

Abstract
Mitochondrial dynamics—fission and fusion—are associated with ischaemic heart disease (IHD). This study explored the protective effect of vagal nerve stimulation (VNS) against isoproterenol (ISO)‐induced myocardial ischaemia in a rat model and tested whether VNS plays a role in preventing disorders of mitochondrial dynamics and function. Isoproterenol not only caused cardiac injury but also increased the expression of mitochondrial fission proteins [dynamin‐related peptide1 (Drp1) and mitochondrial fission protein1 (Fis‐1)) and decreased the expression of fusion proteins (optic atrophy‐1 (OPA1) and mitofusins1/2 (Mfn1/2)], thereby disrupting mitochondrial dynamics and leading to increase in mitochondrial fragments. Interestingly, VNS restored mitochondrial dynamics through regulation of Drp1, Fis‐1, OPA1 and Mfn1/2; enhanced ATP content and mitochondrial membrane potential; reduced mitochondrial permeability transition pore (MPTP) opening; and improved mitochondrial ultrastructure and size. Furthermore, VNS reduced the size of the myocardial infarction and ameliorated cardiomyocyte apoptosis and cardiac dysfunction induced by ISO. Moreover, VNS activated AMP‐activated protein kinase (AMPK), which was accompanied by phosphorylation of Ca2+/calmodulin‐dependent protein kinase kinase β (CaMKKβ) during myocardial ischaemia. Treatment with subtype‐3 of muscarinic acetylcholine receptor (M3R) antagonist 4‐diphenylacetoxy‐N‐methylpiperidine methiodide or AMPK inhibitor Compound C abolished the protective effects of VNS on mitochondrial dynamics and function, suggesting that M3R/CaMKKβ/AMPK signalling are involved in mediating beneficial effects of VNS. This study demonstrates that VNS modulates mitochondrial dynamics and improves mitochondrial function, possibly through the M3R/CaMKKβ/AMPK pathway, to attenuate ISO‐induced cardiac damage in rats. Targeting mitochondrial dynamics may provide a novel therapeutic strategy in IHD.
Keywords: myocardial ischaemia, vagal nerve stimulation, mitochondrial dynamics, mitochondrial function, AMP‐activated protein kinase, subtype‐3 of muscarinic acetylcholine receptor, cardioprotection
Introduction
In ischaemic heart disease (IHD), which continues to be the main cause of the death worldwide, heart mitochondria directly sustain injury 1. Isoproterenol (ISO)‐induced myocardial ischaemia induces myocardial damage similar to that in patients with myocardial infarction (MI) and is commonly used for generating an experimental model in rats 2, 3. Injury to mitochondrial ultrastructure and function occurs during early ischaemia and progresses during sustained ischaemia. Mitochondrial dysfunction plays a major role in myocardial ischaemia 4, with phenotypes including decreased mitochondrial metabolic enzymes and ATP content and opening of the mitochondrial permeability transition pore (MPTP), which results in a burst of reactive oxygen species (ROS) and Ca2+ uptake, leading to apoptosis or necrosis 5, 6. Importantly, mitochondrial function relies heavily on changes to mitochondrial ultrastructure and morphology—the phenomenon of mitochondrial dynamics 7. Dynamic mitochondria constantly undergo fusion and fission, with these two opposing processes regulated by mitochondrial fusion [optic atrophy‐1 (OPA1), and mitofusins1 and 2 (Mfn1/2)] and fission proteins [dynamin‐related peptide1 (Drp1), and mitochondrial fission protein1 (Fis‐1)] respectively 8. Both mitochondrial fission and fusion are essential for cell metabolic function and facilitate segregation of dysfunctional or damaged mitochondria before apoptosis 9, 10. Regulation of proteins mediating mitochondrial dynamics or inhibition of excessive mitochondrial fission attenuates mitochondrial dysfunction to improve MI 11, 12. Therefore, targeting these proteins that regulate mitochondrial dynamics could prevent cardiac injury occurring due to myocardial ischaemia.
Our previous research showed that acetylcholine (ACh), the major neurotransmitter of the vagal nerve, inhibits ROS formation, improves mitochondrial biogenesis and initiates a mitophagy process to mitigate myocardial ischaemia–reperfusion injury (IRI) 13, 14, 15, 16. Interestingly, clinical studies have reported that imbalances in the cardiac autonomic nervous system, especially reduced vagal activity, are relevant to the pathogenesis of IHD 17, thus bringing increased focus on enhancing vagal activity as a potential therapeutic option to cope with IHD 18. Moreover, previous studies have shown that enhanced vagal activity has a positive effect of reducing injury and enhancing recovery of myocardial function in both animal studies and clinical practice 19, 20, 21. Cumulative studies have shown vagal nerve stimulation (VNS) prevents both myocardial ischaemia and burn injury through attenuation of mitochondrial dysfunction and suppression of myocardial apoptosis 22, 23. Although VNS and ACh have beneficial effects on mitochondria that, in turn, have a cardioprotective role, the mechanism by which VNS regulates mitochondrial dynamics following myocardial ischaemia is not fully understood.
AMP‐activated protein kinase (AMPK), a key cellular energy sensor and regulator of metabolic homoeostasis, modulates mitochondrial function, endoplasmic reticulum (ER) stress, autophagy and apoptosis, and prevents myocardial necrosis and contractile dysfunction during MI 24. Furthermore, a previous study from our laboratory showed ACh promotes cell survival via an AMPK‐induced cardiomyocyte autophagy pathway during cardiomyocyte hypoxia/reoxygenation injury 13. Increasing evidence suggests AMPK acts as a hub to bridge mitochondrial dysfunction and IHD 25, 26; however, AMPK's role in mitochondrial dynamics regulation during myocardial ischaemia remains unknown. This study aimed to elucidate this role using ISO‐induced myocardial ischaemia in a rat model by examining the protective effects of VNS on mitochondrial dynamics and function, with specific focus on AMPK‐related pathways.
Materials and methods
Animals and induction of myocardial ischaemia by ISO
Male Sprague–Dawley (SD; 180–200 g) rats were obtained from Xi'an Jiaotong University Laboratorial Animal Center and housed under standard conditions, with aces to food and water ad libitum. Isoproterenol 25 mg/kg (Sigma‐Aldrich, Saint Louis, MO, USA) dissolved in saline was administered through subcutaneous injections to the rats at 24‐hr intervals for 2 days to induce myocardial injury, on the basis of preliminary experimental results discussed previously 27, 28. All experiments in this study conformed to the Guideline on the Care and Use of Laboratory Animals and were approved by the Ethics Committee of Xi'an Jiaotong University.
Vagal nerve stimulation
With subjects under general anaesthesia and mechanical ventilation, the right cervical vagal nerve was identified and transected in the neck region. A pair of platinum wires was placed at the distal end of the vagal nerve for stimulation, and the electrode was connected to an isolated constant voltage stimulator (Power Lab; AD Instruments, Bella Vista, New South Wales, Australia). The vagal nerve was stimulated with electrical rectangular pulses of 2 Hz for 1 msec. Electrical voltage pulses ranging from 2 to 4 V were required to obtain a 10% reduction in basal heart rate (HR) 21. Vagal nerve stimulation was performed 60 min. after the last ISO administration and was continued for 240 min.
Animal experiment protocol
Sprague–Dawley rats were randomized into four groups: (i) vehicle‐treated without VNS (Sham), (ii) vehicle‐treated with VNS (Sham+VNS), (iii) ISO‐treated without VNS (ISO) and (iv) ISO‐treated with VNS (ISO+VNS). Separate sets of rats were studied for in vivo hemodynamics prior to sacrifice and their tissue samples were subject to electron microscope morphological analysis and protein assays (n = 6). Another set of rats was killed after experimental operation for the study of isolated heart mitochondria (n = 6). To examine the effects of the AMPK‐related signalling pathway, another set of rats were administered Compound C (an AMPK inhibitor; Sigma‐Aldrich, 0.25 mg/kg bw through tail vein injection 30 min. before VNS) and 4‐diphenylacetoxy‐N‐methylpiperidine methiodide (4‐DAMP, a selective subtype‐3 of a muscarinic acetylcholine receptor (M3R) antagonist, Sigma‐Aldrich; 0.5 μg/kg bw through tail vein injection 30 min. before VNS) 29. Treatment with Compound C or 4‐DAMP at doses specified did not significantly influence physiological parameters (data not shown). The experimental protocol and study animal disposition is presented in Figure 1.
Figure 1.

Experimental protocol. Sham, vehicle‐treated without vagal nerve stimulation. Sham+VNS: vehicle‐treated with vagal nerve stimulation; ISO, ISO‐treated without vagal nerve stimulation. ISO+VNS, ISO‐treated with vagal nerve stimulation. CC (ISO+VNS+Compound C), the rats were treated with Compound C (0.25 mg/kg, IV) 30 min. prior to VNS treatment. 4‐DAMP (ISO+VNS+4‐DAMP), 4‐DAMP (0.5 μg/kg, IV) was injected 30 min. prior to VNS treatment.
Hemodynamic measurements
Hemodynamic parameters were assessed by invasive LV catheterization and recorded using a polygraph recorder (PowerLab; AD Instruments) as described previously. Briefly, the left ventricle was catheterized with a heparin‐filled polyethylene catheter from the right common carotid artery to measure LV end‐diastolic pressure (LVEDP) and maximal rate of increase/decrease in LV pressure (±dP/dtmax). The right femoral artery was catheterized and connected to a transducer for recording blood pressure and HR during the experiment. In addition, needle electrodes were inserted subcutaneously for the limb lead at position II for electrocardiographic monitoring.
Measurement of myocardial infarct size
Following the experiment, the heart was excised immediately and sliced transversely into 2–3 mm‐thick sections, which were incubated in 1% 2,3,5‐triphenyltetrazolium chloride (Sigma‐Aldrich) solution for 30 min. at 37°C in the dark, fixed in 10% formalin, and photographed using a digital camera. The non‐infarcted myocardium was stained bright red, whereas infarcted myocardium appeared pale grey. Infarct size percentage was calculated for each individual slice by cumulative planimetry using computerized Image‐Pro Plus 6.0 (Media Cybernetics Inc., Silver Spring, MD, USA).
Measurement of serum enzymatic activity/level
After hemodynamic studies, blood samples were rapidly collected and serum was obtained by centrifugation at 4500 × g for 6 min. Serum levels of creatine kinase myocardium (CK‐MB) and lactate dehydrogenase (LDH) were detected with biochemical detecting system (AU2700; Olympus Melville, NY, USA). Serum cardiac troponin I (cTnI), citrate synthase (CS) and cytochrome C oxidase (CCO) activity were measured using a rat ELISA kit (Beyotime Biotech, Haimen, China) according to the manufacturer's instructions.
TUNEL staining
Tissue cryosections were stained using the TUNEL system (Promega, Madison, WI, USA) according to the manufacturer's protocol, and staining was observed under fluorescence microscopy (TE‐2000U; Nikon, Tokyo, Japan). The level of apoptotic cardiomyocytes was shown as a percentage of the number of TUNEL‐positive cells to the number of total cells.
Determination of ATP content
Myocardial tissue ATP content was determined by an Enhanced ATP Assay Kit (Beyotime, China) according to the manufacturer's instructions, and the results are shown in arbitrary units.
Preparation of mitochondrial fractions
Isolation of mitochondria was performed with the Mitochondria Fractionation Kit (Beyotime Biotech, Haimen, China) according to the manufacturer's protocol. Briefly, fresh cardiac tissue was mixed with a mitochondria extraction reagent and stirred in a homogenizer and the suspension was centrifuged at 1000 × g for 5 min. (4°C); the supernatant obtained was centrifuged at 3500 × g for 10 min. (4°C) and the precipitate contained the mitochondrial fraction.
Mitochondrial membrane potential determination
Mitochondrial membrane potential (ψm) was measured using the JC‐1 assay kit (Beyotime Biotech) according to the manufacturer's instructions. Fresh isolated mitochondria were incubated with an equal volume of JC‐1 staining solution (10 mg/ml) for 20 min. at 37°C in the dark and rinsed twice with buffer. JC‐1 fluorescence was measured by a fluorescence microscope under single excitation (488 nm) and dual emission (shift from 530 to 590 nm). The ratio of green and red fluorescent intensities indicated changes in mitochondrial membrane potential.
Assessment of MPTP
Mitochondrial permeability transition pore opening was assessed using a commercially available kit (Genmed Scientifics Inc., Boston, MA, USA) according to the manufacturer's instructions. Briefly, purified mitochondria (10 mg/ml, 20 μl) were transferred to 96‐well plates and incubated with pre‐warmed Reagent A. After 1 min., inducing medium (Reagent B, including CaCl2) was added and absorbance at 540 nm was measured using a multi‐mode microplate reader (Awareness Technology, Palm City, FL, USA), with decrease in absorbance indicating transition in mitochondrial permeability.
Transmission electron microscopy
Fresh LV tissue isolated was fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2 hrs at 4°C. Following fixation with 1% osmium tetroxide in 0.1 M phosphate buffer, the tissue was dehydrated with a graded series of ethanol to 100% and infiltrated with propylene oxide to embedding media (Epon 812 resin). Ultrathin sections were cut with an ultramicrotome, post‐stained with uranyl acetate and lead citrate, and then viewed by transmission electron microscopy (TEM; H‐7650; Hitachi, Tokyo, Japan). Digital images were analysed using Image J to manually generate masks of mitochondrial contours that were then used for the calculation of mitochondrial area, perimeter, maximum diameter and total mitochondrial number.
Examination of mitochondrial size and complexity
Mitochondrial size and complexity were evaluated using flow cytometry 30. MitoTracker Green (Beyotime), which passively accumulates in the lipid environment of intact mitochondria, was used to selectively stain intact mitochondria. Debris in the samples did not have a lipid environment and was excluded to obtain an accurate gating (R1) of the mitochondria. After establishing gating parameters, gated events (10,000/sample) were analysed using the forward scatter detector (FSC) and side scatter detector (SSC), and the results are shown in an FSC versus SSC density plot. The geometric mean (arbitrary units) representing FSC (logarithmic scale) was defined to depict size, whereas data from SSC (logarithmic scale) were considered an indicator of complexity.
Western blot
Cardiac tissue proteins were extracted with a protease inhibitor containing lysis buffer, sample proteins were resolved on SDS‐PAGE, and then transferred to polyvinylidene fluoride membranes to be probed with primary antibody overnight and then washed before incubation with horseradish peroxidase‐linked secondary antibody for 30 min. at room temperature. Bands were visualized with ECL‐Plus reagent (Millipore, Billerica, MA, USA) and detected by a ChemiDoc‐It Imaging System (Upland, CA, USA).
Statistical analysis
Results are presented as the mean ± S.E.M. Data analysis was processed with one‐way anova followed by a Tukey post hoc test or Student's t‐test. P < 0.05 was considered indicative of statistical significance. Statistical analyses were performed using GraphPad Prism Version 5.01 (GraphPad Software, La Jolla, CA, USA).
Results
VNS treatment attenuates damage in ISO‐induced myocardial ischaemia
To verify whether ISO treatment induced myocardial ischaemia, electrocardiographic parameters and serum myocardial enzymes were measured. In the ISO group, a marked elevation of the ST‐segment and increased levels of diagnostic marker enzymes (LDH, CK‐MB and cTnI) was evident, compared to the sham group, clearly suggesting that ISO treatment caused myocardial ischaemia (Fig. 2A and C–E). Vagal nerve stimulation in ISO‐administered rats significantly decreased ST‐segment elevation, infarct size and levels of serum myocardial enzymes, compared with the ISO group (P < 0.05; Fig. 2A–E). Otherwise, compared with the sham group, ISO reduced hemodynamic parameters, mean arterial pressure (MAP), LV developed pressure (LVDP) and ±dP/dtmax, whereas the effect was partially ameliorated after VNS (P < 0.05; Fig. 2F–H). These data indicate that VNS treatment improves cardiac function and reduces myocardial infarct size. Furthermore, to ensure effectiveness of VNS, HR during the stimulation period was monitored. As shown in Figure 2I, a 10% HR reduction was consistent during VNS treatment, and after cessation of VNS, HR showed an upward trend towards the baseline level, which indicated that the heart‐rate reduction was achieved by VNS.
Figure 2.

VNS treatment protects cardiac function against ISO‐induced myocardial damage in a rat model. (A) Electrocardiogram. Arrow denotes elevated ST‐segment in the ISO‐treated group. (B) Measurement of myocardial infarct size. (C–E) Detection of myocardial enzymes in serum. (F–H) Haemodynamics analysis. (I) HR during stimulation period. LDH, lactate dehydrogenase; CK‐MB, creatine kinase‐MB; cTnI, cardiac troponin‐I; MAP, mean arterial pressure; LVDP, LV developed pressure; ±dP/dtmax, maximal rate of the increase/decrease in LV pressure; HR, heart rate. n = 6, **P < 0.01 versus Sham; # P < 0.05 versus ISO; ## P < 0.01 versus ISO.
VNS decreases cardiomyocyte apoptosis induced by ISO
To elucidate whether VNS plays a positive cardioprotective role, cardiomyocyte apoptosis was measured using the TUNEL assay, the Bcl‐2/Bax ratio, and the level of cleaved caspase‐3. Isoproterenol significantly induced myocardial apoptosis compared with sham treatment, indicated by an increase in the number of TUNEL‐positive cells, decrease in the Bcl‐2/Bax ratio and an increase in the level of cleaved‐caspase‐3. Vagal nerve stimulation markedly ameliorated the amount of ISO‐induced TUNEL‐positive cells (P < 0.01; Fig. 3A and B), enhanced the expression of Bcl‐2, an anti‐apoptotic factor, and raised the Bcl‐2/Bax ratio (P < 0.05; Fig. 3C and D), while it decreased the level of cleaved caspase‐3 and downregulated the cleaved caspase‐3 ratio (P < 0.05; Fig. 3C and E). This suggests that VNS treatment reduces ISO‐induced apoptosis of cardiomyocytes.
Figure 3.
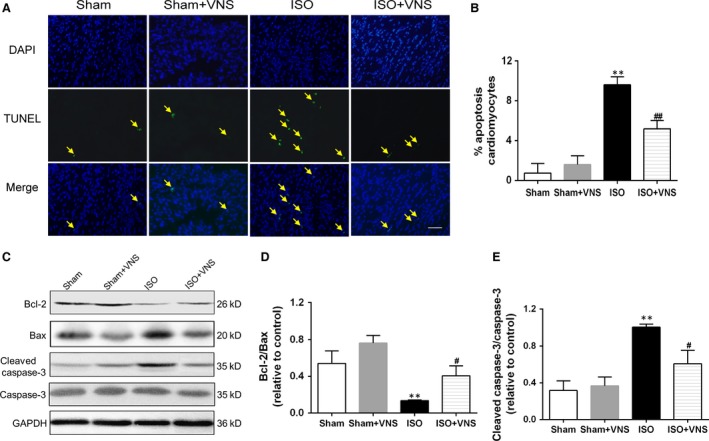
VNS treatment ameliorates ISO‐induced myocardial apoptosis. (A) Fluorescence microscopy images indicating TUNEL (green) and cell nuclei (blue) staining. Arrows indicate TUNEL‐positive cells, scale bar = 100 μm. (B) Quantification of apoptotic cardiomyocytes. (C) Representative immunoblots and (D and E) Western blot analysis of Bcl‐2/Bax and cleaved caspase‐3/caspase‐3 expression changes. n = 6, **P < 0.01 versus Sham; # P < 0.05 versus ISO; ## P < 0.01 versus ISO.
VNS regulates the expression of mitochondrial dynamics proteins in ISO‐induced myocardial ischaemia
To further study whether mitochondria is injured in ISO‐induced myocyte, we examined the major proteins controlling mitochondrial morphology by examining the expression of Fis‐1 and Drp1 (proteins which regulate fission events), OPA1 (which regulates mitochondrial inner membrane fusion), and Mfn1/2 (which controls outer membrane fusion). Phosphorylation of Drp1 at Ser616 promotes translocation of Drp1 from the cytosol to the mitochondrial membrane to mediate fission 31. Compared with the sham group, expression of Fis‐1 and p‐Drp1 increased and of Mfn1/2 and OPA1 decreased in the ISO group. This change manifested as enhanced fission and weakened fusion in the mitochondria, with resultant mitochondrial dysfunction and morphologic changes. Vagal nerve stimulation treatment not only suppressed p‐Drp1 and Fis‐1 expression but also restored Mfn1/2 and OPA1 levels compared to the ISO group (P < 0.05; Fig. 4), suggesting that VNS alleviates the shift in balance between fission and fusion following myocardial ischaemia.
Figure 4.
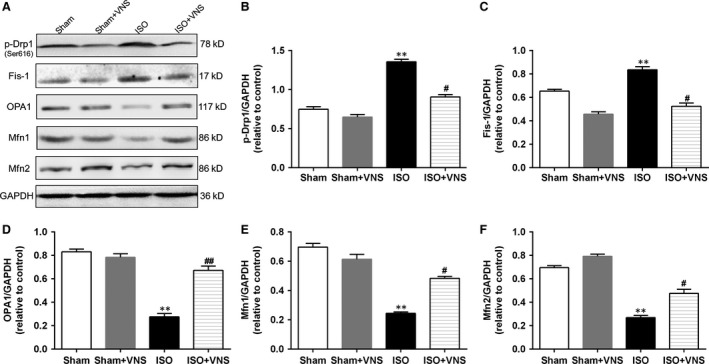
VNS treatment partially restores the expression of ISO‐induced cardiac mitochondrial dynamics proteins. (A) Representative immunoblots and (B–F) Western blot analysis of the mitochondrial dynamics protein expression of p‐Drp1, Fis‐1, OPA1, Mfn1, and Mfn2. n = 6, **P < 0.01 versus Sham; # P < 0.05 versus ISO; ## P < 0.01 versus ISO.
VNS suppresses ISO‐induced cardiac mitochondrial dysfunction and morphological abnormality
Mitochondrial ATP content, membrane potential and MPTP opening were determined to elucidate the effects of VNS on mitochondrial function. In the ISO group, the ATP content and mitochondrial membrane potential decreased dramatically with MPTP opening, as compared to the sham group, and this was reversed by VNS (P < 0.05; Fig. 5A–C). Moreover, cardiac mitochondrial ultrastructure and morphology were evaluated by TEM. In the sham group, mitochondria had an even, elongated shape and were strictly aligned between myofibrils, whereas in the ISO‐induced cardiomyocyte, mitochondrial arrangement was irregular, with clusters of mitochondrial fragments and high diversity in shape and size—an effect partially inhibited by VNS (Fig. 5D, top). We further quantified these morphological changes (Fig. 5D, bottom). Morphological analysis indicated ISO treatment induced a significant reduction in mitochondrial area, perimeter, diameter, and density, compared with the sham group that reverted to baseline after VNS (P < 0.05; Fig. 5E–H). To validate these findings, isolated mitochondrial morphology was analysed using flow cytometry. Mitochondrial size (FSC) and internal complexity (SSC) in the ISO group decreased significantly when compared with the sham group—an effect that was partially ameliorated after VNS during myocardial ischaemia (P < 0.05; Fig. 6). These morphological observations indicate VNS treatment can alleviate myocardial organelle, particularly mitochondrial, damage induced by myocardial ischaemia. Taken together, these data indicate VNS mitigates ISO‐induced cardiac mitochondrial dysfunction and morphological abnormality.
Figure 5.
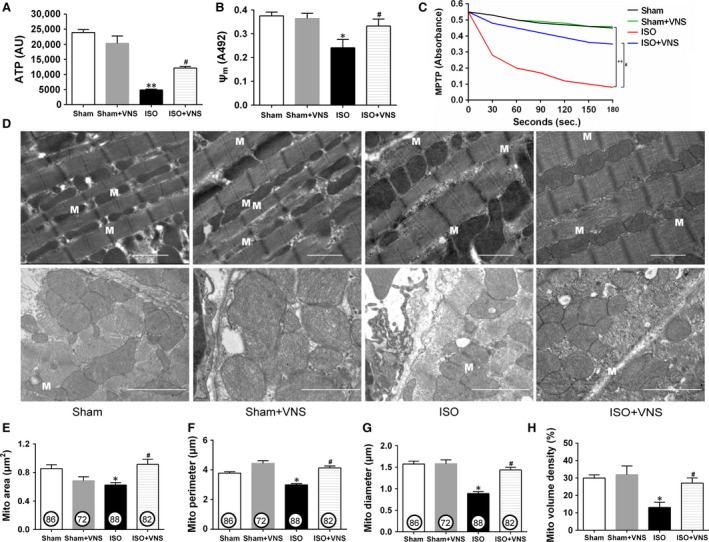
VNS suppresses ISO‐induced mitochondrial dysfunction and morphological damage. (A–C) Levels of myocardial ATP content (A), mitochondrial membrane potential (B), and mitochondrial permeability transition pore opening (C). (D) Transverse (top) and longitudinal (bottom) sections of mitochondria in cardiomyocytes from each experimental rat. M: mitochondrion, scale bar = 2 μm. (E–G) Area, perimeter and diameter of each individual mitochondrion estimated from electron microscopic image. Numbers in circles represent the number of fields analysed per group. (H) Mitochondrial volume density as measured by the grid analysis and expressed as a percentage. n = 6, *P < 0.05 versus Sham; **P < 0.01 versus Sham; # P < 0.05 versus ISO.
Figure 6.
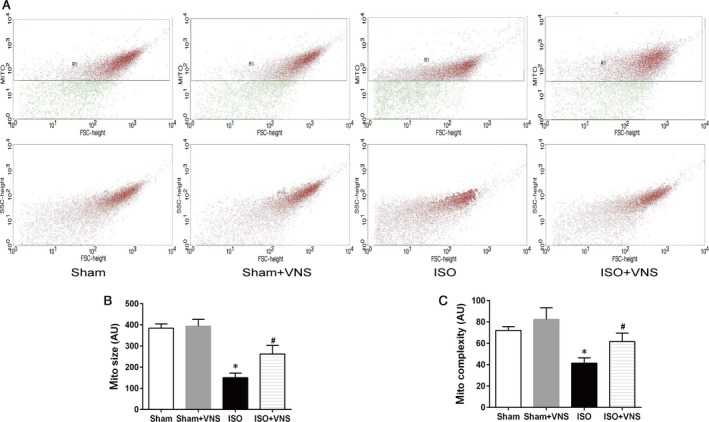
VNS attenuates ISO‐induced mitochondrial size and integrity. Relative size and internal complexity of cardiac mitochondria isolated from each experimental group were determined by flow cytometry. (A) Intact mitochondria are in red and debris and noise are shown in green (top) in representative gated‐density plots (R1 zoom) from each group of rats indicating size (FSC) versus internal complexity (SSC) of isolated mitochondria (bottom). (B) Analysis of isolated cardiac mitochondrial size in each experimental group. (C) Analysis of isolated cardiac mitochondria complexity in each experimental group. FSC, forward scatter; SSC, side scatter; AU, arbitrary units. n = 6, *P < 0.05 versus Sham; # P < 0.05 versus ISO.
VNS activates AMPK through preferred phosphorylation of CaMKKβ, instead of LKB1, in ISO‐induced myocardial ischaemia
To the best of our knowledge, AMPK activation can protect the heart against myocardial IRI through regulation of mitochondrial function 26. Therefore, we aimed to determine whether the AMPK pathway is involved in VNS‐mediated regulation of mitochondrial dynamics. First, we examined the phosphorylation of AMPK and acetyl‐CoA carboxylase (ACC, a substrate of AMPK), and found these were reduced in the ISO group, compared with the sham group, but were restored by VNS treatment (P < 0.05; Fig. 7B and C). Second, we analysed two kinases predicted to be upstream of AMPK—liver kinase B1 (LKB1) and Ca2+/calmodulin‐dependent kinase kinase β (CaMKKβ). As shown in Figure 7D and E, although there was no significant difference in the phosphorylation of LKB1 between any of the four groups, a significant decrease in CaMKKβ phosphorylation was evident in the ISO group. Interestingly, VNS caused CaMKKβ phosphorylation increase in both the VNS alone and the VNS+ISO group (P < 0.05), indicating that VNS may potentially activate AMPK through phosphorylation of CaMKKβ rather than LKB1.
Figure 7.

VNS activates AMPK via CaMKKβ, rather than LKB1, phosphorylation. (A) Representative Western blot showing protein expression of p‐AMPK, AMPK, p‐ACC, ACC, p‐LKB, LKB, p‐CaMKKβ, CaMKKβ, and GAPDH. (B) Quantitative analysis of AMPK phosphorylation. (C–E) Quantitative analysis of ACC phosphorylation (a substrate protein of AMPK) and LKB1 and CaMKKβ phosphorylation (upstream kinases of AMPK). (F and G) Western blot analysis of p‐AMPK/AMPK and p‐CaMKKβ/CaMKKβ. n = 6, *P < 0.05 versus Sham; # P < 0.05 versus ISO ; ^P < 0.05 versus ISO.
Furthermore, 4‐DAMP (a selective M3R antagonist) was used to determine the mechanism by which VNS activates CaMKKβ and its downstream kinases, because other research reported Gq receptors (activated by M1R, M3R and M5R) to be upstream for CaMKKβ/AMPK signalling 32, with choline—a precursor and metabolite of ACh—demonstrated to produce beneficial effects on the heart via cardiac M3R activation 33, 34. As shown in Figure 7F and G, inhibition of M3R decreased expression of p‐AMPK and p‐CaMKKβ, suggesting a possible role of M3R in the activation of the CaMKKβ/AMPK pathway by VNS.
Inhibition of AMPK or M3R compromises VNS‐induced protective effects on mitochondrial dynamics and function
To further determine the role of AMPK and related pathways in the regulation of mitochondrial dynamics following VNS treatment, we used Compound C (an AMPK inhibitor) and 4‐DAMP. Compared with the ISO+VNS group, both AMPK and M3R inhibition increased p‐Drp1 and Fis‐1 expression, whereas expression of OPA1 and Mfn1/2 were significantly reduced (P < 0.05; Fig. 8A–F), indicating AMPK may function as an essential link between VNS treatment and mitochondrial dynamics and M3R acts as a bridge for VNS to activate the CaMKKβ/AMPK pathway. Moreover, we determined the effect of AMPK and M3R inhibition on mitochondrial function. Compared with the ISO+VNS group, AMPK and M3R inhibition led to a reduction in mitochondrial CS and CCO activity (P < 0.01; Fig. 8G and H), corroborating the hypothesis that M3R and AMPK are involved in the regulation of mitochondrial dynamics and function that is induced by VNS during myocardial ischaemia.
Figure 8.

Inhibition of AMPK and M3R reverses the protective effect of VNS on mitochondrial dynamics and function. (A–F) Representative western blot and quantitative analysis measuring the expression of p‐Drp1, Fis‐1, OPA1, Mfn1, Mfn2 and GAPDH. (G and H) Detection of mitochondrial citrate synthase (CS) and cytochrome C oxidase (CCO) activity. n = 6, **P < 0.01 versus Sham; # P < 0.05 versus ISO; ## P < 0.01 versus ISO; ^P < 0.05 versus ISO+VNS; ^^P < 0.01 versus ISO+VNS.
Discussion
Mitochondrial damage contributes to cardiac dysfunction and cardiomyocyte injury via loss of metabolic capacity as well as production and release of toxic products during myocardial ischaemia. This study focused on changes in mitochondrial dynamics and the signalling pathways involved during myocardial ischaemia, with or without VNS treatment. Our results found that: (i) ISO treatment disrupts myocardial mitochondrial dynamics, as evidenced by downregulation of mitochondrial fusion proteins (OPA1 and Mfn1/2) and up‐regulation of mitochondrial fission proteins (p‐Drp1 and Fis‐1) and a large number of mitochondrial fragments in rat heart; (ii) VNS not only alleviated myocardial infarct size, reduced apoptosis and improved cardiac function but also up‐regulated expression of mitochondrial fusion proteins (OPA1 and Mfn1/2) and downregulated the levels of mitochondrial fission proteins (p‐Drp1 and Fis‐1), thereby reversing the damaging effects of ISO‐induced myocardial ischaemia, subsequently leading to improved mitochondrial function and morphology evidenced by increased ATP content and mitochondrial membrane potential and a reduction in MPTP opening and (iii) importantly, VNS activated AMPK through CaMKKβ, but not LKB1, phosphorylation. Treatment with either the M3R blocker 4‐DAMP or AMPK inhibitor Compound C mitigated the positive effects of VNS on mitochondrial dynamic protein expression together with CS and CCO activities. Taken together, these novel findings suggest VNS elicits an improvement in mitochondrial dynamics, possibly through an M3R/CaMKKβ/AMPK signalling pathway during myocardial ischaemia (Fig. 9).
Figure 9.
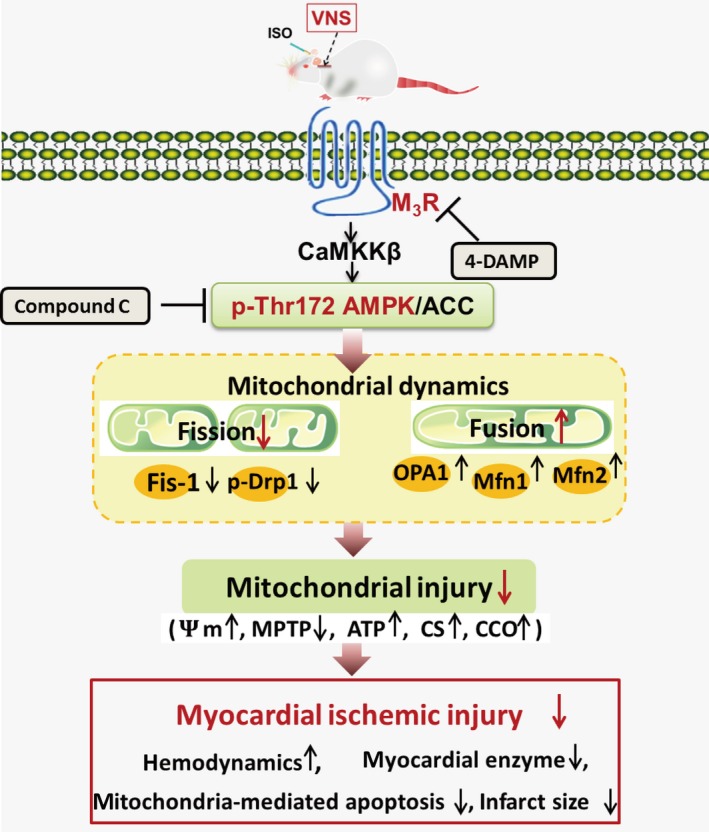
Schematic illustration of VNS regulation of mitochondrial fission and fusion through an M3R/CaMKKβ/AMPK pathway in rats with ISO‐induced myocardial ischaemia. AMPK, AMP‐activated protein kinase; AAC, acetyl‐CoA carboxylase; CaMKKβ, Ca2+/calmodulin‐dependent protein kinase kinase β; CS, citrate synthase; CCO, cytochrome C oxidase; Drp1, dynamin‐related peptide1; Fis‐1, mitochondrial fission protein1; ISO, isoproterenol; MPTP, mitochondrial permeability transition pore; M3R, subtype 3 of muscarinic acetylcholine receptor; Mfn1, mitofusin1; Mfn2, mitofusin2; OPA1, optic atrophy‐1; VNS, vagal nerve stimulation; 4‐DAMP, 4‐diphenylacetoxy‐N‐methypiperidine methiodide; ψm, mitochondrial membrane potential.
Three main methods exist for the development of a MI model in rats: coronary artery ligation, an electrocautery technique applied to the epicardial surface, and administration of ISO 35. Although left coronary artery ligation is most frequently used to induce acute myocardial damage in rat, this surgical procedure has disadvantages of a high mortality rate and a large variation in infarct size 36, 37. The extent of cardiac damage produced by the electrical method, consisting of overlapping burns, is not consistent among laboratories, limiting the reproducibility of the results obtained with this procedure 36. Pharmacological induction of heart damage is achieved by treatment with the β‐adrenergic receptor agonist ISO 38, which is a classic and easy method to induce myocardial ischaemia. In addition, it has reported that ISO has deleterious cardiac effects in rat, including necrosis, apoptosis, mitochondrial alterations, hypertrophy, fibrosis, oxidative damage and inflammatory cell infiltration, which is similar to that in the infarcted human heart 3, 39, 40. Importantly, ISO induction could imitate sympathetic overexcited state in myocardial ischaemia, which is in line with the recent found that imbalanced autonomic nervous system (excessive sympathetic activity and reduced vagal activity) exists in most of the myocardial ischaemic patients 41. In our study, 25 mg/kg ISO induced myocardial injury, demonstrated by elevation of the ST‐segment and levels of LDH, CK‐MB and cTnI. Furthermore, ISO reduced hemodynamic parameters (MAP, LVDP, and ±dP/dtmax) and increased the infarct area. Taken together, ISO induces an acute MI and, additionally, induced mitochondrial dysfunction—including impaired oxidative metabolism, calcium mishandling 42, decreased mitochondrial bioenergetics and enhanced oxidative stress 43, but the effects on mitochondrial dynamics were rare.
Mitochondria are dynamic organs and undergo fusion and division that generate interconnected mitochondrial networks facilitating physiological cell adaptation. Generally, mitochondrial outer and inner membrane fusion events are, respectively, mediated by Mfn1/2 and OPA1. Fused mitochondria are required for transmission of membrane potential to dissipate metabolic energy and to exchange mtDNA products in heteroplasmic cells to defend against ageing 44, 45. In contrast, phosphorylation of Drp1 on Ser616 promotes mitochondrial fission 31. Fis‐1, as a crucial receptor, assists Drp1 to complete the fission event 46. Fission is crucial for mitochondrial inheritance through growth and division, cytochrome C release to promote apoptosis, and turnover of damaged organelles by mitophagy 47, 48. Thus, it is important to define the role of mitochondrial dynamics in cardiovascular diseases. Lam et al. reported that ISO accelerated turnover of proteins mediating mitochondrial dynamics, such as Miro1/2, LONP, PHB and most respiratory chain components; however, Mfn1/2 and Fis‐1 states remained unchanged 49. Our results showed ISO‐induced myocardial ischaemia was followed by a slew of mitochondrial debris in cardiac tissues—an indication of a defect in mitochondrial dynamics in hearts subjected to ischaemia. This result concurs with the study performed in HL‐1 cardiomyocytes 50. Moreover, in the ISO group, the expression of mitochondrial fission proteins p‐Drp1 and Fis‐1 was significantly enhanced whereas that of fusion proteins OPA1 and Mfn1/2 was weakened when compared to the sham group. Thus, it is reasonable to consider that repair of imbalanced mitochondrial dynamics could benefit mitochondrial and cardiac function and might represent a potential strategic target for treatment in myocardial ischaemia.
Emerging evidence supports the idea that improved vagal tone has markedly protective effects on attenuating cardiac mitochondrial ROS generation, decreasing mitochondrial swelling, cytochrome C release, inhibiting MPTP opening and increasing ATP production 22, 51, 52. So, what does VNS modulate to induce these positive effects on mitochondrial function? This study found that VNS rectified the ISO‐induced turbulence in mitochondrial dynamics—it weakened the expression of p‐Drp1 and Fis‐1 and up‐regulated OPA1 and Mfn1/2, subsequently decreasing the amount of mitochondrial fragments and increasing the elongated mitochondrial network. Furthermore, not only were the mitochondrial dynamics proteins involved in mitochondrial morphology but they were also involved in regulating mitochondrial function. Inhibition of Drp1 has been shown to suppress mitochondria‐mediated apoptosis and Bax facilitates Drp1 translocation to the mitochondrial membrane to promote fission events 53, 54. On the other hand, OPA1 has been shown to play a critical role in maintaining cristae junctions, and disruption of OPA1 results in changes to cristae morphology and impaired mitochondrial metabolic ability 55, 56. After VNS rectified mitochondrial dynamics, mitochondrial dysfunction was also reversed in this study. Vagal nerve stimulation decreased p‐Drp1 level, caused recovery of mitochondrial membrane potential, decreased MPTP opening, and reduced the number of injured mitochondrial‐mediated myocardial cells undergoing apoptosis. Moreover, treatment with VNS promoted OPA1 expression and protected mitochondrial ultrastructure and metabolism, including by enhancing the activity of mitochondrial metabolic enzymes (e.g. CS and CCO) and ATP content. Therefore, we theorize VNS regulates mitochondrial dynamics, thereby preventing damage induced by myocardial ischaemia.
AMPK (a conserved energy sensor) plays an important role in regulating cell survival and death in response to pathological stress, including ER, oxidative and osmotic stress 57, 58. This crucial function of AMPK was demonstrated by a study which demonstrated that treatment with AMPK activators increased cell viability 59. Moreover, our previous research demonstrated that AMPK activation played an important role in VNS or ACh ability to protect from mitochondrial biogenesis and antioxidative stress, and provide autophagic cytoprotection during myocardial ischaemia 13, 60, 61. More interestingly, another study demonstrated that AMPK activation prevented mitochondrial fission by decreasing Drp1 and Fis‐1 levels in high glucose‐induced endothelial apoptosis 62. In this study, we showed that VNS treatment activated AMPK and ACC (the substrate of AMPK) in ISO‐induced myocardial damage. Furthermore, inhibition of AMPK by Compound C mitigated the protective effect of VNS, suggesting that AMPK was indeed involved in VNS‐mediated protection of mitochondrial dynamics and function. In mammalian cells, CaMKKβ and LKB1 are thought to be the two major upstream kinases of AMPK 63, 64. This study found that VNS activation of AMPK was accompanied by increased CaMKKβ, but not LKB1, phosphorylation, which may be explained by the change in cytosolic Ca2+ level 65, moreover, it should be noted that CaMKKβ is directly regulated by Ca2+. Our previous study demonstrated that ACh expression inhibited hypoxia/reoxygenation‐induced intracellular Ca2+ overload to prevent mitochondrial damage in vascular endothelial cells 66. Furthermore, Mungai et al. demonstrated that moderate hypoxia‐induced AMPK activation occurred through a calcium‐mediated pathway and was abolished by knockdown of CaMKKβ, but not LKB1, which suggested that a Ca2+/CaMKKβ/AMPK pathway could enhance the ability of mitochondria to protect cells against a more severe insult 67. We hypothesize that, in response to ISO‐induced myocardial ischaemia, VNS stimulates intercellular Ca2+ release to protect mitochondria and VNS activates a CaMKKβ/AMPK pathway to alleviate myocardial ischaemia through mitochondrial protection.
Activation of the CaMKKβ/AMPK pathway is usually induced by Gq‐coupled receptors, including M1,3,5R 32, all of which are involved in the regulation of energy metabolism 68, 69. Further study showed that M3R plays a critical role in glucose homoeostasis 68, 70. Some research reported that M3R activation by choline, an ACh precursor and metabolite, has a protective effect on both cardiac and vascular endothelial cells by enhanced phosphorylation of Connexin‐43, Ca2+/calmodulin‐dependent protein kinase II (CaMK II), and endogenous antioxidant capacity, and diminished Ca2+ overload 33, 34, 71, 72, 73. Moreover, M3R activation decreased Bcl‐2 expression and cytochrome C release and attenuated mitochondria‐mediated apoptosis 22, 72. In this work, the results showed that M3R inhibition decreased p‐AMPK and p‐CaMKKβ expression and blocked the beneficial effects of VNS on mitochondrial dynamics and function during myocardial ischaemia, suggesting the possible involvement of M3R in the activation of the CaMKKβ/AMPK pathway by VNS. Further studies are needed to clarify the role of M3R in the cardioprotective action of VNS.
In summary, our study offers salient evidence that VNS protected mitochondrial fusion and fission, and functions against ISO‐induced myocardial ischaemia. Notably, we indicated that VNS‐mediated mitochondrial protection possibly acts through activation of M3R/CaMKKβ/AMPK pathway. Our findings provide new insights into the mechanism underlying VNS‐mediated cardioprotection, indicating that protection of mitochondria through VNS treatment could be a potential strategy to prevent myocardial ischaemia.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We appreciate the technical support and materials provided by the electron microscope center of Xi'an Jiaotong University. This study was supported by the National Natural Science Foundation of China (Major International Joint Research Project, no. 81120108002; General Project, no. 81473203; Young Program, no. 81302774) and Specialized Research Fund for the Doctoral Program of Higher Education (no. 20130201130008).
Contributor Information
Dong‐Ling Li, Email: lidl@mail.xjtu.edu.cn.
Wei‐Jin Zang, Email: zwj@mail.xjtu.edu.cn.
References
- 1. Dong G, Chen T, Ren X, et al Rg1 prevents myocardial hypoxia/reoxygenation injury by regulating mitochondrial dynamics imbalance via modulation of glutamate dehydrogenase and mitofusin 2. Mitochondrion. 2016; 26: 7–18. [DOI] [PubMed] [Google Scholar]
- 2. Mukherjee D, Ghosh AK, Dutta M, et al Mechanisms of isoproterenol‐induced cardiac mitochondrial damage: protective actions of melatonin. J Pineal Res. 2015; 58: 275–90. [DOI] [PubMed] [Google Scholar]
- 3. Lobo FH, Ferreira NL, Sousa RB, et al Experimental model of myocardial infarction induced by isoproterenol in rats. Rev Bras Cir Cardiovasc. 2011; 26: 469–76. [DOI] [PubMed] [Google Scholar]
- 4. Lesnefsky EJ, Moghaddas S, Tandler B, et al Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001; 33: 1065–89. [DOI] [PubMed] [Google Scholar]
- 5. Nagoor Meeran MF, Jagadeesh GS, Selvaraj P. Thymol, a dietary monoterpene phenol abrogates mitochondrial dysfunction in β‐adrenergic agonist induced myocardial infarcted rats by inhibiting oxidative stress. Chem Biol Interact. 2016; 244: 159–68. [DOI] [PubMed] [Google Scholar]
- 6. Mukherjee D, Ghosh AK, Bandyopadhyay A, et al Melatonin protects against isoproterenol‐induced alterations in cardiac mitochondrial energy‐metabolizing enzymes, apoptotic proteins, and assists in complete recovery from myocardial injury in rats. J Pineal Res. 2012; 53: 166–79. [DOI] [PubMed] [Google Scholar]
- 7. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012; 46: 265–87. [DOI] [PubMed] [Google Scholar]
- 8. Hall AR, Burke N, Dongworth RK, et al Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol. 2014; 171: 1890–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iglewski M, Hill JA, Lavandero S, et al Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hypertens Rep. 2010; 12: 418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010; 88: 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Disatnik MH, Ferreira JCB, Campos JC, et al Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long‐term cardiac dysfunction. J Am Heart Assoc. 2013; 2: e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang H, Wang Y, Sun L, et al Aerobic interval training attenuates mitochondrial dysfunction in rats post‐myocardial infarction: roles of mitochondrial network dynamics. Int J Mol Sci. 2014; 15: 5304–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao M, Sun L, Yu X, et al Acetylcholine mediates AMPK‐dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol Biochem. 2013; 32: 601–13. [DOI] [PubMed] [Google Scholar]
- 14. Sun L, Zhao M, Yang Y, et al Acetylcholine attenuates hypoxia/reoxygenation injury by inducing mitophagy through PINK1/Parkin signal pathway in H9c2 cells. J Cell Physiol. 2016; 231: 1171–81. [DOI] [PubMed] [Google Scholar]
- 15. Miao Y, Zhou J, Zhao M, et al Acetylcholine attenuates hypoxia/reoxygenation‐induced mitochondrial and cytosolic ROS formation in H9c2 cells via M2 acetylcholine receptor. Cell Physiol Biochem. 2013; 31: 189–98. [DOI] [PubMed] [Google Scholar]
- 16. Sun L, Zhao M, Yu X, et al Cardioprotection by acetylcholine: a novel mechanism via mitochondrial biogenesis and function involving the PGC‐1α pathway. J Cell Physiol. 2013; 228: 1238–48. [DOI] [PubMed] [Google Scholar]
- 17. Thayer JF, Lane RD. The role of vagal function in the risk for cardiovascular disease and mortality. Biol Psychol. 2007; 74: 224–42. [DOI] [PubMed] [Google Scholar]
- 18. He X, Zhao M, Bi X, et al Novel strategies and underlying protective mechanisms of modulation of vagal activity in cardiovascular diseases. Br J Pharmacol. 2015; 172: 5489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mastitskaya S, Marina N, Gourine A, et al Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagal pre‐ganglionic neurones. Cardiovasc Res. 2012; 95: 487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hiraki T, Baker W, Greenberg JH. Effect of vagus nerve stimulation during transient focal cerebral ischemia on chronic outcome in rats. J Neurosci Res. 2012; 90: 887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kong SS, Liu JJ, Hwang TC, et al Optimizing the parameters of vagus nerve stimulation by uniform design in rats with acute myocardial infarction. PLoS ONE. 2012; 7: e42799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu X, Costantini T, Lopez NE, et al Vagal nerve stimulation protects cardiac injury by attenuating mitochondrial dysfunction in a murine burn injury model. J Cell Mol Med. 2013; 17: 664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shinlapawittayatorn K, Chinda K, Palee S, et al Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm. 2014; 11: 2278–87. [DOI] [PubMed] [Google Scholar]
- 24. Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends Endocrin Met. 2015; 26: 422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paiva MA, Rutter‐Locher Z, Goncalves LM, et al Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart C. 2011; 300: H2123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zaha VG, Qi D, Su KN, et al AMPK is critical for mitochondrial function during reperfusion after myocardial ischemia. J Mol Cell Cardiol. 2016; 91: 104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mukherjee D, Roy SG, Bandyopadhyay A, et al Melatonin protects against isoproterenol‐induced myocardial injury in the rat: antioxidative mechanisms. J Pineal Res. 2010; 48: 251–62. [DOI] [PubMed] [Google Scholar]
- 28. Li H, Xie YH, Yang Q, et al Cardioprotective effect of paeonol and danshensu combination on isoproterenol‐induced myocardial injury in rats. PLoS ONE. 2012; 7: e48872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhuo X, Wu Y, Ni Y, et al Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation‐mediated endoplasmic reticulum stress. Apoptosis. 2013; 18: 800–10. [DOI] [PubMed] [Google Scholar]
- 30. Williamson CL, Dabkowski ER, Baseler WA, et al Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart C. 2010; 298: H633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang C, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin‐related protein Drp1. Ann N Y Acad Sci. 2010; 1201: 34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Merlin J, Evans BA, Csikasz RI, et al The M3‐muscarinic acetylcholine receptor stimulates glucose uptake in L6 skeletal muscle cells by a CaMKK–AMPK–dependent mechanism. Cell Signal. 2010; 22: 1104–13. [DOI] [PubMed] [Google Scholar]
- 33. Yang B, Lin H, Xu C, et al Choline produces cytoprotective effects against ischemic myocardial injuries: evidence for the role of cardiac m3 subtype muscarinic acetylcholine receptors. Cell Physiol Biochem. 2005; 16: 163–74. [DOI] [PubMed] [Google Scholar]
- 34. Zhao J, Su Y, Zhang Y, et al Activation of cardiac muscarinic M3 receptors induces delayed cardioprotection by preserving phosphorylated connexin43 and up‐regulating cyclooxygenase‐2 expression. Br J Pharmacol. 2010; 159: 1217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patten RD, Hall‐Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circulation. 2009; 2: 138–44. [DOI] [PubMed] [Google Scholar]
- 36. Zaragoza C, Gomez‐Guerrero C, Martin‐Ventura JL, et al Animal models of cardiovascular diseases. J Biomed Biotechnol. 2011; 2011: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hou Y, Huang C, Cai X, et al Improvements in the establishment of a rat myocardial infarction model. J Int Med Res. 2011; 39: 1284–92. [DOI] [PubMed] [Google Scholar]
- 38. Zbinden G, Bagdon RE. Isoproterenol‐induced heart necrosis, an experimental model for the study of angina pectoris and myocardial infarct. Rev Can Biol. 1963; 22: 257–63. [PubMed] [Google Scholar]
- 39. Heather LC, Catchpole AF, Stuckey DJ, et al Isoproterenol induces in vivo functional and metabolic abnormalities; similar to those found in the infarcted rat heart. Acta Physiol Pol. 2009; 12: 31. [PubMed] [Google Scholar]
- 40. Nichtova Z, Novotova M, Kralova E, et al Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. Gen Physiol Biophys. 2012; 31: 141–51. [DOI] [PubMed] [Google Scholar]
- 41. Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension. 2012; 59: 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Willis BC, Salazar‐Cantú A, Silva‐Platas C, et al Impaired oxidative metabolism and calcium mishandling underlie cardiac dysfunction in a rat model of post‐acute isoproterenol‐induced cardiomyopathy. Am J Physiol Heart C. 2015; 308: H467–77. [DOI] [PubMed] [Google Scholar]
- 43. Khatua TN, Dinda AK, Putcha UK, et al Diallyl disulfide ameliorates isoproterenol induced cardiac hypertrophy activating mitochondrial biogenesis via eNOS‐Nrf2‐Tfam pathway in rats. Biochem Biophys Rep. 2016; 5: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zorzano A, Liesa M, Sebastián D, et al Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol. 2010; 21: 566–74. [DOI] [PubMed] [Google Scholar]
- 45. Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010; 1201: 21–5. [DOI] [PubMed] [Google Scholar]
- 46. Loson OC, Song Z, Chen H, et al Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013; 24: 659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landes T, Martinou J. Mitochondrial outer membrane permeabilization during apoptosis: the role of mitochondrial fission. BBA Mol Cell Res. 2011; 1813: 540–5. [DOI] [PubMed] [Google Scholar]
- 48. Shirihai O. Mitochondrial fusion, fission and autophagy: impact of diet on mitochondrial quality control. FASEB J. 2013; 27: 441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lam MP, Wang D, Lau E, et al Protein kinetic signatures of the remodeling heart following isoproterenol stimulation. J Clin Invest. 2014; 124: 1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ong SB, Subrayan S, Lim SY, et al Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010; 121: 2012–22. [DOI] [PubMed] [Google Scholar]
- 51. Katare RG, Ando M, Kakinuma Y, et al Vagal nerve stimulation prevents reperfusion injury through inhibition of opening of mitochondrial permeability transition pore independent of the bradycardiac effect. J Thorac Cardiovasc Surg. 2009; 137: 223–31. [DOI] [PubMed] [Google Scholar]
- 52. Shinlapawittayatorn K, Chinda K, Palee S, et al Low‐amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia‐reperfusion injury. Heart Rhythm. 2013; 10: 1700–7. [DOI] [PubMed] [Google Scholar]
- 53. Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005; 6: 657–63. [DOI] [PubMed] [Google Scholar]
- 54. Rolland SG, Conradt B. New role of the BCL2 family of proteins in the regulation of mitochondrial dynamics. Curr Opin Cell Biol. 2010; 22: 852–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parra V, Verdejo HE, Iglewski M, et al Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt‐mTOR‐NFκB‐Opa‐1 signaling pathway. Diabetes. 2014; 63: 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meeusen S, DeVay R, Block J, et al Mitochondrial inner‐membrane fusion and crista maintenance requires the dynamin‐related GTPase Mgm1. Cell. 2006; 127: 383–95. [DOI] [PubMed] [Google Scholar]
- 57. Bi X, He X, Xu M, et al Acetylcholine ameliorates endoplasmic reticulum stress in endothelial cells after hypoxia/reoxygenation via M3 AChR‐AMPK signaling. Cell Cycle. 2015; 14: 2461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hayashi T, Hirshman MF, Fujii N, et al Metabolic stress and altered glucose transport: activation of AMP‐activated protein kinase as a unifying coupling mechanism. Diabetes. 2000; 49: 527–31. [DOI] [PubMed] [Google Scholar]
- 59. Shin SM, Kim SG. Inhibition of arachidonic acid and iron‐induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1,2‐Dithiole‐3‐thione congeners. Mol Pharmacol. 2008; 75: 242–53. [DOI] [PubMed] [Google Scholar]
- 60. Wu S, Wei Y. AMPK‐mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: implication of the cell survival in mitochondrial diseases. BBA Mol Basis Dis. 2012; 1822: 233–47. [DOI] [PubMed] [Google Scholar]
- 61. Li L, Chen Y, Gibson SB. Starvation‐induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013; 25: 50–65. [DOI] [PubMed] [Google Scholar]
- 62. Bhatt MP, Lim Y, Kim Y, et al C‐Peptide activates AMPKα and prevents ROS‐mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013; 62: 3851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ahn Y, Kim H, Lim H, et al AMP‐activated protein kinase: implications on ischemic diseases. BMB Rep. 2012; 45: 489–95. [DOI] [PubMed] [Google Scholar]
- 64. Hawley SA, Pan DA, Mustard KJ, et al Calmodulin‐dependent protein kinase kinase‐β is an alternative upstream kinase for AMP‐activated protein kinase. Cell Metab. 2005; 2: 9–19. [DOI] [PubMed] [Google Scholar]
- 65. Garcia‐Dorado D, Ruiz‐Meana M, Inserte J, et al Calcium‐mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012; 94: 168–80. [DOI] [PubMed] [Google Scholar]
- 66. He X, Bi X, Lu X, et al Reduction of mitochondria–endoplasmic reticulum interactions by acetylcholine protects human umbilical vein endothelial cells from hypoxia/reoxygenation injurysignificance. Arterioscler Thromb Vasc Biol. 2015; 35: 1623–34. [DOI] [PubMed] [Google Scholar]
- 67. Mungai PT, Waypa GB, Jairaman A, et al Hypoxia triggers AMPK activation through reactive oxygen species‐mediated activation of calcium release‐activated calcium channels. Mol Cell Biol. 2011; 31: 3531–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Azua IRD, Gautam D, Guettier JM, et al Novel insights into the function of β‐cell M3 muscarinic acetylcholine receptors: therapeutic implications. Trends Endocrinol Metab. 2011; 22: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gautam D, Han SJ, Hamdan FF, et al A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo . Cell Metab. 2006; 3: 449–61. [DOI] [PubMed] [Google Scholar]
- 70. Olianas MC, Dedoni S, Onali P. Involvement of store‐operated Ca2+ entry in activation of AMP‐activated protein kinase and stimulation of glucose uptake by M3 muscarinic acetylcholine receptors in human neuroblastoma cells. BBA Mol Cell Res. 2014; 1843: 3004–17. [DOI] [PubMed] [Google Scholar]
- 71. Patanè S. M3 muscarinic acetylcholine receptor in cardiology and oncology. Int J Cardiol. 2014; 177: 646–9. [DOI] [PubMed] [Google Scholar]
- 72. Shi H, Wang H, Li D, et al Differential alterations of receptor densities of three muscarinic acetylcholine receptor subtypes and current densities of the corresponding K+ channels in canine atria with atrial fibrillation induced by experimental congestive heart failure. Cell Physiol Biochem. 2004; 14: 31–40. [DOI] [PubMed] [Google Scholar]
- 73. Lu X, Bi X, He X, et al Activation of M3 cholinoceptors attenuates vascular injury after ischaemia/reperfusion by inhibiting the Ca2+/calmodulin‐dependent protein kinase II pathway. Br J Pharmacol. 2015; 172: 5619–33. [DOI] [PMC free article] [PubMed] [Google Scholar]