

Abstract
Background and Purpose
The sphingosine analogue, FTY720 (GilenyaR), alleviates clinical disease progression in multiple sclerosis. Here, we variously assessed the effects of an azide analogue of (S)‐FTY720 vinylphosphonate (compound 5; a sphingosine kinase 1 activator), (R)‐FTY720 methyl ether (ROMe, a sphingosine kinase 2 inhibitor) and RB‐020 (a sphingosine kinase 1 inhibitor and sphingosine kinase 2 substrate) on IL‐1β formation, sphingosine 1‐phosphate levels and expression of S1P1 receptors. We also assessed the effect of compound 5 and ROMe in an experimental autoimmune encephalomyelitis (EAE) model in mice.
Experimental Approach
We measured IL‐1β formation by macrophages, sphingosine 1‐phosphate levels and expression levels of S1P1 receptors in vitro and clinical score in mice with EAE and the extent of inflammatory cell infiltration into the spinal cord in vivo.
Key Results
Treatment of differentiated U937 macrophages with compound 5, RB‐020 or sphingosine (but not ROMe) enhanced IL‐1β release. These data suggest that these compounds might be pro‐inflammatory in vitro. However, compound 5 or ROMe reduced disease progression and infiltration of inflammatory cells into the spinal cord in EAE, and ROMe induced a reduction in CD4+ and CD8+ T‐cell levels in the blood (lymphopenia). Indeed, ROMe induced a marked decrease in expression of cell surface S1P1 receptors in vitro.
Conclusion and Implications
This is the first demonstration that an activator of sphingosine kinase 1 (compound 5) and an inhibitor of sphingosine kinase 2 (ROMe, which also reduces cell surface S1P1 receptor expression) have an anti‐inflammatory action in EAE.
Abbreviations
- Compound 5
(S,E)‐3‐azido‐3‐(hydroxymethyl)‐5‐(4‐octylphenyl)pent‐1‐enylphosphonic acid
- DAMPs
Danger associated molecular patterns
- EAE
experimental autoimmune encephalomyelitis
- FTY720
2‐amino‐2‐[2‐(4‐ocytlphenyl)ethyl]‐1,3‐propanediol
- NLRP3
NOD‐like receptor family, pyrin domain containing 3
- PAMPs
Pathogen‐associated molecular patterns
- PP2A
protein phosphatase 2A
- RB‐020
1‐(4‐octylphenethyl)piperidin‐4‐yl)methanol
- ROMe
(R)‐FTY720 methyl ether, (R)‐2‐amino‐2‐(methoxymethyl)‐4‐(4‐octylphenyl)butan‐1‐ol
- SK
sphingosine kinase
- S1P
sphingosine 1‐phosphate
- TLR
toll‐like receptor
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | HDAC1 |
| PAMP | HDAC2 |
| DAMP | HSL, hormone sensitive lipase |
| NLRP3 | PKA |
| GPCRs b | Sphingosine kinase 1 |
| S1P1 receptor | Sphingosine kinase 2 |
| Enzymes c | Sphingosine 1‐phosphate lyase |
| Caspase 1 | |
| ERK‐2 |
| LIGANDS | |
|---|---|
| FTY720 | Okadaic acid |
| FTY720 phosphate | PMA, phorbol myristate acetate |
| IL‐1β | PF‐543 |
| IL‐12 | Sphingosine |
| IL‐18 | S1P, sphingosine 1‐phosphate |
| LPS, lipopolysaccharide | SKi, sphingosine kinase inhibitor |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Sphingosine 1‐phosphate (S1P) is formed by the phosphorylation of sphingosine, and this reaction is catalysed by two isoforms of sphingosine kinase (SK1 and SK2), which are encoded by different genes and exhibit distinct subcellular localizations, biochemical properties and functions (see Pyne and Pyne, 2011 for review). Once produced, S1P can either be exported from cells (through transporter proteins e.g. Spns2 and certain ABC transporters) and act as a ligand on a family of five S1P‐specific GPCRs (S1P1–5) (Blaho and Hla, 2014) or, if retained within the cell, bind to and regulate specific intracellular target proteins. For instance, SK2 catalyses the formation of S1P in the nucleus and the subsequently formed S1P inhibits HDAC1/2 activity to induce c‐fos and p21 expression (Hait et al., 2009).
There is a strong link between S1P and multiple sclerosis (MS), which involves reactive T‐lymphocytes in an autoimmune inflammatory demyelinating disease. Indeed, the sphingosine analogue 2‐amino‐2‐[2‐(4‐ocytlphenyl)ethyl]‐1,3‐propanediol (FTY720) has been licensed for oral treatment of relapsing MS under the trade name Gilenya™. FTY720 is a pro‐drug, which is phosphorylated by SK2 and functionally antagonizes the S1P1 receptor, resulting in its proteasomal degradation and removal from T‐lymphocytes (Hla and Brinkmann, 2011). This traps the T‐lymphocytes in lymph nodes as they cannot egress due to their inability to sense an S1P gradient, which requires surface expression of S1P1 receptors on the T‐cell. The consequence of this is to prevent T‐cell action on the CNS in MS.
Innate immunity to pathogens employs pathogen‐associated molecular patterns (PAMPs) in inflammatory cells to stimulate IL‐1β and IL‐18 release (Schroder and Tschopp, 2010; Takeuchi and Akira, 2010). Danger‐associated molecular patterns (DAMPs) that include sphingosine, released from dead cells, also induce innate immune responses (Chen and Nunez, 2010) by activating the NLRP3 inflammasome. This involves assembly of apoptosis‐associated speck‐like protein containing a caspase recruitment domain with the inflammasome. Caspase‐1 is then recruited to the complex where it cleaves pro‐IL‐1β to produce IL‐1β (Brough and Rothwell, 2007). There is also a link between Nlrp3 and MS and between NLRP3 and sphingosine. Thus, IL‐1β levels, Nlrp3 gene mutation and MS‐like lesions are correlated in the CNS (Dodé et al., 2002; Compeyrot‐Lacassagne et al., 2009). Indeed, Nlrp3 −/− mice exhibit milder symptoms in experimental autoimmune encephalomyelitis (EAE), an animal model for MS disease. In addition, sphingosine and the sphingosine analogue, FTY720, stimulate NLRP3‐dependent release of IL‐1β from LPS‐stimulated macrophages (Luheshi et al., 2012). We have also reported that sphingosine stimulates the caspase‐1‐dependent release of IL‐1β from differentiated U937 macrophages via a mechanism that involves cathepsin B and lysosomal destabilization (Boomkamp et al., 2016). The pro‐inflammatory action of FTY720, in terms of promoting IL‐1β release, and its anti‐inflammatory action, in terms of inhibiting T‐cell trafficking therefore appear opposed, although the latter is linked with the clinical efficacy of FTY720 in MS.
We have previously synthesized an azide analogue of (S)‐FTY720 vinylphosphonate (a pan‐S1P receptor antagonist (Valentine et al., 2010)): compound 5 ((S,E)‐3‐azido‐3‐(hydroxymethyl)‐5‐(4‐octylphenyl)pent‐1‐enylphosphonic acid), which activates SK1 in lysates of HEK 293 cells overexpressing this enzyme and therefore is either a direct or indirect effect (Liu et al., 2013). Similarly, we have synthesized (R)‐FTY720 methyl ether (ROMe; (R)‐2‐amino‐2‐(methoxymethyl)‐4‐(4‐octylphenyl)butan‐1‐ol), which is a selective inhibitor of SK2 (Lim et al., 2011) and 1‐(4‐octylphenethyl)piperidin‐4‐yl)methanol (RB‐020), which is a SK1 inhibitor and a SK2 substrate (Baek et al., 2013). As these compounds are FTY720 analogues, we variously assessed their effect on IL‐1β release, S1P levels and S1P1 receptor expression and compared the effect of compound 5 and ROMe in an EAE model of MS. In this regard, we have previously shown that PF‐543 (which is an inhibitor of SK1 (Schnüte et al., 2012)) exacerbated EAE disease progression (Pyne et al., 2016). We have therefore tested the hypothesis that compound 5 (SK1 activator) might inhibit EAE disease progression due to enhanced anti‐inflammatory activity of SK1, particularly as this might offer novel approaches for therapeutic intervention in MS.
Methods
Cell culture
All cells were maintained at 37°C in a humidified atmosphere containing 5% (v/v) CO2. U937 monocytic cells (ATCC, Manassas, Virginia, USA) were maintained in RPMI medium, supplemented with 10% (v/v) fetal bovine serum, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and 2 mM L‐glutamine (complete RPMI) and differentiated to macrophages using phorbol 12‐myristate 13‐acetate (PMA) as previously described (Twomey et al., 1993; Shepherd et al., 2004; Boomkamp et al., 2016). Briefly, cells were plated at 1 × 106 cells·mL−1 in complete RPMI supplemented with 4 nM PMA. Medium was replenished after 2 days. After 4 days, the cells were cultured in complete RPMI in the absence of PMA for a further 24 h. It is standard practice to differentiate U937 myeloid to macrophages to study inflammatory responses, for example, IL‐1β release.
Human lung microvascular endothelial cells (HLMVECs) (Lonza, San Diego, CA, USA) were cultured in complete endothelial basal media (EBM‐2) containing the growth factors and 10% (v/v) fetal bovine serum. Contact‐inhibited monolayers revealed typical cobblestone morphology and stained positive for acetylated LDL uptake.
CCL39 (Chinese hamster fibroblast) cells that were stably transfected with myc‐tagged S1P1 receptors (Rutherford et al., 2013; a gift from Dr T Palmer, University of Glasgow) were maintained in DMEM with GlutaMAX™ supplemented with 10% (v/v) fetal bovine serum, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Cells grown to confluence on 12‐well plates were quiesced for 24 h prior to treatment with FTY720 (100 nM) or compound 5 (10 μM) or RB‐020 (0.1–10 μM) or ROMe (0.1–10 μM) for 24 h. Cell lysates were analysed by SDS‐PAGE and Western blotted for proteins of interest (see below). CCL39 cells are used as standard for overexpression of recombinant proteins (S1P1) in order to allow testing of compounds for target engagement.
LNCaP‐AI (androgen‐independent) prostate cancer cells (a gift from Dr H Leung, Beatson Institute, Glasgow) were maintained in RPMI supplemented with 1% (v/v) L‐glutamine, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and 10% (v/v) lipid‐stripped fetal calf serum. LNCaP‐AI cells are used as a model system, as the effect of SK1 and SK2 inhibitors on SK1 expression in these cells is well established (Tonelli et al., 2010; McNaughton et al., 2016).
IL‐1β release
U937 cells were either untreated or treated with the indicated compounds alone for 1 h or stimulated with 1 μg·mL−1 LPS for 2 h followed by the compounds, as indicated, for 1 h. A series of experiments was performed with matched control and LPS treatments (where indicated) but in which not all compounds were necessarily assessed at the same time. Supernatants were collected and assayed for IL‐1β protein expression by elisa according to the manufacturer's instructions (Boomkamp et al., 2016).
Western blotting
Following treatment, cells were lysed in sample buffer (62.5 mM Tris‐HCl (pH 6.7), 0.5 M sodium pyrophosphate, 1.25 mM EDTA, 1.25% (w/v) SDS, 0.06% (w/v) bromophenol blue, 12.5% (v/v) glycerol and 50 mM dithiothreitol). Proteins were separated on a 10% (v/v) acrylamide/bisacrylamide gel and transferred to a nitrocellulose Hybond membrane. Membranes were blocked in 5% (w/v) BSA in TBST buffer (composition: 20 mM Tris‐HCl (pH 7.5), 48 mM NaCl, 0.1% (v/v) Tween20) for 1 h at room temperature prior to incubation with anti‐myc tag primary antibody (diluted in blocking buffer) to detect myc‐tagged S1P1 receptors overnight at 4°C. Following three washes in TBST, membranes were incubated with HRP‐conjugated anti‐IgG secondary antibody (diluted in blocking buffer) for 1 h at room temperature. Immunoreactive protein bands were visualized using enhanced chemiluminescence. Blots were stripped and re‐probed for ERK‐2 or actin to ensure equal protein loading (Boomkamp et al., 2016).
FACS analysis
Peripheral blood from naive, control or compound 5‐ or ROMe‐treated mice was collected into EDTA to prevent clotting, and red blood cells were removed with RBC lysis buffer (eBioscience, Altrincham, UK). Cells were added to FACS tubes (0.5 × 106 cells per tube) and incubated (30 min in the dark at 4°C) with FITC‐conjugated anti‐CD4 and PerCP‐Cy5.5‐conjugated anti‐CD8 (1:500 dilution). Cells were washed and resuspended in 0.5 mL FACS buffer (1% (w/v) BSA in PBS) prior to quantification using a BD FACSCanto system and BD FACSDiva software (BD Biosciences, Oxford, UK). Similarly, CCL39 cells, grown in 12‐well plates, were treated with vehicle (DMSO) or compounds, as described in the figure legends. These cells were added to FACS tubes (0.2 × 105 cells per tube) and incubated with allophycocyanin (APC)‐conjugated S1P1 receptor antibody (1:50 dilution; 30 min in the dark at 4°C). Cells were washed twice with FACS buffer, resuspended in 0.25 mL FACS buffer with 12.5 μL propidium iodide (to allow exclusion of dead cells) and quantified.
S1P measurement
HLMVECs, cultured in 35 mm dishes to ~90% confluence, were labelled with 40 μCi·mL−1 of [32P] orthophosphate in phosphate‐free DMEM medium for 3 h. The radioactive medium was aspirated, and cells were incubated with vehicle (DMSO) or the compounds for 1 h in phosphate‐free DMEM medium containing 0.1% (w/v) fatty acid free BSA (1 mL final volume) for 1 h prior to addition of exogenous sphingosine (2 μM) in 0.1% (w/v) fatty acid‐free BSA for an additional 1 h. Lipids were extracted by addition of 1 mL methanol containing 1% (v/v) 12 N HCl, scraped into tubes followed by an additional 1 mL of methanol containing 1% (v/v) 12 N HCl. To this methanolic extract, 2 mL of CHCl3 and 1 mL of 0.1N NaOH was added and vortexed, and the lower CHCl3 layer was collected, dried under N2 and subjected to TLC for S1P separation and quantification (Tonelli et al., 2013).
EAE mouse model
All animal care and experimental procedures complied with the guidelines of the UK Home Office Animals (Scientific Procedures) Act 1986, and were approved by the University of Strathclyde Ethical Review Panel. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). C57BL/6 mice were purchased from Harlan (Oxford, UK) and maintained at the Biological Procedure Unit, University of Strathclyde. Female mice between 7–12 weeks old were used in all experiments.
EAE induction and clinical evaluation
EAE was induced as previously described (Jiang et al., 2009). Mice were immunized subcutaneously on the back with 100 μg of myelin oligodendrocyte glycoprotein peptide fragment 35‐55 (MOG35‐55) in 100 μL of PBS emulsified with an equal volume of complete Freund's adjuvant (CFA, total 350 μg of Mycobacterium tuberculosis, strain H37RA). Each mouse also received i.p. 100 ng·100 μl−1 of Pertussis toxin (PTX) in PBS on days 0 and 2 post immunization. EAE was scored according to a 0–5 scale as follows: 0, no clinical sign; 1, complete loss of tail tone; 2, hind limb weakness; 3, hind limb paralysis; 4, forelimb involvement; and 5, moribund. In a previous study (in house), the maximum clinical score in our model of EAE mice was 2.625 ± 0.7048 (mean ± SD, n = 20). Assuming a change in clinical score of 1.4 to be of clinical relevance, the number of mice required to detect this at 80% statistical power (two‐sided, 5% significance) was calculated to be six mice per group. Eighteen females from separate litters born within 5 days of each other were used. These were then randomly divided them into three cages of 6 and each cage randomly designated to a specific treatment (cage A = compound 5, cage B = ROMe and cage C = vehicle) in a non‐blinded manner (due to available expertise).
To confirm that the infiltration of immune cells in the spinal cord tissue and subsequent inflammation and demyelination was EAE‐specific, control mice were immunized with a similar protocol including CFA and PTX but with PBS in place of MOG35‐55 peptide. These mice displayed no signs of clinical disease, sickness or discomfort, and they gained weight throughout the course at a rate similar to naïve mice. The total number of mice employed in the study was 28.
Preparation of compounds, dosage and route of administration
All compounds were dissolved in a vehicle solution of 20% cyclodextrin in PBS. Compound 5 was prepared as a 1.55 mg·mL−1 stock. The dose given was 12.4 mg·kg−1 (the approximate in vivo concentration is 30 μM assuming 100% bioavailability). ROMe was prepared as a 0.404 mg·mL−1 stock. The dose given was 3.234 mg·kg−1 (the approximate in vivo concentration is 10 μM assuming 100% bioavailability). The doses of compound 5 and ROMe used in the EAE model was based on the EC50/IC50 for SK activation/inhibition, respectively (Lim et al., 2011; Liu et al., 2013), and assumed 100% bioavailability. The compounds were all administered i.p. daily from day 0 of MOG33‐55 immunization at the above doses. The EAE vehicle control group received i.p. injection daily of the 20% cyclodextrin vehicle.
Immunohistochemical staining
Mice were killed in a CO2 chamber, and their spinal cords flushed out with PBS by hydrostatic pressure using a 19G syringe. Tissues were immediately frozen in OCT, and 8‐μm‐thick sections were stained with specific primary antibodies for CD4, CD45 and F4/80 overnight. Sections were then washed and incubated with biotinylated secondary antibody and streptavidin‐HRP before being treated with ImmPACT AMEC red peroxidase substrate. Sections were then washed in distilled water and counterstained with haematoxylin. Isotypes with matching IgG were used as negative control and showed no staining in all tissues.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Statistical analysis employed unpaired t‐test, one‐way ANOVA with Bonferroni post test, two‐way ANOVA with Bonferroni post test or repeated measures ANOVA (using GraphPad Prism TM, V4.0, (GraphPad Software, San Diego)), as appropriate and as indicated in the figure legends. Data were considered to be significant when P < 0.05. Values of n for in vitro experiments are derived from independent experiments rather than within‐experiment replicates. Densitometric values (presented as mean ± SD) were normalized using the corresponding data for actin for the same samples and were obtained from five independent experiments. Statistical analysis was undertaken using one‐way ANOVA with Tukey's post hoc test.
Materials
Cell culture media (RPMI and DMEM with GlutaMAX) and supplements (penicillin, streptomycin and L‐glutamine) were obtained from Life Technologies (Paisley, UK). Fetal bovine serum was from Seralabs (Sussex, UK). Other chemicals including phorbol 12‐myristate 13‐acetate (PMA), LPS, the caspase‐1 inhibitor (Ac‐YVAD‐CHO), protein phosphatase 2A (PP2A) inhibitor (okadaic acid), Pertussis toxin, cyclodextrin, HRP‐conjugated anti‐mouse IgG secondary antibody and haematoxylin were from Sigma (Poole, UK); the SK1/2 inhibitor SKi, (2‐(p‐hydroxyanilino)‐4‐(p‐chlorophenyl)thiazole) was from Merck Chemicals Ltd. (Nottingham, UK); propidium iodide was from eBioscience (Altrincham, UK). Antibodies employed included anti‐myc (to detect myc‐tagged S1P1 receptors) (# sc‐40, Santa Cruz, Wembley, UK), ERK‐2 (# 610104, Transduction Laboratories, Oxford, UK), actin (# A2066, Sigma, Poole, UK), anti‐S1P1‐APC (# FAB7089A, R&D Systems, UK), biotinylated secondary antibody, anti‐CD4, anti‐CD45 and anti‐F4/80 and streptavidin‐HRP (eBioscience, Hatfield, UK). ImmPACT AMEC red peroxidase substrate was from Vector Laboratories, Peterborough, UK. Dithiothreitol was purchased from Enzo (Exeter, UK), nitrocellulose Hybond membrane from GE Healthcare (Little Chalfont, UK) and IL‐1β protein elisa kits from R&D Systems (Abingdon, UK), myelin oligodendrocyte glycoprotein peptide fragment 35‐55 (MOG35‐55) (Sigma Genosys, Haverhill, UK) and complete Freund's adjuvant from Difco (Detroit, MI). Compound 5, RB‐020 and ROMe were synthesized as described previously (Lim et al., 2011; Baek et al., 2013; Liu et al., 2013). Compound 5 was a gift from Young Ah Kim (City University of New York).
Results
Effect of compound 5 and RB‐020 on IL‐1β release
We have previously shown that the azide analogue of (S)‐FTY720 vinylphosphonate (compound 5) is an activator of SK1 and is not a substrate for either SK1 or SK2 (Liu et al., 2013). We have also shown that RB‐020 is an SK1 inhibitor and SK2 substrate (Baek et al., 2013) and that ROMe has a Ki = 16 μM for SK2 inhibition (Lim et al., 2011) with no inhibitory effect on SK1 at concentrations of 100 μM. See Figure 1A for chemical structures. In addition, we have previously shown that SK1 inhibitors induce the proteasomal degradation of SK1 to create SK1 null cells (Tonelli et al., 2010; McNaughton et al., 2016). In androgen‐sensitive prostate cancer cells, this can lead to apoptosis (Loveridge et al., 2010). Therefore, we tested the effect of compound 5, RB‐020 and ROMe on SK1 expression levels in androgen‐independent LNCaP‐AI prostate cancer cells. We show here that the SK1 inhibitors RB‐020 and PF543 induce a decrease in SK1 expression in LNCaP‐AI cells, but, as expected, the SK1 activator compound 5 (10 μM) and the SK2 inhibitor ROMe (0 μM) were without effect on SK1 expression (Figure 1B). These findings are consistent with previous findings that demonstrate that the proteasomal degradation of SK1 is associated with SK1 inhibitors.
Figure 1.
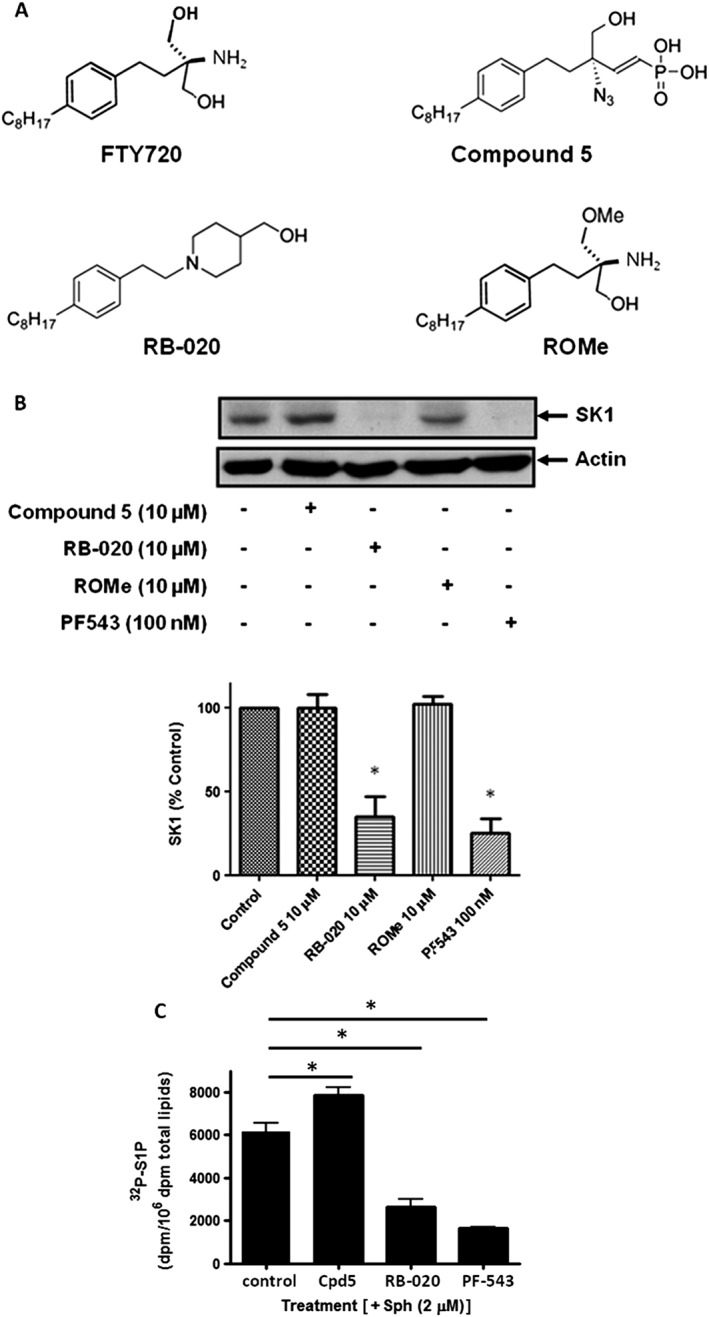
Chemical structures and effects on SK1 expression and S1P formation in cells. (A) Chemical structures of FTY720, compound 5, RB‐020 and ROMe. (B) Western blot showing the effect of compound 5 (Cpd5; 10 μM), RB‐020 (10 μM), PF543 (100 nM) and ROMe (10 μM) on the expression levels of SK1 in LNCaP‐AI cells. Actin was used as a loading control. Results are representative of five independent experiments. Also shown is the densitometric quantification represented as a bar graph. *P < 0.05, significantly different from control. (C) Bar graphs showing the effect of compound 5 (30 μM), RB‐020 (10 μM) and PF‐543 (100 nM) on S1P formation from exogenously added sphingosine in HLMVECs. Results are expressed as means ± SD for three independent experiments *P < 0. 05, significantly different from control; one‐way ANOVA with Bonferroni post test. Control values are: basal [32P]S1P = 718 + 169 dpm; plus sphingosine [32P]S1P = 6154 + 569 dpm.
We have previously shown that ROMe modulates cellular sphingolipid levels with a reduction in S1P and an increase sphingosine levels (Watson et al., 2013). Here, we show that the SK1 inhibitor/SK2 substrate RB‐020 and the SK1 selective nanomolar potent inhibitor, PF‐543 (Schnüte et al., 2012) reduce intracellular S1P, formed from exogenously added sphingosine in HLMVECs (Figure 1C). In contrast, compound 5 increased S1P formation from exogenously added sphingosine in HLMVECs (Figure 1C), consistent with this compound functioning as an activator of SK1 in these cells. Compound 5 activates recombinant SK1 by ~50% at 30 μM in lysates from HEK 293 cells overexpressing this enzyme (Liu et al., 2013), and this is consistent with the 28% increase in S1P formed from exogenously added sphingosine (Figure 1C). Interestingly, RB‐020 is very heavily phosphorylated in HLMVECs (phosphorylated RB‐020: 30527 ± 2304 dpm per 106 dpm in total phospholipids), consistent with our previous finding that RB‐020 is a substrate for purified SK2 (Figure 1C, Baek et al., 2013).
Given their chemical similarity with FTY720, we investigated the effect of compound 5 or sphingosine or RB‐020 on IL‐1β release from macrophages. We show here that treatment of differentiated U937 cells with compound 5 (10 μM) or FTY720 (10 μM) or sphingosine (20 μM) significantly increased IL‐1β release and IL‐1β release showed an upward trend in the presence of RB‐020 (10 μM) (P = 0.06, Figure 2A). There is evidence that PAMPs, such as LPS and the toll‐like receptor 4 (TLR4) and DAMPs, such as sphingosine, can functionally interact to regulate NLRP inflammasome activity and IL‐1β formation (Netea et al., 2009; Luheshi et al., 2012; Escamilla‐Tilch et al., 2013). Therefore, we tested the effect of the compounds in the presence of LPS. LPS stimulated IL‐1β release in differentiated U937 macrophages (Figure 2B). Moreover, treatment of these cells with compound 5 (10 μM), FTY720 (10 μM) or RB‐020 (10 μM) increased IL‐1β release in the presence of LPS (Figure 2B, upper panel). The effects of compound 5 was not additive with sphingosine (Figure 3), suggesting a common mechanism of action for this effect. To investigate whether sphingosine kinase inhibitors increase IL‐1β release, we tested the effect of the SK2 selective inhibitor, ROMe and the SK1/2 inhibitor SKi, (2‐(p‐hydroxyanilino)‐4‐(p‐chlorophenyl)thiazole). We have previously demonstrated that ROMe (SK2 inhibitor) has no effect on LPS‐induced IL‐1β release (Boomkamp et al., 2016), and this is shown here (Figure 2B, lower panel). In addition, SKi had no effect on IL‐1β release in the presence of LPS (Figure 2B, lower panel). The effect of compound 5 on IL‐1β release in the presence of LPS was reduced by pretreatment of cells with the caspase‐1 inhibitor, Ac‐YVAD‐CHO (Figure 4A), thereby confirming a role for the inflammasome. Finally, treatment of cells with the PP2A inhibitor, okadaic acid (0.5 μM), had no effect on the ability of compound 5 to increaseIL‐1β release in the presence of LPS (Figure 4B).
Figure 2.
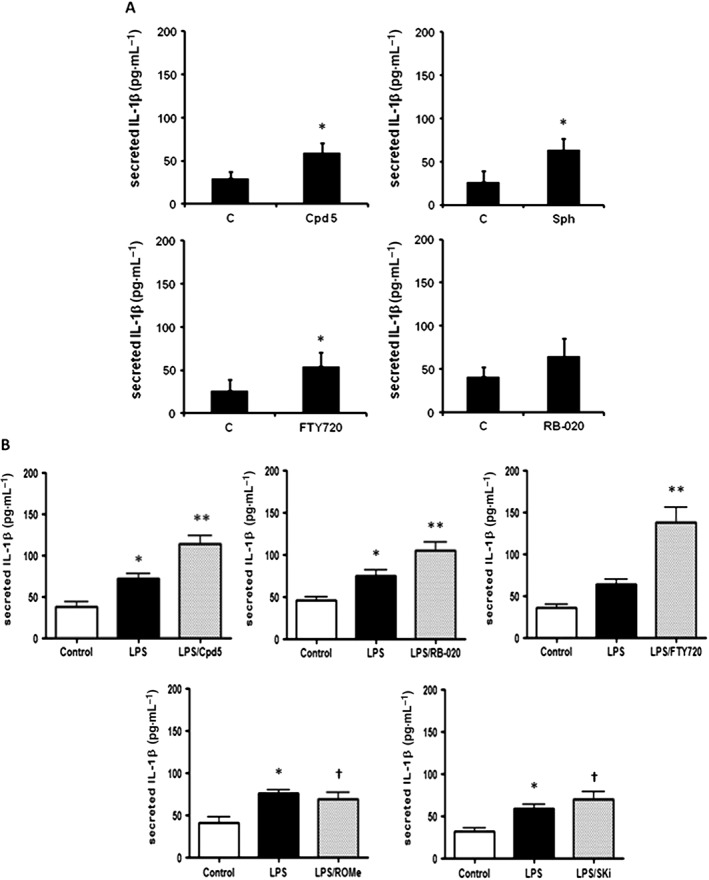
Compound 5, RB‐020 and FTY720, but not ROMe or SKi, enhance IL‐1β release in the presence and absence of LPS in differentiated U937 cells. Differentiated U937 cells were treated with (A) sphingosine (Sph; 20 μM) , compound 5 (Cpd5), FTY720 or RB‐020 (all at 10 μM) alone for 2 h or (B) LPS (1 μg·mL−1) for 2 h and compound 5, RB‐020, FTY720, ROMe or SKi (all at 10 μM) for a further 1 h and the supernatants assayed by elisa for IL‐1β released (pg mL−1). (A) Results are expressed as means ± SEM for independent experiments with matched controls (n = 5 for Sph, FTY720 and RB‐020 and n = 6 for compound 5); *P < 0.05, significantly different from control; unpaired t‐test. (B) Results are expressed as means ± SEM for independent experiments with matched controls and LPS (n = 5, ROMe panel; n = 6, compound 5, RB‐020 and SKi panels; n = 8; FTY720 panel); *P < 0.05, significantly different from control; one‐way ANOVA with Dunnett's post test; **P < 0.05, significantly different from LPS; † not significantly different from LPS; one‐way ANOVA with Bonferroni post test.
Figure 3.
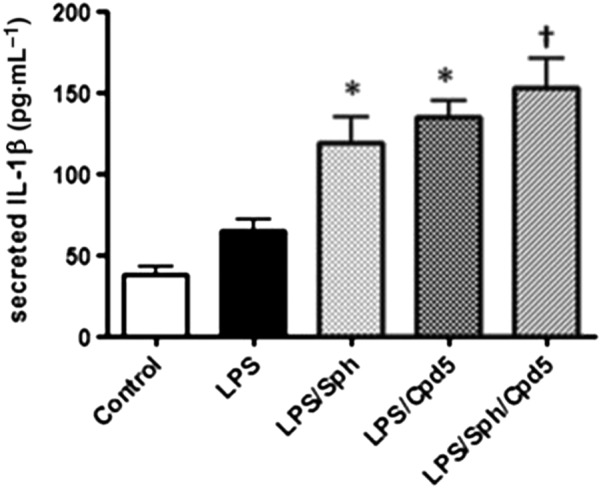
Sphingosine or compound 5 enhances IL‐1β release in the presence of LPS in a non‐additive manner. Differentiated U937 cells were treated with LPS (1 μg·mL−1) for 2 h and then with sphingosine (Sph; 20 μM) or compound 5 (Cpd5; 10 μM) or both sphingosine and compound 5 for 1 h and the supernatants assayed for IL‐1β by elisa. Results are expressed as means ± SEM for six independent experiments. *P < 0.05, significantly different from LPS alone; † not significantly different from LPS/Sph or LPS/compound 5; one‐way ANOVA with Bonferroni post test.
Figure 4.
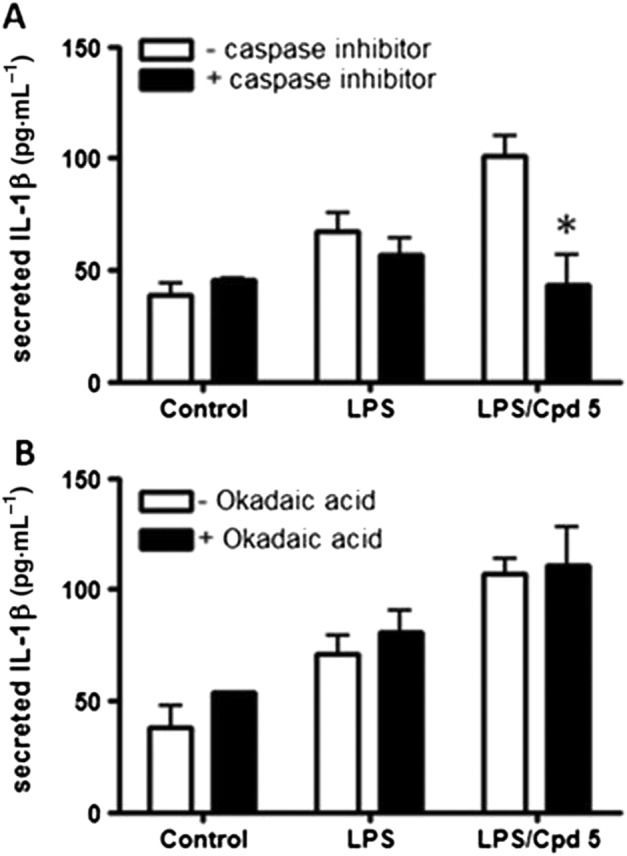
The effect of the caspase‐1 inhibitor Ac‐YVAD‐CHO or the PP2A inhibitor okadaic acid on the increase in IL‐1β release by compound 5 (Cpd5) in the presence of LPS. Differentiated U937 cells were treated with vehicle or Ac‐YVAD‐CHO (10 μM) or okadaic acid (0.5 μM) for 15 min, stimulated with LPS (1 μg·mL−1) for 2 h prior to treatment with compound 5 (10 μM) for 1 h and the supernatants assayed for IL‐1β by elisa. (A) The effect of AC‐YVAD‐CHO on the increase in IL‐1β release by compound 5 in the presence of LPS. (B) The lack of effect of okadaic acid on the increase in IL‐1β release induced by compound 5 in the presence of LPS. Results are expressed as means ± SEM for four (caspase inhibitor) or three (okadaic acid) independent experiments. *P < 0.05, significant effect of caspase inhibitor (Ac‐YVAD‐CHO); two‐way ANOVA with Bonferroni post test.
Effect of compound 5 and ROMe on disease progression in EAE
We have previously shown that PF‐543 (an inhibitor of SK1) exacerbates EAE disease progression (Pyne et al., 2016). Therefore, we tested the effect of the SK1 activator, compound 5, on EAE disease progression and compared it with the SK2 inhibitor, ROMe. Both compound 5 and ROMe markedly reduced EAE disease progression (see clinical score in Figure 5A). The efficacy of these compounds is remarkable with 11 out of 12 mice exhibiting no symptoms whatsoever. EAE mice have a massive infiltration of immune cells in the spinal cord, which was not evident in PBS control tissues (Figure 5B), confirming that the presence of immune cells in the CNS was EAE‐specific. Compound 5 or ROMe induced a considerable decrease in the infiltration of leukocytes, including CD45+ leukocytes, CD4+ T‐cells and F4/80+ macrophages in the spinal cord of EAE mice (Figure 5B). Immunohistochemical staining of spinal sections demonstrated that there are no infiltrating inflammatory cells in mice treated with compound 5 or ROMe (Figure 5B). In addition, ROMe (but not compound 5) induced a reduction in CD4+ and CD8+ T‐cell levels in the blood (lymphopenia) (Figure 5C).
Figure 5.
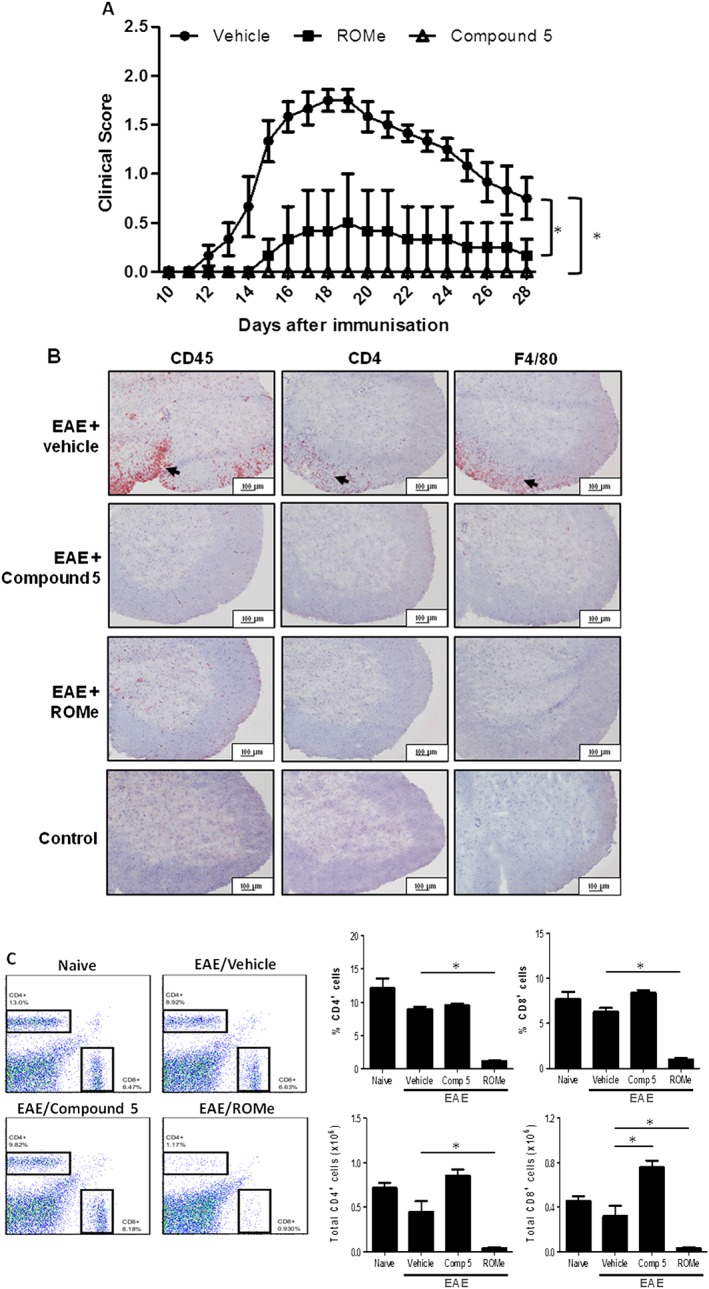
Compound 5 or ROMe attenuate disease progression in EAE. EAE‐treated mice were treated with vehicle alone or dosed with compound 5 (12.4 mg·kg−1) or ROME (3.2 mg·kg−1). (A) Clinical scores are plotted against time and are represented as means ± SEM for n = 6 mice per treatment. *P < 0.05 for compound 5 or ROMe, significantly different from vehicle; repeated measures ANOVA. (B) Immunohistochemical staining of representative spinal sections with specific anti‐CD4, anti‐CD45 or anti‐F4/80 antibodies. Arrows denote infiltrating cells. Results are representative of n = 6 mice per treatment. We have included immunohistochemical staining of spinal sections from control mice (representative of results from 10 naïve mice) to show that there are no infiltrating inflammatory cells. (C) FACS analysis of CD4+ and CD8+ cells. FACS plots are representative of n = 6 animals per treatment group for vehicle, compound 5 and ROMe and 3 for naïve animals. Graphs show combined data; bars represent mean ± SEM. *P < 0.05, significantly different from vehicle; one‐way ANOVA.
Effect of compound 5 and RB‐020 on expression of S1P1 receptors
As FTY720, when converted by SK2 to FTY720‐phosphate, is a functional antagonist of S1P1 receptors (by inducing proteasomal degradation of S1P1) and this underlies part of its action in alleviating MS disease progression (Bigaud et al., 2014), we assessed the effect of compound 5, RB‐020 or ROMe on expression of S1P1 receptors in CCL39 cells that stably overexpress myc‐tagged S1P1 receptors (Boomkamp et al., 2016). This expression system is used for analysing agents that modify S1P1 receptors, as it is very well established that FTY720 (specifically FTY720 phosphate) induces proteasomal degradation of this receptor (Oo et al., 2007). In common with FTY720, which significantly reduced EAE severity in mice and S1P1 receptor levels in CCL39 cells (Boomkamp et al., 2016), compound 5, RB‐020 or ROMe (0.1–10 μM) reduced the levels of this receptor (Figure S1). Indeed, proteolytic fragments of the S1P1 receptor were detected in cells treated with compound 5 or RB‐020 (Figure S1). We also assessed the cell surface expression of S1P1 receptors on CCL39 cells by FACS analysis. Treatment of CCL39 cells with RB‐020, ROMe (0.1–10 μM) or FTY720 induced a 70–80% reduction of the cell surface expression of S1P1 receptors, while compound 5 induced an ~40% reduction (Figure 6). Therefore, the ROMe‐induced reduction in CD4+ and CD8+ T‐cell levels in the blood might be determined by a threshold expression of cell surface S1P1 receptors.
Figure 6.
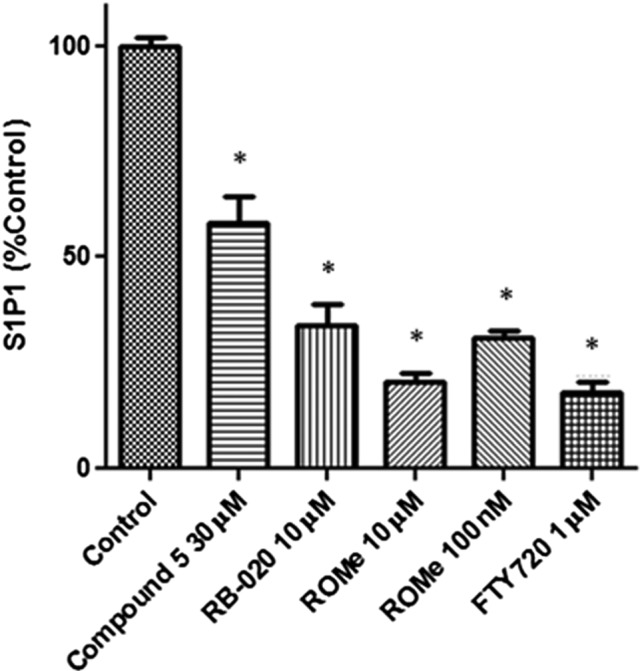
Analysis of cell surface expression of S1P1 receptors in CCL39 cells. FACS analysis of CCL39 cells pretreated with compound 5 (30 μM), RB‐020 (10 μM), ROMe (100 nM or 10 μM), or FTY720 (1 μM), on cell surface expression of S1P1 receptors. Graphs show combined data; bars represent mean ± SEM for five (or for 100 nM ROMe, three) independent experiments. *P < 0.05, significantly different from control; one‐way ANOVA.
Discussion
We have presented in vivo data to show that ROMe and compound 5 exhibit remarkable efficacy in reducing disease progression in an EAE model. However, the mechanisms by which this occurs differ. We demonstrate that compound 5 (unlike ROMe) does not induce a reduction in CD4+ and CD8+ T‐cell levels in the blood. Indeed, ROMe reproduces the effect of FTY720 (in clinical use for MS as GilenyaR) in substantially reducing (>70%) cell surface expression of S1P1 receptors in CCL39 cells. Therefore, it is possible that the effect of ROMe on EAE progression involves modulation of S1P1 receptors and inhibition of T‐lymphocyte trafficking from lymph nodes. However, the effects on EAE disease progression might also be related to the ability of ROMe to inhibit SK2 (Figure 7), which is known to affect gene expression (Hait et al., 2009), as this compound induces a marked reduction in the infiltration of leukocytes and F4/80 macrophages into the spinal cord of EAE mice. Indeed, SK2 knockout mice are protected from EAE, and this is associated with a partial lymphopenia (Imeri et al., 2016). In addition, S1P enhances the development of Th17 cells (Liao et al., 2007), which are involved in the pathogenesis of MS, and it is possible that SK2 regulates this compartment‐specific pool of S1P. Th17 cells were found primarily within central memory T‐cells, and FTY720 reduced blood central memory T‐cells, including RORγt and IL‐17‐producing T‐cells, by >90% (Mehling et al., 2010). S1P also inhibits the function of regulatory T‐cells (Tregs), thereby preventing the suppressive effect of Tregs on Th17 formation (Liu et al., 2009). Therefore, inhibition of S1P formed by SK2 might block these processes to alleviate EAE disease progression. There is also a close link between Th17 and Th1 cells and Th1 cells can be formed from Th17 cells (Harbour et al., 2015). In this regard, IL‐12 is a Th1 cytokine and the IL‐12β1 receptor associates with SK2 and increases IL‐12‐induced STAT4‐mediated transcriptional activity. In addition, dominant negative SK2 in Th1 cells reduces IL‐12 stimulated IFNγ formation whereas WT SK2 enhances it (Yoshimoto et al., 2003). Finally, the SK2 inhibitor, ABC294640, has been shown to be anti‐inflammatory, consistent with the findings here with ROMe. ABC294640 reduced TLR4 expression, NF‐κB activation, pro‐inflammatory cytokine/chemokine production, adhesion molecule expression and the infiltration of monocytes/macrophages and neutrophils (Liu et al., 2012).
Figure 7.
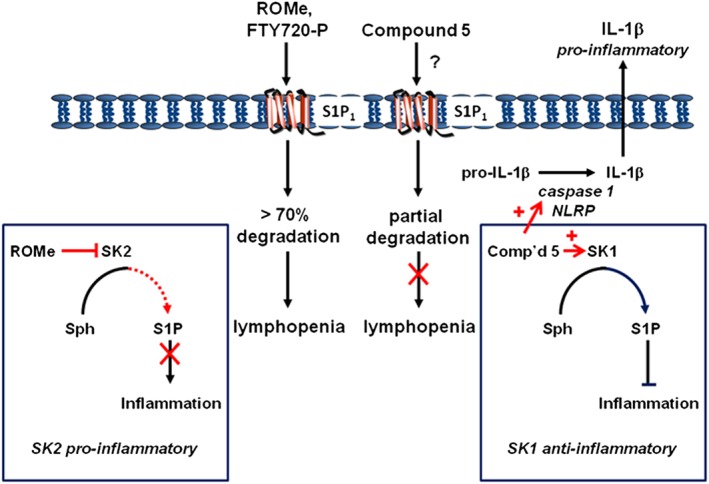
Diagram summarizing the observed effects of ROMe and compound 5 in inflammatory cells involved in EAE and MS disease progression.
The lack of effect of the SK1 activator, compound 5 on levels of CD4+ and CD8+ cells in the blood is consistent with its lesser effect on cell surface expression of S1P1 receptors in CCL39 cells; that is, these receptors may not fall below a threshold level required to prevent egress from lymph nodes in vivo. The mechanism by which compound 5 affects the expression of S1P1 receptors might involve direct binding of this compound to the receptor and subsequent degradation of the receptor (Figure 7). This is based on the finding that the parent compound, (S)‐FTY720 vinylphosphonate, is able to bind to and antagonize S1P1 receptors (Valentine et al., 2010). However, the effect of compound 5 in vivo involves a novel anti‐inflammatory mechanism of action that is distinct from that of FTY720, whereby SK1 regulates a compartment‐specific pool of S1P that functions in an anti‐inflammatory manner (Figure 7). This is supported by the demonstration that SK1 is a negative regulator of Th1 and Th0 cells and siRNA knockdown of SK1 enhances IL‐2, TNF‐α and IFN‐γ release in response to T cell receptor (TCR) stimulation (Yang et al., 2005). Moreover, overexpression of SK1 reduces these effects in response to TCR stimulation (Yang et al., 2005). In addition, the loss of SK1 potentiates induction of the pro‐inflammatory chemokine CCL5 and several other chemokines and cytokines (Adada et al., 2013). Significantly, our findings are the first to demonstrate that an activator of SK1 has this (or any) effect in vivo.
The effect of compound 5 on disease progression in the EAE model does not appear to be hindered by its stimulatory effects on IL‐1β release, as detected in an in vitro cellular system (Figure 7). Indeed, the major effect of RB‐020 and compound 5 on IL‐1β release is more likely to relate to their ability to mimic sphingosine and FTY720, and this is supported by the finding that both sphingosine and FTY720 enhance LPS‐stimulated IL‐1β release. However, SK inhibitors such as ROMe or SKi do not increase IL‐1β. One possible explanation is that these SK inhibitors might not induce sufficient accumulation of sphingosine and/or do not promote sphingosine formation in the correct cellular compartment where IL‐1β processing occurs. In addition, despite structural similarity to FTY720, the failure of ROMe to induce IL‐1β release suggests that the 3‐OH group in FTY720 and sphingosine is required for inflammasome‐dependent stimulation of IL‐1β release.
The effectiveness of ROMe and compound 5 in reducing EAE disease progression is consistent with a pharmacological mode of action on S1P signalling. We therefore conclude that combined modulation of SK1 (activation) and SK2 activity (inhibition) and S1P1 receptors (down‐regulation) might provide an effective anti‐inflammatory action to decrease disease progression in MS. Our data are also consistent with the partial protection from EAE observed in inducible Sgpl1 (S1P lyase) knockout mice (Billich et al., 2013) and the anti‐inflammatory effect of S1P lyase inhibitors (Bagdanoff et al., 2010).
In conclusion, we have demonstrated that compound 5 exhibits a novel mechanism of action compared with FTY720 and this provides the rationale for future development of SK1 activators in the treatment of MS.
Author contributions
S.P., N.J.P. and H.R.J. worked on conception and design; acquisition of data and analysis was conducted by S.P., S.D.B., M.B., N.M. and M.M.; N.J.P., S.P. and H‐R.J. did the data interpretation; and drafting the work. The MS was critically revised by N.J.P., S.P., H.R.J., M.B. and S.D.B.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The effects of compound 5, ROMe and RB‐020 on myc‐tagged S1P1 receptor expression in CCL39 cells. CCL39 cells stably overexpressing myc‐tagged S1P1 receptors were treated with FTY720 (100 nM), compound 5 (10 μM), ROMe (0.1–10 μM) or RB‐020 (0.1–10 μM) for 24 h. Cell lysates were then Western blotted with anti‐myc antibody. ERK‐2 or actin was used as a protein loading control. Results are representative of 3 independent experiments.
Supporting info item
Barbour, M. , McNaughton, M. , Boomkamp, S. D. , MacRitchie, N. , Jiang, H. ‐R. , Pyne, N. J. , and Pyne, S. (2017) Effect of sphingosine kinase modulators on interleukin‐1β release, sphingosine 1‐phosphate receptor 1 expression and experimental autoimmune encephalomyelitis. British Journal of Pharmacology, 174: 210–222. doi: 10.1111/bph.13670.
References
- Adada MM, Orr‐Gandy KA, Snider AJ, Canals D, Hannun YA, Obeid LM et al. (2013). Sphingosine kinase 1 regulates tumor necrosis factor‐mediated RANTES induction through p38 mitogen‐activated protein kinase but independently of nuclear factor κB activation. J Biol Chem 288: 27667–27679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek DJ, MacRitchie N, Pyne NJ, Pyne S, Bittman R (2013). Synthesis of selective inhibitors of sphingosine kinase 1. Chem Commun (Camb) 49: 2136–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdanoff JT, Donoviel MS, Nouraldeen A, Carlsen M, Jessop TC, Tarver J et al. (2010). Inhibition of sphingosine 1‐phosphate lyase for the treatment of rheumatoid arthritis: discovery of (E)‐1‐(4‐((1R,2S,3R)‐1,2,3,4‐tetrahydroxybutyl)‐1H‐imidazol‐2‐yl)ethanone oxime (LX2931) and (1R,2S,3R)‐1‐(2‐(isoxazol‐3‐yl)‐1H‐imidazol‐4‐yl)butane‐1,2,3,4‐tetraol (LX2932). J Med Chem 53: 8650–8662. [DOI] [PubMed] [Google Scholar]
- Bigaud M, Guerini D, Billich A, Bassilana F, Brinkmann V (2014). Second generation S1P pathway modulators: research strategies and clinical developments. Biochim Biophys Acta 1841: 745–758. [DOI] [PubMed] [Google Scholar]
- Billich A, Baumruker T, Beerli C, Bigaud M, Bruns C, Calzascia T et al. (2013). Partial deficiency of sphingosine‐1‐phosphate lyase confers protection in experimental autoimmune encephalomyelitis. PLoS One 8: e59630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho VA, Hla T (2014). An update on the biology of sphingosine 1‐phosphate receptors. J Lipid Res 55: 1596–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomkamp SD, Byun HS, Ubhi S, Jiang H‐R, Pyne S, Bittman R et al. (2016). Effect of ether glycerol lipids on interleukin‐1β release and experimental autoimmune encephalomyelitis. Chem Phys Lipids 194: 2–11. [DOI] [PubMed] [Google Scholar]
- Brough D, Rothwell NJ (2007). Caspase‐1‐dependent processing of prointerleukin‐1 beta is cytosolic and precedes cell death. J Cell Sci 120: 772–781. [DOI] [PubMed] [Google Scholar]
- Chen GY, Nunez G (2010). Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeyrot‐Lacassagne S, Tran TA, Guillaume‐Czitrom S, Marie I, Koné‐Paut I (2009). Brain multiple sclerosis‐like lesions in a patient with Muckle–Wells syndrome. Rheumatology 48: 1618–1619. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodé C, Le Dû N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G et al. (2002). New mutations of CIAS1 that are responsible for Muckle‐Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 70: 1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla‐Tilch M, Filio‐Rodríguez G, García‐Rocha R, Mancilla‐Herrera I, Mitchison NA, Ruiz‐Pacheco JA et al. (2013). The interplay between pathogen‐associated and danger‐associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol 91: 601–610. [DOI] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine‐1‐phosphate. Science 325: 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT (2015). Th17 cells give risee to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A 112: 7061–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T, Brinkmann V (2011). Sphingosine 1‐phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology 76: S3–S8. [DOI] [PubMed] [Google Scholar]
- Imeri F, Schwalm S, Lyck R, Zivkovic A, Stark H, Engelhardt B et al. (2016). Sphingosine kinase 2 deficient mice exhibit reduced experimental autoimmune encephalomyelitis: resistance to FTY720 but not ST‐968 treatments. Neuropharmacology 105: 341–350. [DOI] [PubMed] [Google Scholar]
- Jiang HR, Al Rasebi Z, Mensah‐Brown E, Shahin A, Xu D, Goodyear CS et al. (2009). Galectin‐3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J Immunol 182: 1167–1173. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JJ, Huang MC, Goetzl EJ (2007). Cutting edge: Alternative signaling of Th17 cell development by sphingosine 1‐phosphate. J Immunol 178: 5425–5428. [DOI] [PubMed] [Google Scholar]
- Lim KG, Sun C, Bittman R, Pyne NJ, Pyne S (2011). (R)‐FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF‐7 breast cancer cells. Cell Signal 23: 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA et al. (2009). The receptor S1P1 overrides regulatory T cell‐mediated immune suppression through Akt–mTOR. Nat Immunol 10: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Rehman H, Shi Y, Krishnasamy Y, Lemasters JJ, Smith CD et al. (2012). Inhibition of sphingosine kinase‐2 suppresses inflammation and attenuates graft injury after liver transplantation in rats. PLoS One 7: e41834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, MacRitchie N, Pyne S, Pyne NJ, Bittman R (2013). Synthesis of (S)‐FTY720 vinylphosphonate analogues and evaluation of their potential as sphingosine kinase 1 inhibitors and activators. Bioorg Med Chem 21: 2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveridge C, Tonelli F, Leclercq T, Lim KG, Long JS, Berdyshev E et al. (2010). The sphingosine kinase 1 inhibitor 2‐(p‐hydroxyanilino)‐4‐(p‐chlorophenyl)thiazole induces proteasomal degradation of sphingosine kinase 1 in mammalian cells. J Biol Chem 285: 38841–38852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luheshi NM, Giles JA, Lopez‐Castejon G, Brough D (2012). Sphingosine regulates the NLRP3 inflammasome and IL‐1β release from macrophages. Eur J Immunol 42: 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton M, Pitman M, Pitson SM, Pyne NJ, Pyne S (2016). Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen‐independent LNCaP‐AI prostate cancer cells. Oncotarget 7: 16663–16675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehling M, Lindberg R, Raulf F, Kuhle J, Hess C, Kappos L et al. (2010). Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology 75: 403–410. [DOI] [PubMed] [Google Scholar]
- Netea MG, Nold‐Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH et al. (2009). Differential requirement for the activation of the inflammasome for processing and release of IL‐1beta in monocytes and macrophages. Blood 113: 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR et al. (2007). Immunosuppressive and anti‐angiogenic sphingosine 1‐phosphate receptor‐1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem 282: 9082–9089. [DOI] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ (2011). Translational aspects of sphingosine 1‐phosphate biology. Trends Mol Med 17: 463–472. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, McNaughton M, Boomkamp S, MacRitchie N, Evangelisti C, Martelli AM et al. (2016). Role of sphingosine 1‐phosphate receptors, sphingosine kinase and sphingosine in cancer and inflammation. Adv Biol Regul 60: 151–159. [DOI] [PubMed] [Google Scholar]
- Rutherford C, Childs S, Ohotski J, McGlynn L, Riddick M, MacFarlane S et al. (2013). Regulation of cell survival by sphingosine 1‐phosphate receptor 1 via reciprocal ERK‐dependent suppression of BIM and PI‐3‐kinase/protein kinase C‐mediated up‐regulation of MCL‐1. Cell Death Dis 4: E927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnüte ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW et al. (2012). Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase‐1. Biochem J 444: 79–88. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010). The inflammasomes. Cell 140: 821–832. [DOI] [PubMed] [Google Scholar]
- Shepherd MC, Baillie GS, Stirling DI, Houslay MD (2004). Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte–macrophage differentiation of human U937 cells. Br J Pharmacol 142: 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010). Pattern recognition receptors and inflammation. Cell 140: 805–820. [DOI] [PubMed] [Google Scholar]
- Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G et al. (2010). FTY720 and (S)‐FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen‐independent prostate cancer cells. Cell Signal 22: 1536–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli F, Alossaimi M, Natarajan V, Gorshkova I, Berdyshev E, Bittman R et al. (2013). The roles of sphingosine kinase 1 and 2 in regulating the metabolome and survival of prostate cancer cells. Biomolecules 3: 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey BM, McCallum S, Isenberg DA, Latchman DS (1993). Elevation of heat shock protein synthesis and hsp gene transcription during monocyte to macrophage differentiation of U937 cells. Clin Exp Immunol 93: 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine WJ, Kiss GN, Liu J, E S, Gotoh M, Murakami‐Murofushi K et al. (2010). (S)‐FTY720‐vinylphosphonate, an analogue of the immunosuppressive agent FTY720, is a pan‐antagonist of sphingosine 1‐phosphate GPCR signaling and inhibits autotaxin activity. Cell Signal 22: 1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson DG, Tonelli F, Alossaimi M, Williamson L, Chan E, Gorshkova I et al. (2013). The roles of sphingosine kinases 1 and 2 in regulating the Warburg effect in prostate cancer cells. Cell Signal 25: 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Castle BE, Hanidu A, Stevens L, Yu Y, Li X et al. (2005). J. Sphingosine kinase 1 is a negative regulator of CD4+ Th1 cells. J Immunol 175: 6580–6588. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Furuhata M, Kamiya S, Hisada M, Miyaji H, Magami Y et al. (2003). Positive modulation of IL‐12 signaling by sphingosine kinase 2 associating with the IL‐12 receptor beta 1 cytoplasmic region. J Immunol 171: 1352–1359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The effects of compound 5, ROMe and RB‐020 on myc‐tagged S1P1 receptor expression in CCL39 cells. CCL39 cells stably overexpressing myc‐tagged S1P1 receptors were treated with FTY720 (100 nM), compound 5 (10 μM), ROMe (0.1–10 μM) or RB‐020 (0.1–10 μM) for 24 h. Cell lysates were then Western blotted with anti‐myc antibody. ERK‐2 or actin was used as a protein loading control. Results are representative of 3 independent experiments.
Supporting info item