug treatment through 4 weeks of amylin, leptin or amylin/leptin infusion expressed as change in fat mass in grams. Change in fat mass for the calorie‐restricted groups (D) cohort 2 and (E) cohort 3 showing fat mass (g) at baseline, after calorie restriction (post‐calorie restriction [CR]) and at termination. * p < 0.05 vs. vehicle; ^p < 0.05 vs. leptin monotherapy.
Calorie restriction (50–75%) for 15 days induced 12.4 ± 0.3% reduction in body weight (mean body weight of 621 ± 12 g). CR‐induced weight loss in C2 was associated with reduced fat mass (Figure 3D) and lower fat‐free mass expressed in grams but not as a percent of body weight (Table 1). Rats in this cohort (C2) subsequently allowed ad libitum access to HFD and administered vehicle for 4 weeks regained all of the lost body weight (change in weight from pre‐CR phase of 7.4 ± 5.9 g equating to final body weight 1.1 ± 0.8% above initial pre‐CR body weight), with most of the weight being regained in the first week (Figure 1). Weight regain was associated with a regain in fat mass (Figure 3B) and increased fat‐free mass (Table 1). Leptin alone did not prevent the rapid body weight gain (change in weight from pre‐CR phase of −10.2 ± 6.6 g equating to −1.3 ± 1.0% lower than initial pre‐CR body weight) or alter body composition (Figure 3A,B,D). Leptin‐treated rats consumed slightly more food than vehicle controls (Table 1). Amylin administration significantly prevented body weight gain that was not further enhanced by the co‐administration of leptin. Amylin‐mediated prevention of weight regain (−43.1 ± 4.3 g or −6.2 ± 0.7% from pre‐CR weight) was associated with reductions in fat mass (Figure 3B) and inhibition of cumulative food intake over the 4‐week treatment period (Table 1). In the amylin/leptin combination group, body weight regain was inhibited (−58.5 ± 11.2 g, or −8.3 ± 1.5% from pre‐CR weight) in concert with reduced fat mass and lower cumulative food intake compared with vehicle‐treated or leptin‐treated rats (Table 1). There were no significant differences between C2 rats treated with either amylin alone or combination of amylin/leptin.
Further CR until day 35 in the C3 cohort led to overall weight reduction of 24.9 ± 0.3% (mean body weight of 519 ± 11 g), corresponding to the typical DIO weight status in previous studies exploring amylin/leptin synergy 11, 15. Weight loss of this extent was associated with a dramatic reduction in fat mass (Figure 3E) and fat‐free mass (Table 1). Normalized fat‐free mass actually increased after caloric restriction, because of the dramatic loss of fat mass in this cohort (Table 1). Rats in C3 allowed ad libitum access to HFD, and administered vehicle for 4 weeks regained almost all of their lost weight (change in weight from pre‐CR phase of −17.4 ± 6.4 g equating to final body weight 2.3 ± 0.8% below initial pre‐CR body weight; Figure 1). Weight regain was associated with increased fat mass (Figure 3C,E) and fat‐free mass (Table 1). Similar to the effects observed in the C2 cohort, leptin monotherapy had no impact on preventing weight regain (change in weight from pre‐CR phase of −16.0 ± 6.6 g or −2.3 ± 0.9%; Figure 1), regain of fat mass (Figure 3C,E), fat‐free mass changes or food intake (Table 1). Amylin monotherapy significantly prevented body weight regain (change in weight from pre‐CR phase of −77.3 ± 6.6 g equating to −11.2 ± 0.7% lower than initial pre‐CR body weight; Figure 1) and inhibited regain of adipose tissue (Figure 3C) without significantly affecting fat‐free mass when expressed relative to body weight (Table 1). Amylin‐mediated prevention of body weight was associated with significant inhibition of cumulative food intake (Table 1). Effects on body weight, body composition and food intake in the amylin/leptin combination were similar to those observed with amylin alone. Body weight regain was significantly inhibited (−72.9 ± 5.2 g, or −10.7 ± 0.8% from pre‐CR weight; Figure 1) but was not different from amylin monotherapy. Likewise, gains in fat mass were inhibited (Figure 3C,E), there was no impact on percent fat‐free mass and food intake was inhibited, but none of these effects were different from amylin monotherapy group (Table 1).
To more clearly show the impact of pharmacotherapy, the percentage change in body weight relative to vehicle control groups was analysed for each cohort (Figure 2). In all three cohorts, the impact of pharmacotherapy was extremely similar irrespective of starting body weight or nutritive status. Leptin as a monotherapy was ineffective irrespective of the weight status of the cohorts. Amylin monotherapy induced significant weight loss in C1 at the same magnitude as observed in typical DIO rats previously 18 and reduced the change in weight in C2 and C3 significantly relative to vehicle and leptin‐treated rats. Amylin/leptin co‐administration did not elicit any additional weight loss, in any cohort, beyond that seen with amylin alone (Figure 2).
Measurement of terminal plasma concentrations of glucose, total cholesterol and triglycerides revealed no significant effect of pharmacotherapy on any parameter within any cohort (Table 2). There was a trend for reduced plasma triglycerides in amylin‐treated and amylin/leptin‐treated groups, irrespective of cohort, compared with vehicle controls, but this did not achieve statistical significance.
Table 2.
Terminal plasma glucose, total cholesterol or triglycerides of non‐fasted DIO rats at termination after 28 days of vehicle or drug treatment
| Cohort | Drug treatment | Plasma glucose (mg dL−1) | Cholesterol (mg dL−1) | Triglycerides (mg dL−1) |
|---|---|---|---|---|
| Cohort 1 | Vehicle | 156 ± 3 | 123 ± 6 | 238 ± 44 |
| Amylin | 149 ± 5 | 106 ± 8 | 132 ± 17 | |
| Leptin | 152 ± 4 | 116 ± 11 | 166 ± 46 | |
| Amylin/leptin | 153 ± 3 | 109 ± 7 | 110 ± 26 | |
| Cohort 2 | Vehicle | 163 ± 4 | 121 ± 12 | 224 ± 41 |
| Amylin | 155 ± 4 | 102 ± 3 | 129 ± 27 | |
| Leptin | 154 ± 3 | 94 ± 3 | 213 ± 24 | |
| Amylin/leptin | 151 ± 4 | 110 ± 8 | 142 ± 24 | |
| Cohort 3 | Vehicle | 155 ± 5 | 112 ± 13 | 185 ± 28 |
| Amylin | 162 ± 10 | 94 ± 5 | 139 ± 33 | |
| Leptin | 147 ± 1 | 113 ± 5 | 182 ± 32 | |
| Amylin/leptin | 149 ± 4 | 100 ± 8 | 127 ± 22 |
Discussion
Two potential ways to overcome obesity‐related leptin resistance are by co‐administering a second agent (a ‘leptin sensitizer’) and/or to use diet‐induced weight loss to drive down endogenous leptin levels. The present study examined both of these strategies. Co‐administration of amylin and leptin induces synergistic reductions in body weight in DIO rats and obese/overweight humans 11, 12, 13. Subsequent clinical and preclinical studies revealed that synergy may be conditional upon the initial weight status of the animal, with reduced synergy evident in rats or humans starting with higher than average weight 15. The present studies confirmed these findings by showing that dose regimens of amylin/leptin that synergized in DIO rats (mean weight of 476 g; 12) failed to synergize in the ad libitum very DIO group (mean weight of 696 g). Of note, the relative efficacy of amylin monotherapy to decrease food intake and body weight does not appear to be attenuated by prevailing body weight or nutritive status. In all three cohorts, amylin exerted a similar level of vehicle‐corrected weight loss on the order of 6–8%. These findings are in line with studies in high‐fat‐fed female rats showing that amylin's anorexigenic and fat‐specific weight‐lowering effects were unaltered across a variety of nutritive states 18, 19.
In human subjects that have undergone CR to induce 10% body weight loss, leptin administration can attenuate or prevent counter‐regulatory metabolic responses 20 and significantly increased satiation 21. These data suggest that in obese weight‐reduced humans, some aspects of leptin responsiveness are present. The overall neuroendocrinological and physiological state induced by CR‐mediated weight reduction is that of leptin insufficiency or leptin deficiency. Such data led to the speculation that leptin may be an effective weight loss maintenance molecule. Our data suggest that leptin administration alone, or in combination with amylin, is unable to override the orexigenic drive that results from prolonged CR to prevent rebound hyperphagia and subsequent weight regain. Our results clearly indicate that, at least in DIO rats, restoration of amylin/leptin synergy does not occur after CR‐mediated weight loss and sheds insight into the regulation of a body weight ‘set point’ related to leptin resistance. These conclusions can only be considered in the scope of the degree of initial obesity, magnitude of restriction and doses of amylin and leptin tested.
Recently, the glucagon‐like peptide‐1 receptor agonist exenatide has been proposed to exert leptin‐sensitizing actions. DIO mice treated with exenatide to induce robust 28% weight loss (in conjunction with switching from high‐fat to low‐fat diet) were further weight reduced by the addition of a leptin analogue suggesting exenatide‐mediated enhancements in leptin sensitivity 22. Conversely, DIO mice weight matched via CR did not respond to subsequent leptin analogue administration 22. Similar results were also obtained when fibroblast growth factor‐21 (FGF‐21) was utilized in combination with diet switching to induce significant weight loss. Concomitant co‐administration of the leptin analogue with FGF‐21 yielded superior body weight loss compared with FGF‐21 alone, and leptin alone was ineffective 22. Interestingly, in DIO mice maintained on HFD throughout the treatment period, there was no additional benefit of leptin in combination with either exenatide or FGF‐21 22. In DIO mice, it appears that exenatide or FGF‐21, but not weight loss, can restore some degree of leptin sensitization but only in the context of removing HFD and after ~28% weight loss, a highly contrived metabolic state that is unlikely to be relevant to clinical treatment of obesity. In obese human subjects that had lost 30.8% of body weight following gastric bypass surgery, leptin administration had no effect on body weight 23. Overall, the extant data imply that pharmacological interventions may hold promise for restoration of leptin sensitization but not weight loss per se and that a nutritional component and/or its influence upon underlying neuronal metabolic circuitries may be involved. To what extent switching extremely obese rats from an HFD to a low‐fat chow diet will enable restoration of amylin‐mediated leptin re‐sensitization has not been reported.
Although our pharmacological study did not include detailed mechanistic endpoints, one possibility for why leptin sensitivity was not restored is that the threshold for leptin sensitivity is continually (upwardly) re‐set through long‐term hyperleptinemia induced by exposure to HFD 24. At states of typical obesity, amylin agonism may be able to alter this threshold such that leptin responsiveness is restored, but that prolonged CR appears to be insufficient to enable a lowering of the leptin sensitivity threshold to a degree such that a level of amylin agonism that would normally engage leptin functionality does so in the extremely obese state. How amylin engages leptin responsiveness intrinsically, or in typical obesity, is unknown, although it is almost certainly occurring within the brain. Amylin administration enhances leptin signalling in the hypothalamus of DIO rats 11, 13. More recently, it has been shown that amylin and leptin work cooperatively in the hypothalamus to regulate feeding behaviour 25 and that leptin and amylin modulate the firing rate of a distinct population of neurons in the area postrema of the brainstem 26. Within the ventromedial hypothalamus, where sustained amylin infusion has been shown to enhance leptin binding, amylin agonism has been linked with interleukin‐6 production and enhanced leptin signalling (27). Taken together, while still not clearly defined, the neural substrates connecting amylin to leptin (and leptin resistance) are being identified and seem to encompass the primary neuronal centres responsible for feeding behaviour. The regulation of hypothalamic interleukin‐6 production, and potentially other central mediators of amylin/leptin interaction, in extreme obesity is an important question in the context of leptin sensitivity and whether or not it can be restored via pharmacological mechanism/s.
The formal possibility also remains that synergy can be restored with higher doses/exposures of leptin and/or amylin, or by utilizing optimized analogues of amylin and/or leptin. The dose of leptin administered in the present studies has been shown to at least replace the decline in endogenous leptin levels elicited by amylin and amylin/leptin‐induced weight and fat loss in DIO rats at baseline 12. Here, terminal levels of plasma leptin from C1 were 30 ng mL−1, which is within the physiological range for obese rodents and humans (28), and in the leptin alone or amylin/leptin group, leptin concentrations were 80–100 ng mL−1 (data not shown). These observations suggest that our very DIO rats were not in a state of leptin deficiency. Nevertheless, the possibility remains that we had achieved higher plasma levels of leptin by administering a larger dose, or administered more potent leptin analogues; we could have restored leptin responsiveness. Alternatively, the possibility remains that in states of extreme obesity, the leptin sensitivity threshold remains irreversibly perturbed.
In conclusion, we demonstrate that analogous to clinical observations with pramlintide and metreleptin in individuals with a higher body mass index that amylin/leptin synergy is absent in extremely DIO rats. Synergy, or even additivity, was clearly not rescued by reducing body weight via CR. Despite extreme obesity, amylin continued to elicit significant reductions in body weight and adiposity and prevent weight gain significantly in CR DIO rats. The failure of CR‐mediated weight loss to restore amylin‐induced leptin sensitization suggests that the threshold for leptin sensitivity (or resistance) may be further increased in the face of continued exposure to HFD and hyperleptinemia and cannot be readily restored to normal levels by food restriction.
Conflict of Interest Statement
At the time when this work was performed, all authors were employed at Amylin Pharmaceuticals, LLC. All authors held stock in Amylin Pharmaceuticals, LLC. J. L. T. is a current employee of MedImmune/AstraZeneca, which retains ownership of pramlintide, marketed as SYMLIN.
Acknowledgements
The authors wish to thank Jim Napora and the staff of the Comparative Medicine facility at Amylin Pharmaceuticals for assistance with animal husbandry and care.
Trevaskis, J. L. , Wittmer, C. , Athanacio, J. , Griffin, P. S. , Parkes, D. G. , and Roth, J. D. (2016) Amylin/leptin synergy is absent in extreme obesity and not restored by calorie restriction‐induced weight loss in rats. Obesity Science & Practice, 2: 385–391. doi: 10.1002/osp4.62.
References
- 1. Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 2. Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose‐escalation trial. JAMA 1999; 282: 1568–1575. [DOI] [PubMed] [Google Scholar]
- 3. Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 2002; 110: 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Halaas JL, Gajiwala KS, Maffei M, et al. Weight‐reducing effects of the plasma protein encoded by the obese gene. Science 1995; 269: 543–546. [DOI] [PubMed] [Google Scholar]
- 5. Chan JL, Lutz K, Cochran E, et al. Clinical effects of long‐term metreleptin treatment in patients with lipodystrophy. Endocr Pract 2011; 17: 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Halaas JL, Boozer C, Blair‐West J, et al. Physiological response to long‐term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A 1997; 94: 8878–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Santoro A, Mattace Raso G, Meli R. Drug targeting of leptin resistance. Life Sci 2015; 140: 64–74. [DOI] [PubMed] [Google Scholar]
- 8. Lutz TA. Amylinergic control of food intake. Physiol Behav 2006; 89: 465–471. [DOI] [PubMed] [Google Scholar]
- 9. Aronne L, Fujioka K, Aroda V, et al. Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo‐controlled, dose‐escalation study. J Clin Endocrinol Metab 2007; 92: 2977–2983. [DOI] [PubMed] [Google Scholar]
- 10. Smith SR, Aronne LJ, Burns CM, et al. Sustained weight loss following 12‐month pramlintide treatment as an adjunct to lifestyle intervention in obesity. Diabetes Care 2008; 31: 1816–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roth JD, Roland BL, Cole RL, et al. Leptin responsiveness restored by amylin agonism in diet‐induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A 2008; 105: 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trevaskis JL, Coffey T, Cole R, et al. Amylin‐mediated restoration of leptin responsiveness in diet‐induced obesity: magnitude and mechanisms. Endocrinology 2008; 149: 5679–5687. [DOI] [PubMed] [Google Scholar]
- 13. Turek VF, Trevaskis JL, Levin BE, et al. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology 2010; 151: 143–152. [DOI] [PubMed] [Google Scholar]
- 14. Osto M, Wielinga PY, Alder B, Walser N, Lutz TA. Modulation of the satiating effect of amylin by central ghrelin, leptin and insulin. Physiol Behav 2007; 91: 566–572. [DOI] [PubMed] [Google Scholar]
- 15. Trevaskis JL, Parkes DG, Roth JD. Insights into amylin‐leptin synergy. Trends Endocrinol Metab 2010; 21: 473–479. [DOI] [PubMed] [Google Scholar]
- 16. Amylin Pharmaceuticals press release. 2010; Available at: http://www.takeda.com/press/article_35851.html, 2015.
- 17. Ludwig DS. Lifespan Weighed Down by Diet. JAMA 2016; 315: 2269–2270. [DOI] [PubMed] [Google Scholar]
- 18. Roth JD, Hughes H, Kendall E, Baron AD, Anderson CM. Antiobesity effects of the beta‐cell hormone amylin in diet‐induced obese rats: effects on food intake, body weight, composition, energy expenditure, and gene expression. Endocrinology 2006; 147: 5855–5864. [DOI] [PubMed] [Google Scholar]
- 19. Roth JD, Hughes H, Coffey T, et al. Effects of prior or concurrent food restriction on amylin‐induced changes in body weight and body composition in high‐fat‐fed female rats. Am J Physiol Endocrinol Metab 2007; 293: E1112–E1117. [DOI] [PubMed] [Google Scholar]
- 20. Rosenbaum M, Goldsmith R, Bloomfield D, et al. Low‐dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest 2005; 115: 3579–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kissileff HR, Thornton JC, Torres MI, et al. Leptin reverses declines in satiation in weight‐reduced obese humans. Am J Clin Nutr 2012; 95: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Muller TD, Sullivan LM, Habegger K, et al. Restoration of leptin responsiveness in diet‐induced obese mice using an optimized leptin analog in combination with exendin‐4 or FGF21. J Pept Sci 2012; 18: 383–393. [DOI] [PubMed] [Google Scholar]
- 23. Korner J, Conroy R, Febres G, et al. Randomized double‐blind placebo‐controlled study of leptin administration after gastric bypass. Obesity (Silver Spring) 2013; 21: 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS One 2010; 5: e11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Z, Kelly L, Heiman M, Greengard P, Friedman JM. Hypothalamic Amylin Acts in Concert with Leptin to Regulate Food Intake. Cell Metab. 2015; 22: 1059–1067. [DOI] [PubMed] [Google Scholar]
- 26. Smith PM, Brzezinska P, Hubert F, et al. Leptin influences the excitability of area postrema neurons. Am J Physiol Regul Integr Comp Physiol 2016; 310: R440–R448 [DOI] [PubMed] [Google Scholar]
- 27. Le Foll C, Johnson MD, Dunn‐Meynell AA, et al. Amylin‐induced central IL‐6 production enhances ventromedial hypothalamic leptin signaling. Diabetes 2015; 64: 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive‐leptin concentrations in normal‐weight and obese humans. N Engl J Med 1996; 334: 292–295. [DOI] [PubMed] [Google Scholar]