

Abstract
In recent years, defined progress has been made in understanding the mechanisms of hemodynamic disturbances occurring in liver cirrhosis, which are based on portal hypertension. In addition to pathophysiological disorders related to endothelial dysfunction, it was revealed: There is the restructuring of the vasculature, which includes vascular remodeling and angiogenesis. In spite of the fact that these changes are the compensatory-adaptive response to the deteriorating conditions of blood circulation, taken together, they contribute to the development and progression of portal hypertension causing severe complications such as bleeding from esophageal varices. Disruption of systemic and organ hemodynamics and the formation of portosystemic collaterals in portal hypertension commence with neovascularization and splanchnic vasodilation due to the hypoxia of the small intestine mucosa. In this regard, the goal of comprehensive treatment may be to influence on the chemokines, proinflammatory cytokines, and angiogenic factors (vascular endothelial growth factor, placental growth factor, platelet-derived growth factor and others) that lead to the development of these disorders. This review is to describe the mechanisms of restructuring of the vascular bed in response to hemodynamic disturbances in portal hypertension. Development of pathogenetic methods, which allow correcting portal hypertension, will improve the efficiency of conservative therapy aimed at prevention and treatment of its inherent complications.
Keywords: Portal hypertension, Vascular remodeling, Angiogenesis, Pathogenesis, Liver cirrhosis
Core tip: The purpose of the review is to describe the mechanisms of restructuring of the vascular bed in response to hemodynamic disturbances in portal hypertension. In addition to pathophysiological disorders related to endothelial dysfunction, it was revealed: There is the restructuring of the vasculature, which includes vascular remodeling and angiogenesis. In spite of the fact that these changes are the compensatory-adaptive response to the deteriorating conditions of blood circulation, taken together, they contribute to the development and progression of portal hypertension causing severe complications such as bleeding from esophageal varices.
INTRODUCTION
The majority of severe complications of liver cirrhosis are directly related to its characteristic hemodynamic disturbances, and portal hypertension is at their core[1]. Due to various anatomical and functional factors, the increase in hepatic vascular resistance causes the development of portal hypertension. The synthesis of extracellular matrix components leads to serious changes in the liver cytoarchitectonics. Accompanying this process, hypoxia with the participation of vascular endothelial growth factor (VEGF) induce pathological angiogenesis. In addition, sinusoidal endothelial dysfunction in liver cirrhosis disturbs the balance between vasoconstrictors and vasodilators produced by the endothelium. There are endothelin-1 (ET-1) and nitrogen oxide (NO), the most studied at present[2].
In spite of the formation of portosystemic shunts, the subsequent development of hyperdynamic circulatory syndrome contributes to the progression of portal hypertension. Appearing in this case circulatory disorders are caused by endothelial dysfunction and restructuring of the vascular bed that includes vascular remodeling and angiogenesis[3] (Figure 1).
Figure 1.
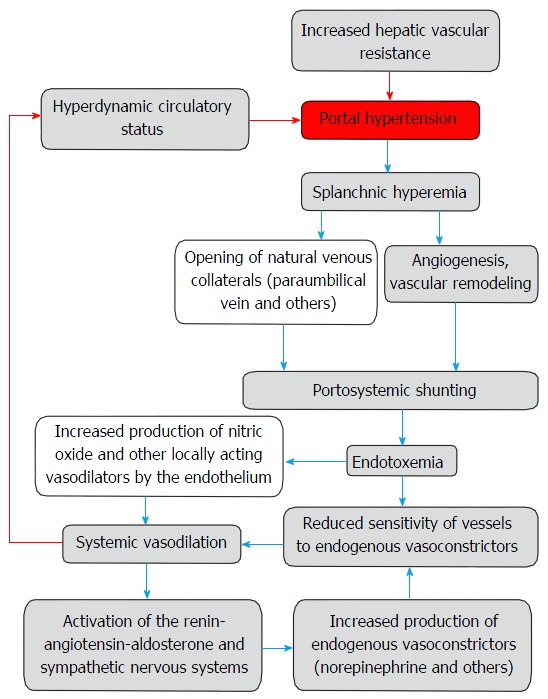
Potential mechanisms of the pathogenesis of portal hypertension in liver cirrhosis.
The purpose of the review is to describe the mechanisms of restructuring of the vascular bed in response to hemodynamic disturbances in portal hypertension.
CURRENT CONCEPT OF VASCULAR REMODELING
The term “remodeling” started to be used in the 1980s of the last century, mainly in cardiology. In the strict interpretation, it means the process of reorganization of the existing structure, during which it joins a new material or it is entirely changed. Vascular remodeling is an adaptive response to long-term hemodynamic disturbances. This process includes several stages[4]: (1) perception of signals about modified conditions of blood circulation and circulating humoral factors; (2) signal transmission within a cell and between adjacent cells; (3) synthesis, activation, or release of substances affecting cell growth, death, migration, or extracellular matrix construction; and (4) structural changes of the vascular wall (both cellular and extracellular components), its mechanics, and function.
Endothelial cells are the main sensors perceiving changes in blood flow and the impact of various humoral factors. They are constantly activated by mechanical stimuli, such as shear stress and intravascular pressure, which are transformed into intracellular and extracellular chemical signals within endothelial cells. These changes occur in the early stage of the mechanotransduction process[5]. It involves a variety of physiological elements including ion channels, molecules of cell-matrix and cell-cell interactions [integrins, platelet-endothelial cell adhesion molecule-1 (PECAM-1) or CD31, adherent compounds], tyrosine kinase receptors, caveolae, G protein-coupled receptors and G-proteins, glycocalyx, endothelial cell cytoskeletal components, and others. In response to mechanotransduction, there are formation and secretion of biologically active substances produced by the endothelium. They regulate the development of extracellular matrix, proliferation, migration, and organization of endothelial and smooth muscle cells, as well as sensitivity to growth factors - the key events in the vascular remodeling. Currently, the most studied ones are the vasodilators NO and prostacyclin (PGI2), and the vasoconstrictor ET-1[6].
Structural changes of blood vessels consist in eutrophic, hypertrophic, and hypotrophic remodeling. In inward eutrophic remodeling, outer and lumen diameters are reduced, and media cross-sectional area is unaltered. Inward hypertrophic remodeling is characterized by an increase in media/lumen ratio owing to media cross-sectional area increasing. Outward hypotrophic remodeling refers to an increase in lumen diameter and a decrease in cross-sectional area[7].
Distensibility is an important mechanical characteristic of the blood vessels defined as the percentage of change of their volume for each 1 mmHg change in intraluminal pressure. It depends primarily on the stiffness of the vascular wall, which mainly consists of collagen, elastin, and smooth muscles. It has been shown on the experimental model of spontaneously hypertensive rats that reduced distensibility of major arteries is related to increased quantity and changing structure of elastin, whereas its reduction in mesenteric resistance vessels mainly results from the modification of the elastin structure and possibly collagen accumulation[8].
MOLECULAR MECHANISMS OF THE ANGIOGENIC PROCESS
Angiogenesis is the complicated physiological process of creating new blood vessels. It occurs via activation of endothelial cells, expression of proteases in them, extracellular matrix destruction, proliferation, migration of endothelial cells, and creation of the highly permeable primary vascular structures, which is reconstructed into the three-dimensional vascular network after stabilization and “maturation” by attracting pericytes and smooth muscle cells[9].
The major inductor of angiogenesis in both pathological and physiological conditions is hypoxia. The cells react to lack of oxygen by several mechanisms including accumulation of hypoxia-inducible factors (HIFs). They are activated in the physiologically important sites of regulation of the oxygen pathways to provide quick and adequate responses to hypoxic stress and activate genes that regulate the angiogenesis process, vasomotor control, energy metabolism, erythropoiesis, and apoptosis[10].
The HIFs family include three α-subunits, which are connected with a β-subunit (HIF-1β). HIF-1α is ubiquitously expressed, whereas HIF-2α is found in a more limited set of cell types, particularly in vascular ECs, type II pneumocytes, hepatocytes, and macrophages. The role of HIF-3α is less well understood in hypoxic responses[11].
HIFs are considered to be the major transcriptional regulators of genes involved in response to the lack of oxygen, whereas miRNAs regulate gene expression post-transcriptionally[12].
The most investigated angiogenic growth factors are the family of VEGF. It consists of five homologs: VEGF-A, B, C, D and placental growth factor (PlGF). All VEGFs binds to various related receptors: VEGFR-1, VEGFR-2, VEGFR-3. Only the first two of them are responsible for transmission of angiogenic signals. What is more, the binding of VEGF-A to VEGFR-2 and the increase of vascular permeability under the influence of nitric oxide are the mechanisms triggering vasculogenesis and angiogenesis[13].
Members of the fibroblast growth factor (FGF) family are also capable of stimulating angiogenesis. Cellular response to the impact of FGFs develops via specific binding to FGF-receptors (FGFR), which have internal tyrosine kinase activity. Dimerization of FGFR is a precondition for activation and phosphorylation of signaling molecules occurring with the participation of heparin-binding proteins. The process stimulates cell differentiation, proliferation, migration, and destruction of the extracellular matrix. It is important to note that while members of the VEGF family are involved mostly in the capillary formation, the FGF mainly involved in arteriogenesis[14].
Although the platelet-derived growth factor (PDGF) influence on angiogenesis is not so explicit in contrast with VEGF and FGF, studies in vivo have shown that it is capable of causing the formation of blood vessels and regulating their tone[15].
Tie-2 (Tek), a tyrosine kinase receptor expressed by endothelial cells, and its ligands, the angiopoietins (Ang), play an important role in the coordination of the angiogenesis process. Angiopoetin-1 (Ang-1) promotes stabilization of vessels via inhibiting endothelial cells apoptosis and stimulating its formation. In contrast, Ang-2, an antagonist of Ang-1, results in destabilization of vessels by converting the endothelial cells to the proliferative phenotype from the stable state. However, Ang-2 is also able to stimulate angiogenesis with the participation of VEGF[16].
Integrin alpha v beta 3 (αvβ3) and alpha v beta 5 (αvβ5) were regarded as positive regulators of angiogenesis for a long time. Nevertheless, the recent studies have shown their inhibitory role in this process[17].
Vascular endothelial (VE)-cadherin, an endothelial-specific adhesion molecule, promotes cell-cell junctions during neovascularization and manages the transport of molecules through the endothelial lining[18].
Thrombospondin-1, an antiangiogenic protein, has a direct impact on migration and apoptosis of endothelial cells. Furthermore, it prevents the release of VEGF from the extracellular matrix by suppressing the activation of matrix metalloproteinases (MMPs)[19]. Endostatin, a fragment of the C-terminal part of the collagen XVIII α1-chain, and angiostatin, a plasminogen degradation product, are also inhibitors of angiogenesis[20].
The first step in the formation of new blood vessels is vasodilatation. Ang-2 and VEGF affect the formation of endothelial fenestrae. Increased vascular permeability causes extravasation of plasma proteins, which will further serve as a scaffold for migrating endothelial cells. Meanwhile, integrins provide them information about the presence of the angiogenic sites. The next step is the destruction of the basement membrane and the extracellular matrix by activated MMPs. This induces the subsequent migration and proliferation of endothelial cells with the participation of angiogenic growth factors such as VEGF, FGF, and epidermal growth factor (EGF). One of the regulators of this process is a transmembrane protein ESDN - endothelial and smooth muscle cell-derived neuropilin-like protein[21].
Endothelial progenitor cells differentiate into endothelial cells. VE-cadherin and integrins coordinate their binding, and tumor necrosis factor α (TNF-α), FGF, and PDGF create conditions for the formation of new capillaries. Endothelial cells form the new basement membrane and the extracellular matrix with the participation of surrounding pericytes. Ang-1 provides final vascular stabilization[9].
MECHANISM OF PORTAL-SYSTEMIC COLLATERALS FORMATION
In the early stages of the development of portal hypertension, a moderate increase in the portal pressure leads to a redistribution of blood flow toward the muscle layer of the small intestine. The appearance of mucosal hypoxia causes a significant increase in NAD(P)H oxidase activity, the main source of reactive oxygen species (ROS) in the mucous membrane, and also leads to increased production of VEGF and NO by arterioles, contributing splanchnic vasodilation[22]. In addition, multiple signaling pathways are stimulated, such as mitogen-activated protein kinases, tyrosine kinases, and transcription factors that are involved in VEGF-induced neovascularization[23]. It was shown that overexpression of Kruppel-like factor 2 in duodenal tissue with the assistance of microRNAs causes hemodynamic stimuli integration and VEGF-driven angiogenesis in patients with liver cirrhosis[24]. Besides the wall of the small intestine[25], the elevated levels of VEGF, VEGFR-2, and CD31 (PECAM-1) is observed in the mesentery[26].
These pathophysiological disturbances may be the initial step in the development of portosystemic collateral circulation in portal hypertension[27]. Monocytes adhere to the surface of activated endothelial cells and produce growth factors and proteases, such as urokinase plasminogen activator and MMPs, promoting the division and migration of smooth muscle cells. Proinflammatory cytokines [macrophage chemotactic protein-1, granulocyte-macrophage colony-stimulating factor, transforming growth factor β1 (TGF-β1), TNF-alpha] also promote the growth of blood vessels, as well as growth factors such PlGF, which stimulates the growth of endothelial and smooth muscle cells, FGF - through upregulated expression of PDGF receptor, and VEGF by reaction with Ang-1. At the same time, anti-inflammatory cytokines (e.g., interleukin-10) inhibit the process[28].
It was shown in animal model of prehepatic portal hypertension induced by partial portal vein ligation, that the blockade of VEGFR-2 with anti-VEGFR-2 monoclonal antibody for 5-7 d and inhibition of VEGF/VEGFR-2 signalization using autophosphorylation inhibitor VEGFR-2 for 5 d after the operation resulted in 50% reduction of portosystemic collateral vessel formation[29,30]. Blockade of NAD(P)H also contributed to this owing to the reduced splanchnic expression of VEGF, VEGFR-2 and CD31[31].
It should be noted that the emerging shunts are very dynamic vascular bed because of the expression of various receptor types on the surface of the endothelial lining, for example, α and β adrenoreceptors, 5-HT2 receptors. Furthermore, vasoactive substances such as NO, ET-1, prostaglandins can affect their tonus[32]. In particular, it was noted that excessive discharge of blood through portosystemic collaterals because of postprandial splanchnic hyperemia promotes their dilation due to shear stress, which in turn induces the overproduction of NO by endothelial cells[33]. Although natural portosystemic anastomoses are found in all patients with portal hypertension, they acquire the highest clinical significance in the development of gastroesophageal varices, because their rupture leads to life-threatening bleeding. The determining factor in their formation is the type of hepatofugal blood flow, and a gastroesophageal drainage path is the most important in this situation. The left gastric vein plays the main role in this path. It drains blood from both surfaces of the stomach, ascends from right to left along the lesser curvature into the lesser omentum, to the esophageal opening of the diaphragm, where it receives esophageal veins. It then turns backward and passes from left to right behind the omental bursa and drains into the portal vein. Anastomoses between the left and right gastric veins and the left and short gastric veins, respectively indicated by terms “coronary vein” and “posterior gastric vein”, have clinical significance only in portal hypertension, because they are involved in the formation of esophageal and related with them paraesophageal varices[34].
Immunohistochemical studies, which was conducted in patients with portal hypertension, revealed the existence of the pronounced expression of PDGF, basic FGF-2, EGF and TGF-α in the wall of the coronary vein of the stomach. This fact shows that the increase in pressure in this vein activates smooth muscle cells and induces the release of growth factors that stimulate their proliferation, differentiation, and migration, as well as contribute to the disruption of the metabolism of collagen and elastin fibers. Phenotypic changes of smooth muscle cells is a response to chronic mechanical stimuli. They are lead to thickening of the venous wall and reduce its elasticity[35].
VASCULAR STRUCTURE OF THE LOWER ESOPHAGUS IN CLINICAL PORTAL HYPERTENSION
The venous system of the distal portion of the esophagus includes intraepithelial, subepithelial superficial, deep submucosal and adventitial veins. The largest varices are generally localized 2-3 cm above and 2 cm below the cardia, mainly in the lamina propria of the mucous membrane. They have two types of vascular structure: Palisading type and bar type. The palisading type has dilated intraepithelial channels and numerous small superficial collateral veins. The bar type has triply dilated subepithelial superficial veins and deep submucosal veins which erode the epithelium[36]. Structural changes in the veins of the distal portion of the esophagus in portal hypertension are characterized by thickening of the medial layer because of hyperplasia of elastic and collagen fibers. Elastic fibers become fragmented and sharply tortuous directly in the varicose veins of the esophagus in the background of increasing sclerosis of the vascular wall[37].
Four distinct intramural vascular zones of the gastroesophageal junction were defined as follows: Gastric zone, palisade zone, perforating zone, and truncal zone. Portacaval shunts in this area are formed because of increased pressure in the portal venous system[38].
Gastric zone
The longitudinal veins of the gastric area are located in the submucosa and the lamina propria of the proximal portion of the stomach. They are more abundant near the esophagus, have a small diameter, and form a group of several longitudinal vessels. The veins merge in the submucosa of the distal part of the gastric zone and form large tortuous trunks draining blood into the portal vein system.
Palisade zone
The palisade zone is an extension of the gastric zone. It begins in the projection of the gastroesophageal junction and ends 2-3 cm above it. Veins in that zone are located randomly, close to each other, and are arranged longitudinally and in parallel as a palisade.
Numerous anastomoses are identified between vessels of both gastric and palisade zones. They are localized in the submucosa of the gastroesophageal junction, penetrate the muscularis mucosa, and pass into the lamina propria mainly in a longitudinal direction.
Veins of a proximal portion of the palisade zone simultaneously converge at one point and, perforating the muscularis mucosa, pass into the submucosa again as four or five big trunks. There are arched transverse anastomoses between them. Veins perforating the muscular layer of the esophagus were not detected in this zone.
Perforating zone
Veins of the perforating zone, which is located 3-5 cm above the gastroesophageal junction, are not so homogeneous and constant. Vessels form five polygonal networks in the lamina propria of the esophageal mucosa (as a continuation of the veins of the palisade zone) and perforate the muscular layer, communicating with adventitial veins located on the outer esophageal surface. They were referred to as (treble clef) veins because of their similarity with music symbols.
The perforating zone is the “critical area” for variceal rupture in portal hypertension. This is due to increased resistance to blood flow in this anatomical area, as well as increased fragility and superficial location of perforating veins[39].
Truncal zone
The truncal zone is a region from 8 to 10 cm in length with the bottom edge 5 cm above the gastroesophageal junction. Large longitudinal venous trunks, discovered here in the lamina propria, constitute a continuation of the polygon vascular networks of the perforating zone. They have a small diameter in the proximal portion. Between them, there are several transversely oriented anastomoses. Perforating veins, locating randomly along the zone, pass from the submucosa of the esophagus to its outer surface and communicate with adventitial veins.
In physiological terms, palisade zone is the most important part of the vascular structure of the gastroesophageal junction. Veins are located there mainly in the lamina propria. Their superficial location decreases venous blood flow resistance to a minimum, which would otherwise arise in the high-pressure zone in the area of the lower esophageal sphincter.
A large number of small caliber vessels in the palisade zone with a longitudinal stroke and parallel to each other perfectly adapted to the physiological pressure variations that leads to a bi-directional flow during breathing. When the venous outflow is carried out in the caudal direction, the gastric zone collects and drains the blood into the portal vein system.
Deep submucosal veins are enlarged because of the blood outflow in the cranial direction in portal hypertension. They drain the blood into the enlarged adventitial veins (periesophageal collateral veins) through the numerous veins perforating the esophageal smooth muscle layer in the perforating zone. Adventitial veins, in turn, communicate with paraesophageal collateral veins, which are located in the posterior mediastinum. The blood flows from them usually into the azygos vein[40], which structural changes in response to increased blood flow are characterized by focal destruction, hyperplasia and chaotic arrangement of elastic fibers[37].
THE SYSTEMIC AND SPLANCHNIC ADAPTIVE RESPONSE OF VASCULAR BED TO HEMODYNAMIC DISTURBANCES IN PORTAL HYPERTENSION
The development of portosystemic collateral circulation is a compensatory mechanism, which purpose is decompression of increased portal pressure. However, this does not happen. Conversely, there is a hyperdynamic circulatory state accompanied by increased cardiac output, decreased peripheral vascular resistance, and the opening of arteriovenous communications, which exacerbates portal hypertension. The cause of these disorders may be the flow of vasodilator substances (e.g., glucagon, endocannabinoid, atrial natriuretic peptide, bacterial endotoxin) through the network of portosystemic shunts, as well as increased production of topical vasodilators by endothelium, such as NO, carbon monoxide, PGI2, endothelium-derived hyperpolarizing factor, adrenomedullin, hydrogen sulfide. Furthermore, in spite of increased circulating levels of endogenous vasoconstrictors (noradrenaline, ET-1, angiotensin II), vascular sensitivity to them is significantly reduced[41].
Abdominal aorta
Adaptive response of the abdominal aorta to shear stress, induced by the blood flow in the conditions of the hyperdynamic circulation, may be associated with oxidative stress. Production of ROS, such as superoxide and hydrogen peroxide, which are toxic metabolic products of the cells, leads to non-specific damage of nucleic acids, proteins, lipids, and its other components. ROS regulate vascular tone, endothelial cells sensitivity to oxygen, their growth, proliferation, and apoptosis. Furthermore, they promote the expression of inducible genes by transcription factors, including NF-κB. These genes contribute to the synthesis of chemokines, chemokine receptors, proinflammatory cytokines, and adhesion molecules, inducing an inflammatory response. Potential sources of ROS are various enzyme systems: NAD(P)H oxidase, xanthine oxidase, enzymes of arachidonic acid metabolism (cyclooxygenase and lipoxygenase), and the mitochondrial respiratory chain[42].
Increased levels of TNF-α, IL-1β, and IL-6 in the aorta, as a result of oxidative stress, plays an important role in the induction of immune-mediated systemic vascular process in portal hypertension. Particularly, TNF-α induces activation and translocation of NF-κB to the nucleus with activation of NF-κB-dependent target genes. The subsequent increase in expression of connective tissue growth factor may enhance the synthesis of extracellular matrix proteins, particularly, collagen I type, whereas the decrease of the level of MMP-2/TIMP-2 complex (tissue inhibitor of metalloproteinase-2) will contribute to reducing the degradation of extracellular matrix proteins. These processes lead to significant histological changes in the aorta. Its wall thickness decreases, as well as the ratio of medial layer thickness to lumen diameter. Elastic fibers lose their ordered arrangement, and well-marked collagen fibers become more narrow and separated because of the increase of the extracellular matrix in the interstitium of media with a significant decrease in the number of smooth muscle cells[43,44].
The left gastric artery is the first branch of the celiac artery. It is assumed that the hemodynamics in the left gastric artery in portal hypertension may act as the initiator of variceal formation, showing close linkage with variceal recurrence[45].
Mesenteric resistance arteries
Similar infringements also occur in mesenteric resistance arteries. The mechanical stimuli, generated by shear stress, activate endothelial cells and induce hyperproduction of NO and prostaglandins, causing vasodilation[46]. The significantly reduced isometric stiffness of blood vessels and their increased elongation may cause structural changes in the internal elastic membrane and increase fenestrations in it[47]. This contributes to excessive NO-mediated vascular permeability and angiogenic processes in the mesentery of the small intestine because of the high VEGF and eNOS expression in microvessels located there[48].
Portal vein and hepatic artery
Splanchnic hyperemia leads to increased portal venous inflow. The portal vein becomes dilated under the influence of shear stress. Its intima and media are thickened due to the high content of collagen fibers here, hypertrophy, and hyperplasia of smooth muscle cells, which significantly reduce the vascular wall elasticity[49]. At the same time, portal blood flow, supplying the liver, decreases because of collateral circulation, and so-called hepatic arterial buffer response maintains hepatic perfusion constancy. This phenomenon, first described by Lautt[50], was identified in physiological conditions and in various pathological conditions. In liver cirrhosis, it is caused by intrahepatic hypoxia. Oxidative stress contributes to hepatic artery remodeling, which is accompanied by its dilation, decreased elasticity and thinning of the wall, as well as increased expression of adenosine and NO[51]. This reduces hepatic arterial vascular resistance and allows to maintain oxygen supply to the liver, providing protection of the organ structure and function[52].
Splenic artery and vein
Significant histopathological changes also occur in the blood vessels of the spleen. Damaged splenic artery intima becomes thicker, and smooth muscle cells grow into it. The internal elastic lamina is stratified, that is accompanied by the destruction of both included in its structure and localized in media elastic fibers.
Smooth muscle cells, randomly located in media, have a different size and morphology, and the content of separating them collagen fibers, as well as the extracellular matrix, increases significantly, causing the “collagenization” of the vascular wall, thickening, and rigidity[53]. The splenic vein expanding and its intima and media thickening is due to high content of collagen fibers, hypertrophy, and hyperplasia of smooth muscle cells[54]. These pathologic changes in the blood vessels of the spleen lead to a significant reduction of their flexibility.
CONCLUSION
In recent years, defined progress has been made in understanding the mechanisms of hemodynamic disturbances occurring in liver cirrhosis, which are based on portal hypertension. In addition to pathophysiological disorders related to endothelial dysfunction, it was revealed: There is the restructuring of the vasculature, which includes vascular remodeling and angiogenesis. In spite of the fact that these changes are the compensatory-adaptive response to the violated conditions of blood circulation, taken together, they promote the development and progression of portal hypertension causing severe complications such as bleeding from esophageal varices. Disruption of systemic and organ hemodynamics and the formation of portosystemic collaterals in portal hypertension commence with neovascularization and splanchnic vasodilation due to the hypoxia of the small intestine mucosa. In this regard, the goal of comprehensive treatment may be to influence on the chemokines, proinflammatory cytokines, and angiogenic factors (VEGF, PIGF, PDGF and others) that lead to the development of these disorders. Although pathogenetically reasonable methods of correction of portal hypertension are studied mainly at the molecular, cellular level, and in animal experiments, it can be expected that their clinical implementation will improve the efficiency of conservative therapy aimed at prevention and treatment of its inherent complications.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Russia
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Peer-review started: August 19, 2016
First decision: September 13, 2016
Article in press: November 2, 2016
P- Reviewer: Dong L, Mortensen C S- Editor: Gong ZM L- Editor: A E- Editor: Li D
References
- 1.Garbuzenko DV. [Multiorganic hemodynamic disorders in hepatic cirrhosis] Ter Arkh. 2007;79:73–77. [PubMed] [Google Scholar]
- 2.Garbuzenko DV, Arefyev NO, Belov DV. Mechanisms of adaptation of the hepatic vasculature to the deteriorating conditions of blood circulation in liver cirrhosis. World J Hepatol. 2016;8:665–672. doi: 10.4254/wjh.v8.i16.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez M. Molecular pathophysiology of portal hypertension. Hepatology. 2015;61:1406–1415. doi: 10.1002/hep.27343. [DOI] [PubMed] [Google Scholar]
- 4.Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 5.Davies PF, Barbee KA, Volin MV, Robotewskyj A, Chen J, Joseph L, Griem ML, Wernick MN, Jacobs E, Polacek DC, et al. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Annu Rev Physiol. 1997;59:527–549. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- 6.Ngai CY, Yao X. Vascular responses to shear stress: the involvement of mechanosensors in endothelial cells. Open Circ Vasc J. 2010;3:85–94. [Google Scholar]
- 7.Mulvany MJ. Small artery remodelling in hypertension. Basic Clin Pharmacol Toxicol. 2012;110:49–55. doi: 10.1111/j.1742-7843.2011.00758.x. [DOI] [PubMed] [Google Scholar]
- 8.Briones AM, González JM, Somoza B, Giraldo J, Daly CJ, Vila E, González MC, McGrath JC, Arribas SM. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J Physiol. 2003;552:185–195. doi: 10.1113/jphysiol.2003.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J Appl Physiol (1985) 2004;96:1173–1177; discussion 1170-1172. doi: 10.1152/japplphysiol.00770.2003. [DOI] [PubMed] [Google Scholar]
- 11.Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL, Runge A, Liu L, Kim MN, Liang J, et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J Clin Invest. 2012;122:1427–1443. doi: 10.1172/JCI57322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Z, Lai TC, Jan YH, Lin FM, Wang WC, Xiao H, Wang YT, Sun W, Cui X, Li YS, et al. Hypoxia-responsive miRNAs target argonaute 1 to promote angiogenesis. J Clin Invest. 2013;123:1057–1067. doi: 10.1172/JCI65344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS. 2005;(94):209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 14.Klein S, Roghani M, Rifkin DB. Fibroblast growth factors as angiogenesis factors: new insights into their mechanism of action. EXS. 1997;79:159–192. doi: 10.1007/978-3-0348-9006-9_7. [DOI] [PubMed] [Google Scholar]
- 15.Hellberg C, Ostman A, Heldin CH. PDGF and vessel maturation. Recent Results Cancer Res. 2010;180:103–114. doi: 10.1007/978-3-540-78281-0_7. [DOI] [PubMed] [Google Scholar]
- 16.Kim I, Moon SO, Koh KN, Kim H, Uhm CS, Kwak HJ, Kim NG, Koh GY. Molecular cloning, expression, and characterization of angiopoietin-related protein. angiopoietin-related protein induces endothelial cell sprouting. J Biol Chem. 1999;274:26523–26528. doi: 10.1074/jbc.274.37.26523. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, Huang X, Sheppard D, Hynes RO, Hodivala-Dilke KM. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- 18.Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- 19.Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1) J Cell Physiol. 2007;210:807–818. doi: 10.1002/jcp.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eriksson K, Magnusson P, Dixelius J, Claesson-Welsh L, Cross MJ. Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett. 2003;536:19–24. doi: 10.1016/s0014-5793(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 21.Nie L, Guo X, Esmailzadeh L, Zhang J, Asadi A, Collinge M, Li X, Kim JD, Woolls M, Jin SW, et al. Transmembrane protein ESDN promotes endothelial VEGF signaling and regulates angiogenesis. J Clin Invest. 2013;123:5082–5097. doi: 10.1172/JCI67752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraldes JG, Iwakiri Y, Loureiro-Silva M, Haq O, Sessa WC, Groszmann RJ. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state. Am J Physiol Gastrointest Liver Physiol. 2006;290:G980–G987. doi: 10.1152/ajpgi.00336.2005. [DOI] [PubMed] [Google Scholar]
- 23.Angermayr B, Mejias M, Gracia-Sancho J, Garcia-Pagan JC, Bosch J, Fernandez M. Heme oxygenase attenuates oxidative stress and inflammation, and increases VEGF expression in portal hypertensive rats. J Hepatol. 2006;44:1033–1039. doi: 10.1016/j.jhep.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 24.Kobus K, Kopycinska J, Kozlowska-Wiechowska A, Urasinska E, Kempinska-Podhorodecka A, Haas TL, Milkiewicz P, Milkiewicz M. Angiogenesis within the duodenum of patients with cirrhosis is modulated by mechanosensitive Kruppel-like factor 2 and microRNA-126. Liver Int. 2012;32:1222–1232. doi: 10.1111/j.1478-3231.2012.02791.x. [DOI] [PubMed] [Google Scholar]
- 25.Huang HC, Haq O, Utsumi T, Sethasine S, Abraldes JG, Groszmann RJ, Iwakiri Y. Intestinal and plasma VEGF levels in cirrhosis: the role of portal pressure. J Cell Mol Med. 2012;16:1125–1133. doi: 10.1111/j.1582-4934.2011.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–620. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Gana JC, Serrano CA, Ling SC. Angiogenesis and portal-systemic collaterals in portal hypertension. Ann Hepatol. 2016;15:303–313. doi: 10.5604/16652681.1198799. [DOI] [PubMed] [Google Scholar]
- 28.Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255:538–561. doi: 10.1111/j.1365-2796.2003.01297.x. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez M, Vizzutti F, Garcia-Pagan JC, Rodes J, Bosch J. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterology. 2004;126:886–894. doi: 10.1053/j.gastro.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez M, Mejias M, Angermayr B, Garcia-Pagan JC, Rodés J, Bosch J. Inhibition of VEGF receptor-2 decreases the development of hyperdynamic splanchnic circulation and portal-systemic collateral vessels in portal hypertensive rats. J Hepatol. 2005;43:98–103. doi: 10.1016/j.jhep.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Angermayr B, Fernandez M, Mejias M, Gracia-Sancho J, Garcia-Pagan JC, Bosch J. NAD(P)H oxidase modulates angiogenesis and the development of portosystemic collaterals and splanchnic hyperaemia in portal hypertensive rats. Gut. 2007;56:560–564. doi: 10.1136/gut.2005.088013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan CC. Portal-systemic collaterals and angiogenesis. J Chin Med Assoc. 2009;72:223–224. doi: 10.1016/S1726-4901(09)70060-7. [DOI] [PubMed] [Google Scholar]
- 33.Albillos A, Bañares R, González M, Catalina MV, Pastor O, Gonzalez R, Ripoll C, Bosch J. The extent of the collateral circulation influences the postprandial increase in portal pressure in patients with cirrhosis. Gut. 2007;56:259–264. doi: 10.1136/gut.2006.095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arora A, Rajesh S, Meenakshi YS, Sureka B, Bansal K, Sarin SK. Spectrum of hepatofugal collateral pathways in portal hypertension: an illustrated radiological review. Insights Imaging. 2015;6:559–572. doi: 10.1007/s13244-015-0419-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, Tian L, Peng L, Qiu F. Immunohistochemical analysis of growth factor expression and localization in gastric coronary vein of cirrhotic patients. J Tongji Med Univ. 1996;16:229–233. doi: 10.1007/BF02888113. [DOI] [PubMed] [Google Scholar]
- 36.Hashizume M, Kitano S, Sugimachi K, Sueishi K. Three-dimensional view of the vascular structure of the lower esophagus in clinical portal hypertension. Hepatology. 1988;8:1482–1487. doi: 10.1002/hep.1840080603. [DOI] [PubMed] [Google Scholar]
- 37.Turmakhanov ST, Asadulaev ShM, Akhmetkaliev MN. [Morpho-functional Changes of the Azygos Vein and Other Veins of the Gastroesophageal Zone in Portal Hypertension] Ann hir gepatol. 2008;13:58–65. [Google Scholar]
- 38.Vianna A, Hayes PC, Moscoso G, Driver M, Portmann B, Westaby D, Williams R. Normal venous circulation of the gastroesophageal junction. A route to understanding varices. Gastroenterology. 1987;93:876–889. doi: 10.1016/0016-5085(87)90453-7. [DOI] [PubMed] [Google Scholar]
- 39.Noda T. Angioarchitectural study of esophageal varices. With special reference to variceal rupture. Virchows Arch A Pathol Anat Histopathol. 1984;404:381–392. doi: 10.1007/BF00695222. [DOI] [PubMed] [Google Scholar]
- 40.Gaba RC, Couture PM, Lakhoo J. Gastroesophageal Variceal Filling and Drainage Pathways: An Angiographic Description of Afferent and Efferent Venous Anatomic Patterns. J Clin Imaging Sci. 2015;5:61. doi: 10.4103/2156-7514.170730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gracia-Sancho J, Maeso-Díaz R, Bosch J. Pathophysiology and a Rational Basis of Therapy. Dig Dis. 2015;33:508–514. doi: 10.1159/000374099. [DOI] [PubMed] [Google Scholar]
- 42.Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65:S140–S146. doi: 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 43.de Las Heras N, Aller MA, Martín-Fernández B, Miana M, Ballesteros S, Regadera J, Cachofeiro V, Arias J, Lahera V. A wound-like inflammatory aortic response in chronic portal hypertensive rats. Mol Immunol. 2012;51:177–187. doi: 10.1016/j.molimm.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 44.Fernández-Varo G, Ros J, Morales-Ruiz M, Cejudo-Martín P, Arroyo V, Solé M, Rivera F, Rodés J, Jiménez W. Nitric oxide synthase 3-dependent vascular remodeling and circulatory dysfunction in cirrhosis. Am J Pathol. 2003;162:1985–1993. doi: 10.1016/S0002-9440(10)64331-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiyono S, Maruyama H, Kondo T, Sekimoto T, Shimada T, Takahashi M, Yokosuka O. Hemodynamic effect of the left gastric artery on esophageal varices in patients with cirrhosis. J Gastroenterol. 2016;51:900–909. doi: 10.1007/s00535-015-1157-x. [DOI] [PubMed] [Google Scholar]
- 46.Piva A, Zampieri F, Di Pascoli M, Gatta A, Sacerdoti D, Bolognesi M. Mesenteric arteries responsiveness to acute variations of wall shear stress is impaired in rats with liver cirrhosis. Scand J Gastroenterol. 2012;47:1003–1013. doi: 10.3109/00365521.2012.703231. [DOI] [PubMed] [Google Scholar]
- 47.Resch M, Wiest R, Moleda L, Fredersdorf S, Stoelcker B, Schroeder JA, Schölmerich J, Endemann DH. Alterations in mechanical properties of mesenteric resistance arteries in experimental portal hypertension. Am J Physiol Gastrointest Liver Physiol. 2009;297:G849–G857. doi: 10.1152/ajpgi.00084.2009. [DOI] [PubMed] [Google Scholar]
- 48.Geerts AM, De Vriese AS, Vanheule E, Van Vlierberghe H, Mortier S, Cheung KJ, Demetter P, Lameire N, De Vos M, Colle I. Increased angiogenesis and permeability in the mesenteric microvasculature of rats with cirrhosis and portal hypertension: an in vivo study. Liver Int. 2006;26:889–898. doi: 10.1111/j.1478-3231.2006.01308.x. [DOI] [PubMed] [Google Scholar]
- 49.Wen B, Liang J, Deng X, Chen R, Peng P. Effect of fluid shear stress on portal vein remodeling in a rat model of portal hypertension. Gastroenterol Res Pract. 2015;2015:545018. doi: 10.1155/2015/545018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lautt WW. Mechanism and role of intrinsic regulation of hepatic arterial blood flow: hepatic arterial buffer response. Am J Physiol. 1985;249:G549–G556. doi: 10.1152/ajpgi.1985.249.5.G549. [DOI] [PubMed] [Google Scholar]
- 51.Moeller M, Thonig A, Pohl S, Ripoll C, Zipprich A. Hepatic arterial vasodilation is independent of portal hypertension in early stages of cirrhosis. PLoS One. 2015;10:e0121229. doi: 10.1371/journal.pone.0121229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eipel C, Abshagen K, Vollmar B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World J Gastroenterol. 2010;16:6046–6057. doi: 10.3748/wjg.v16.i48.6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li T, Ni JY, Qi YW, Li HY, Zhang T, Yang Z. Splenic vasculopathy in portal hypertension patients. World J Gastroenterol. 2006;12:2737–2741. doi: 10.3748/wjg.v12.i17.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Z, Zhang L, Li D, Qiu F. Pathological morphology alteration of the splanchnic vascular wall in portal hypertensive patients. Chin Med J (Engl) 2002;115:559–562. [PubMed] [Google Scholar]