

Abstract
Increased aortic stiffness is a biomarker for subsequent adverse cardiovascular events. We have previously reported that vascular smooth muscle Src‐dependent cytoskeletal remodelling, which contributes to aortic plasticity, is impaired with ageing. Here, we use a multi‐scale approach to determine the molecular mechanisms behind defective Src‐dependent signalling in an aged C57BL/6 male mouse model. Increased aortic stiffness, as measured in vivo by pulse wave velocity, was found to have a comparable time course to that in humans. Bioinformatic analyses predicted several miRs to regulate Src‐dependent cytoskeletal remodelling. qRT‐PCR was used to determine the relative levels of predicted miRs in aortas and, notably, the expression of miR‐203 increased almost twofold in aged aorta. Increased miR‐203 expression was associated with a decrease in both mRNA and protein expression of Src, caveolin‐1 and paxillin in aged aorta. Probing with phospho‐specific antibodies confirmed that overexpression of miR‐203 significantly attenuated Src and extracellular signal regulated kinase (ERK) signalling, which we have previously found to regulate vascular smooth muscle stiffness. In addition, transfection of miR‐203 into aortic tissue from young mice increased phenylephrine‐induced aortic stiffness ex vivo, mimicking the aged phenotype. Upstream of miR‐203, we found that DNA methyltransferases (DNMT) 1, 3a, and 3b are also significantly decreased in the aged mouse aorta and that DNMT inhibition significantly increases miR‐203 expression. Thus, the age‐induced increase in miR‐203 may be caused by epigenetic promoter hypomethylation in the aorta. These findings indicate that miR‐203 promotes a re‐programming of Src/ERK signalling pathways in vascular smooth muscle, impairing the regulation of stiffness in aged aorta.
Keywords: focal adhesion, aortic stiffness, microRNA, vascular smooth muscle, cytoskeleton
Introduction
Cardiovascular diseases are the leading cause of death worldwide 1. In the vascular system, the proximal aorta plays a critical role as a shock absorber against the intermittent pulsatile output from the heart, and provides a steady flow to smaller vessels downstream 2, 3. With age, the proximal aorta stiffens and this shock absorption function is lost, sending higher pressures to small downstream vessels in the kidney, brain and heart, which predicts for hypertension and end organ damage 4, 5, 6, 7, 8.
Vascular smooth muscle cell (VSMC) contraction is predominantly initiated by Ca2+‐dependent activation of myosin light chain kinase, which phosphorylates the myosin light chains, leading to formation of actomyosin cross‐bridges and increased VSMC stiffness 9. It is now well‐established, however, that the actin cytoskeleton 10, 11, 12, 13, 14, 15 and focal adhesion (FA) complexes 2, 15, 16, 17, 18, 19 influence the output of the contractile machinery to regulate VSMC stiffness. This is achieved through the connection between the FAs and the actin cytoskeleton, which, through the integrins, allow force to be transmitted to the extracellular matrix (ECM) and the tissue as a whole 20, 21. In recent years, we and others have determined a role for FA/actin signalling and remodelling in the regulation of VSMC stiffness, which has emerged as an important contributor to aortic stiffness 2, 22, 23, 24, 25, 26, 27. This process is regulated through activation of the non‐receptor tyrosine kinase Src, which promotes tyrosine phosphorylation of focal adhesion kinase (FAK), paxillin and crk‐associated substrate (CAS), leading to FA remodelling and recycling through an endocytic pathway 17, 24, 28. Furthermore, FA proteins act as extracellular signal regulated kinase (ERK) scaffolds, mediating ERK activation, which was recently shown to be involved in VSMC stiffness development 24. Importantly, previous studies from our laboratory demonstrated that Src‐dependent tyrosine phosphorylation of FA proteins is essentially abolished in aged mouse aorta 2. Moreover, although Src inhibition dramatically decreases agonist‐induced VSMC contractility and stiffness in young mice, it has no such effect in the aorta from aged mice. These results suggest that this Src‐dependent FA pathway is lost in aged aorta, interfering with the dynamic regulation of stiffness 2. The molecular basis behind this age‐related impairment of FA signalling in vascular smooth muscle has not been determined.
Recently, microRNAs (miRs) have emerged as important regulators of VSMC development and function 29, 30, 31, 32, 33, 34, 35, 36, 37, 38. MicroRNAs are a family of short (21–25 nucleotide) RNAs, which typically negatively regulate protein translation of mRNA targets by repressing translation and/or promoting mRNA degradation 39. Mammalian miRs interact with the 3′‐untranslated region (3′‐UTR) of mRNA through a short (6‐8 nucleotide) ‘seed’ sequence, and may therefore regulate the expression of hundreds of targets 39 and drive an entire program of phenotypic function. Critically, miRs have been reported to be dysregulated in cardiovascular diseases, such as hypertension, atherosclerosis and aortic aneurysms 30, 40.
In the current study, we sought to determine whether aberrant expression of miRs could play a role in the age‐related impairment of Src‐mediated plasticity of aortic smooth muscle. We tested the hypothesis that miRs regulate the expression of members of a Src signalling program in contractile vascular smooth muscle, contributing to the aged phenotype. We report here that age‐related upregulation of miR‐203, in a cause‐and‐effect manner, impairs key phosphorylation events downstream of Src tyrosine kinase, which is associated with increased tissue stiffness. We conclude that changes in miR‐203 expression play a pivotal role in defective regulation of stiffness in aged mouse aorta.
Materials and methods
Ethical approval
All procedures were performed in accordance to protocols approved by the Institutional Animal Care and Use Committee of Boston University (Permit Number: A3316‐01). The animals were maintained according to the guidelines set out by the NIH Guide for the Care and Use of Laboratory Animals, and were obtained and used in compliance with federal, state, and local laws. Mice were killed by cervical dislocation following anaesthetization by isoflurane inhalation. For the pulse wave velocity (PWV) measurements, mice were deeply anaesthetized using isoflurane and maintained horizontally on a heated pad (38°C).
In vivo pulse wave velocity measurements
Pulse wave velocity, the standard in vivo measure of arterial stiffness, was performed as previously established 41 in 4‐, 10‐, 18‐ and 24‐month‐old male C57BL/6J mice. Briefly, PWV was measured by acquiring flow pressure waveforms from 2 locations along the aorta, one proximal, (at the level of the renal vein crossing over the aorta) and one approximately 1 cm distal, using high‐resolution Doppler echocardiography (VEVO770; FujiFilm, VisualSonics, Ontario, Canada). This procedure has been validated to record similar PWV measurements from the proximal aortic arch downwards 41. Waveforms were automatically documented for 20‐sec. of continuous recordings with simultaneous electrocardiogram (ECG), allowing assessment of the foot‐to‐foot transit times (TT; Fig. 1A). Pulse wave velocity was calculated by dividing the distance between the proximal and distal locations (in mm) by the difference in the proximal and distal TT of the waveforms (in msec.).
Figure 1.
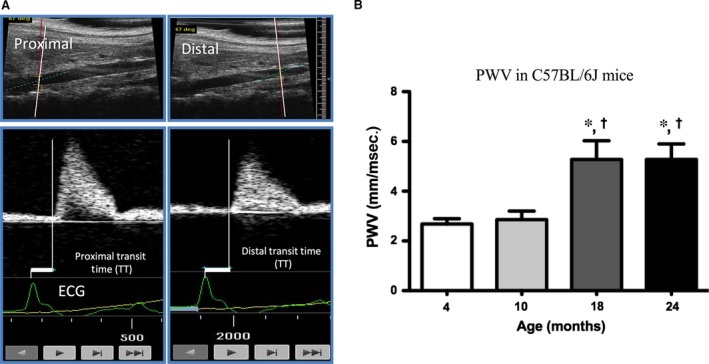
Aortic stiffness is increased in aged mice. Aortic stiffness assessed in vivo by pulse wave velocity (PWV). (A) Blood flow waveforms were continuously recorded for 20 sec. at a proximal and a distal location along the aorta with simultaneous ECG. PWV was calculated by dividing the distance between the proximal and distal locations by the difference between the proximal and distal transit times and expressed in mm/msec. (B) PWV was measured in 4‐ (n = 11), 10‐ (n = 9), 18‐ (n = 8) and 24‐month‐old (n = 7) male mice to obtain a time course of aortic stiffness development. P < 0.05 compared to 4 months (*) and 10 months (†) (two‐tailed Student's t‐test).
Bioinformatic prediction tools
We employed a bioinformatics methodology to determine miRs of interest, using two miR prediction tools, Target Scan (http://www.targetscan.org) and miRbase (http://www.mirbase.org). These tools search for matches between the seed sequence of miRs and the 3′‐UTRs of mRNA targets.
Preparation of aortic samples
Following euthanasia, aortas were quickly excised from young (3–4 months) and aged (24–29 months) mice and placed directly in ice‐cold tissue collection buffer (TCB; modified Krebs solution ‐ in mM: 154 NaCl, 5.4 KCl, 1.2 MgSO4, 10 MOPS, 5.5 glucose, and 1.6 CaCl2; pH = 7.4) 42. Vessels were cleaned from excess perivascular fat and 4–5 mm axial length rings were isolated from the proximal end of the thoracic aorta in preparation for stiffness measurements (see below). For biochemical analyses, tissues were quick‐frozen in an acetone‐dry ice slurry containing 10 mM dithiothreitol (for RNA‐based studies) or 10 mM dithiothreitol and 10% trichloroacetic acid (for western blot studies), as described previously 43.
Measurement of aortic geometry
Axial length, diameter and wall thickness were recorded prior to each experiment for calculation of vessel cross‐sectional area (CSA), which was subsequently used to measure vessel stress. Axial length and diameter were measured under a light microscope (×4) after aortic dissection of unloaded segments in TCB. For wall thickness measurements, ~1 mm length rings were cut at the proximal and distal end of each aortic ring and incubated in nuclear stain (NucBlue; Life Technologies, Carlsbad, CA, USA) for 30 min. After incubation, individual rings were imaged with fluorescence microscopy (×20) for autofluorescence of medial elastin fibres and NucBlue stain via NIS‐Elements software (Nikon Instruments, Melville, NY, USA). Approximately 15–20 measurements were recorded for each ring to calculate the average wall thickness. Subsequently, the averages for the proximal and distal rings for each strip were calculated to obtain the wall thickness for the upper and lower thoracic aorta.
Cell culture
A7r5 rat aortic smooth muscle cells (ATCC, Manassas, VA, USA) were cultured in DMEM high glucose with 10% foetal calf serum, 1% glutamine, 50 units/ml penicillin and 50 μg/ml streptomycin. When A7r5 cells are serum‐starved, they express many smooth muscle‐specific markers, such as α‐actin, smooth muscle myosin, smooth muscle tropomyosin isoforms, h1 calponin and SM22α 44, 45. We favour this approach to the use of primary cultured cells due to the variability of differentiation states with passages. Freshly dissociated cells were not an option for these experiments since they only live 6–9 hrs after isolation. Cells were grown to confluency and then serum‐starved for 24 hrs prior to experimentation.
Transfection of cells
A7r5 cells were seeded at a density of 2 × 105 in 6‐well pates. Cells were transfected using 0.2% Lipofectamine RNAiMAX 3000 reagent, mixed with hsa‐miR‐203 miRvana miR mimic or scrambled miR mimic negative control for 24 (RNA quantification) or 72 hrs (protein quantification). Both hsa‐miR‐203 mimic and scrambled control were diluted in Opti‐Mem medium (Gibco, ThermoScientific, Cambridge, MA, USA) at a final concentration of 5 nM. For results presented in Figure 2, cells were stimulated with 12‐deoxyphorbol 13‐isobutylate 20‐acetate (DPBA) for 10 min. (as described previously Ref. 46). Since DPBA was diluted in dimethylsulphoxide (DMSO), which may influence ERK1/2 phosphorylation 47, 48, 49, equimolar DMSO (0.03%) was added to unstimulated cells as a vehicle control.
Figure 2.
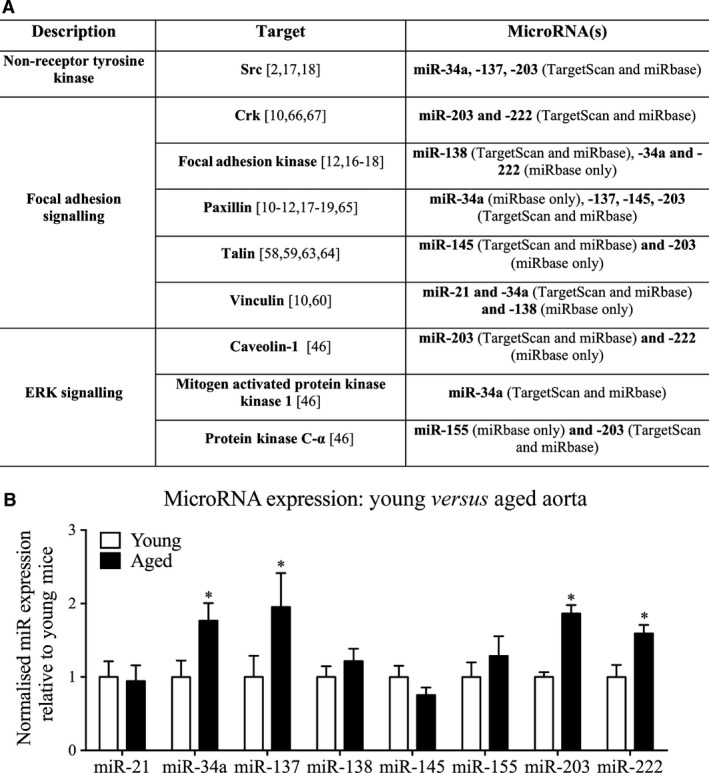
Ageing alters the expression of actin cytoskeletal‐ and focal adhesion‐regulating microRNAs in the mouse thoracic aorta. (A) TargetScan and miRbase were used to identify miRs that could potentially regulate expression of genes involved in FA and ERK signalling pathways, by reverse target prediction (i.e. using the gene targets to select the miRs of interest). (B) The expression level of miRs identified in the above table were compared from young (3 months) and aged (24–29 months) mice (n = 12 mice). *P < 0.05 (two‐tailed Student's t‐test).
Transfection of aortic tissue
Aortas were harvested from 3‐month‐old mice, cut into 2–3 mm pieces and placed in 24 well plates with 1 ml Opti‐Mem medium. Rings were transfected overnight in Opti‐Mem supplemented with 0.2% Lipofectamine RNAiMAX 3000 reagent and hsa‐miR‐203 miRvana miR mimic or scrambled miR mimic negative control (100 nM). The following day, rings were transferred into new 24 well plates containing a 1:1 mixture of TCB and Opti‐Mem in the presence of penicillin (25 U/ml) and streptomycin (25 mg/ml) and the medium was changed every day. On the fourth day of serum‐free organ culture, rings were prepared for measurement of ex vivo aortic stiffness (see below).
Ex vivo aortic stiffness measurement with high‐frequency, low‐amplitude stretches
Aortic rings were mounted with two triangular pieces of wire (0.01 inch diameter). The lower triangle was attached to a micrometer (allowing adjustment of stretch during normalization) and the upper triangle was attached to a force transducer (which recorded changes in vessel wall tension). Once mounted, the vessels were incubated in organ baths containing oxygenated (95% O2 – 5% CO2) physiological salt solution (PSS; Krebs solution – in mM: 120 NaCl, 5.9 KCl, 1.2 NaH2PO4, 25 NaHCO3, 11.5 dextrose, 1 CaCl2, and 1.4 MgCl2; pH = 7.4). Stretch was monitored by the model 300C Dual‐ Mode Lever Arm System by Aurora Scientific (Aurora, ON, Canada) and performed using an electronic input to the lever arm motor via a function generator. Chart software (AD Instruments, Sydney, Australia) was used for data acquisition from the system. Each ring was stretched to optimal length LO (1.8× slack length) 2 and left to equilibrate for 30 min. Vascular smooth muscle viability was confirmed by depolarization for 15 min. through the addition of PSS, in which 51 mM NaCl had been replaced by KCl, followed by return to PSS for 30 min. Since we were interested in activated smooth muscle cell stiffness, measurements at LO were collected at baseline and 15 min. after stimulation with the α‐adrenoreceptor agonist phenylephrine (PE; 10 μM).
Ex vivo aortic stiffness is defined in this study as the material stiffness of aortic tissue, equivalent to the elastic modulus calculated from circumferential force responses to small‐length oscillations (as described previously Refs 2, 18, 50, 51). This method has previously been determined to result in a negligible phase lag of the force response, and does not break actomyosin crossbridges in the aortic tissue 50. Briefly, high frequency (40 Hz), low amplitude (+1% strain from LO) sinusoidal stretching was applied to produce a force response from which the change in stress could be measured as the amplitude of force normalized to CSA. Stiffness (E, kPa), can then be calculated as the change in stress divided by the change in strain (+1% strain from LO): E = (ΔF/A)/(ΔL/L0), where ΔF is the amplitude of the force response to the cyclic stretches, A is the CSA, ΔL is the amplitude of the cyclic stretches, and L0 is the optimal length.
5‐aza‐2′‐deoxycytidine assay
To test if promoter demethylation can cause miR‐203 overexpression, we treated cells with a hypomethylating agent (as described previously Refs. 52, 53). A7r5 cells were seeded into 6‐well plates at a density of 2 × 105 and treated with 1.5 μM of the DNA methyltransferase (DNMT) inhibitor 5‐aza‐2′‐deoxycytidine (5‐Aza) or vehicle control (DMSO) for 72 hrs. Cells were then harvested for quantitative RT‐PCR analysis of miR‐203 expression (see below).
RNA quantification
RNA was isolated with a Trizol‐based protocol at 4°C. For tissue, quick‐frozen aortic strips were homogenized three times at 2500 g for 15 sec. by a beat‐beating technology (Precellys 24; Bertin Technologies, Montigny‐le‐Bretonneux, France). For cell culture, cells were scraped from the cell culture plates directly in Trizol and then mixed several times. RNA was quantified by NanoDrop (Agilent Technologies, Santa Clara, CA, USA) and a 260/280 ratio of >1.8 was required for further study. RNA was reverse transcribed by the TaqMan MicroRNA Reverse Transcription or TaqMan Reverse Transcription kits for the quantification of miR and gene expression, respectively, according to the manufacturer's instructions. TaqMan miR assay kits were used for miR‐21 (5′‐UAGCUUAUCAGACUGAUGUUGA‐3′), miR‐34a (5′‐UGGCAGUGUCUUAGCUGGUUGU‐3′), miR‐137 (5′‐UUAUUGCUUAAGAAUACGCGUAG‐3′), miR‐138 (5′‐AGCUGGUGUUGUGAAUCAGGCCG‐3′), miR‐145 (5′‐GUCCAGUUUUCCCAGGAAUCCCU‐3′), miR‐155 (5′‐UUAAUGCUAAUUGUGAUAGGGGU‐3′), miR‐203 (5′‐GUGAAAUGUUUAGGACCACUAG‐3′), miR‐222 (5′‐AGCUACAUCUGGCUACUGGGU‐3′) and RNU6 (control). For mRNA analysis, TaqMan mouse gene expression assays were used for caveolin‐1 (Mm00483057_m1), Crk (Mm00467065_m1), Dnmt1 (Mm01151063_m1), Dnmt3a (Mm00432881_m1), Dnmt3b (Mm01240113_m1), paxillin (Mm00448533_m1), protein kinase C‐α (Mm00440858_m1), Src (Mm00436785_m1) and talin‐2 (Tln2; Mm00659397_m1). Gene expression was normalized to GAPDH (for tissue) or 18S (for cell culture). Quantitative reverse transcription polymerase chain reaction (qRT‐PCR) was performed on the StepOnePlus Real‐Time PCR system (Applied Biosystems, Waltham, MA, USA). All fold changes were calculated by the ΔΔCt method as described previously 54 and compared with either young mice (in ageing comparisons), scrambled control (in miR mimic comparisons) or vehicle control (in 5‐Aza comparisons).
Western blotting
Mouse aortic tissue was homogenized in a buffer containing 20 mM MOPS, 4% SDS, 10% glycerol, 10 mM DTT, 20 mM β‐glycerophosphate, 5.5 μM leupeptin, 100 μM pepstatin, 20 KIU aprotinin, 2 mM Na3VO4, 1 mM NaF, 100 μM ZnCl2, 20 μM AEBSF, and 5 mM EGTA. Quick‐frozen aortic strips were homogenized ten times at 1500 g for 25 sec. (see above section). Samples were placed on ice between cycles. Cells were harvested on ice for 30 min. by cell‐scraping directly in lysis buffer (mmol/l: 140 NaCl, 3 MgCl2, 1 dithiothreitol and 0.5% Nonidet‐P40 in a 20 mmol/l sodium phosphate buffer, pH 8.0) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN, USA), and mixed several times. Cell lysates were cleared by centrifugation (16,100 g 10 min. at 4°C). Fifteen μg of protein was resolved by SDS‐PAGE, transferred to a nitrocellulose membrane for western blotting with the following antibodies: Caveolin‐1, paxillin, Src, CAS p130, p‐ERK1/2, p‐FAK Tyr925, p‐Paxillin Tyr118 or GAPDH and viewed following incubation with IRDye 680 or IRDye 800CW labelled goat anti‐rabbit or goat‐anti‐mouse IgGs. Protein bands were visualized on an Odyssey infrared imaging system (LI‐COR, Lincoln, NE, USA) and densitometric analysis was performed with the Odyssey 2.1 software (LI‐COR, Lincoln, NE, USA). Intensity was adjusted for display purposes but all analysis was performed on the raw data. For analysis of all protein expression, bands of interest were normalized to GAPDH. Bands were further normalized to young aorta (Fig. 4), scrambled miR mimic control (Fig. 3) or unstimulated cells (Fig. 5).
Figure 4.
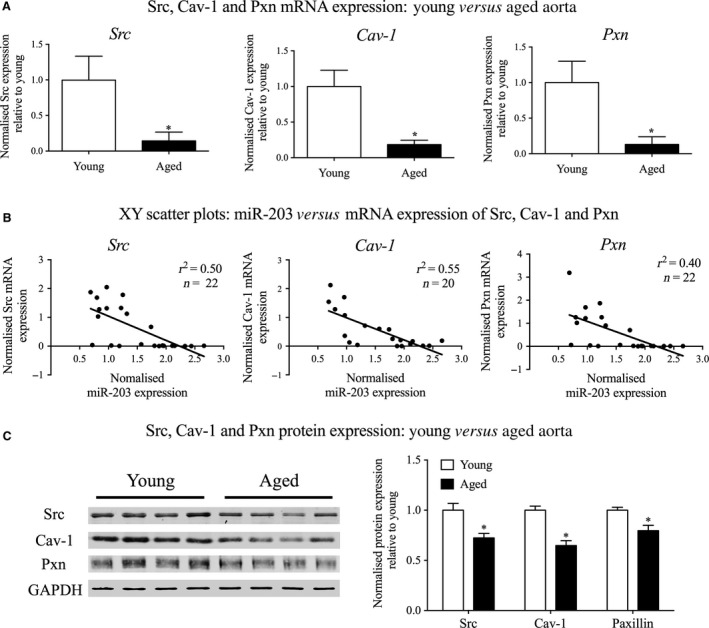
Ageing regulates the expression of miR‐203 targets in mouse aorta. mRNA and protein expression of the validated miR‐203 targets were analysed to determine if ageing regulated their expression in the thoracic aorta. (A) mRNA expression levels of Src, Cav‐1 and Pxn compared in the thoracic aorta from young and aged mice (n = 10–11 mice). (B) XY scatter plots generated to compare the levels of miR‐203 with the mRNA levels of its targets in the aorta from each individual mouse. (C) Typical western blots depicting Src, Cav‐1 and paxillin protein expression in young and aged aorta are shown on the left, with the subsequent quantification comparing their normalized expressions on the right (n = 8 mice). *P < 0.05 (two‐tailed Student's t‐test).
Figure 3.
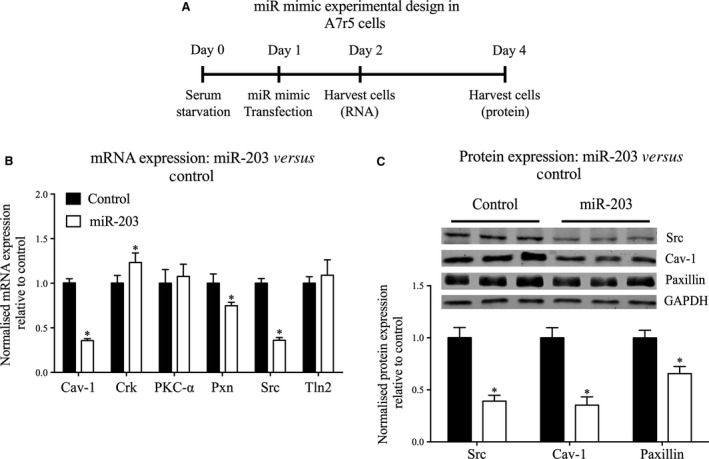
Overexpression of miR‐203 decreases both mRNA and protein levels of predicted targets in A7r5 cells. (A) Schematic of the protocol for transfection of A7r5 cells with miR‐203 mimic or control mimic. (B) mRNA expression levels for Cav‐1, Crk, PKC‐α, Pxn, Src and Tln2 compared in cells transfected with miR‐203 mimic or control (n = 6 experiments). (C) (Top) Representative western blots depicting Src, Cav‐1 and paxillin expression in control and miR‐203 mimic transfected cells. (Bottom) Normalized expression of Src, Cav‐1, and paxillin (n = 12 experiments). *P < 0.05 (two‐tailed Student's t‐test).
Figure 5.
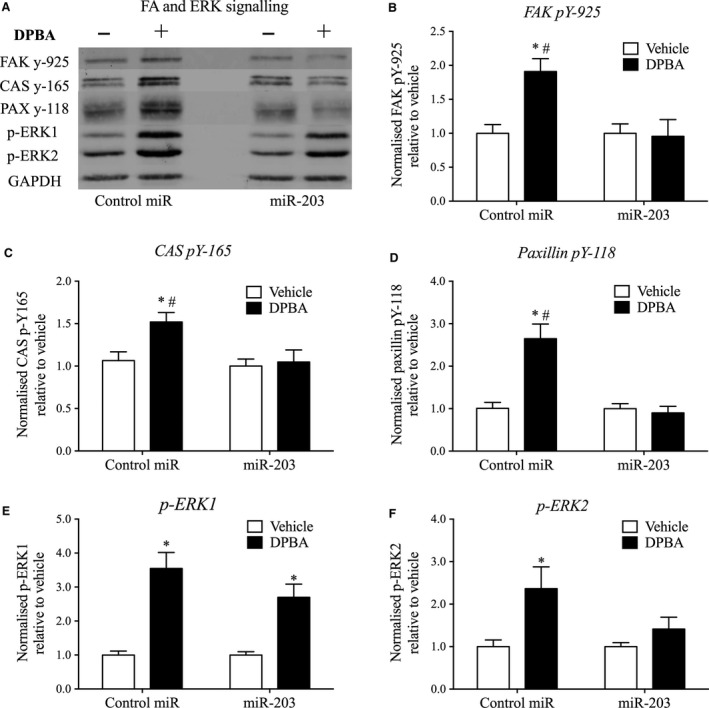
miR‐203 overexpression downregulates agonist‐induced FA and ERK signalling. A7r5 cells were stimulated with DPBA (3 μM for 10 min.) 72 hrs post‐transfection to determine the influence of miR‐203 on signalling pathways downstream of Src. (A) Typical western blots depicting FA signalling in cells transfected with miR‐203 or control mimic. DPBA‐induced phosphorylation of FAK pY‐925 (B), CAS pY‐165 (C), paxillin pY‐118 (D), p‐ERK1 (E) and p‐ERK2 (F) compared in A7r5 cells (n = 7 experiments) transfected with miR‐203 or control mimic. P < 0.05 compared to vehicle control (*) and negative miR mimic control (#) (two‐way anova).
Reagents and antibodies
General laboratory reagents were obtained at analytic grade from Sigma‐Aldrich (St. Louis, MO, USA) and Bio‐Rad (Hercules, CA, USA). The α‐adrenoreceptor agonist PE and hypomethylating agent 5‐Aza were purchased from Sigma‐Aldrich. Cell culture reagents were acquired from Invitrogen and Gibco (ThermoScientific, Cambridge, MA, USA). For the stimulation of A7r5 cells, DPBA (LC Laboratories, Woburn, MA, USA) was used at a final concentration of 3 μM. The following antibodies were used: caveolin‐1 (Cav‐1; #D46G3 rabbit monoclonal 1:2000), p‐ERK1/2 (#T202/Y204 rabbit 1:1000), p‐FAK Tyr925 (#Y925 rabbit 1:200), p‐Paxillin Tyr118 (#Y118 rabbit 1:200) (from Cell Signaling, Danvers, MA, USA), paxillin (#610052 mouse monoclonal 1:1000), CAS p130 (#610271 mouse 1:250) (from BD Transductions, San Jose, CA, USA), Src (#sc‐18 rabbit 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA; for aortic tissue) or (#AM77189 mouse monoclonal 1:500; Abgent, San Diego, CA, USA; for A7r5 cells) and GAPDH (#G9545 rabbit 1:20,000; Sigma‐Aldrich). IRDye labelled secondary antibodies were purchased from LI‐COR (1:1000). RNA‐based experimental reagents, including Trizol, Taqman and miR mimic kits were acquired from Life Technologies, ThermoScientific.
Statistics
All values are presented as mean ± S.E.M. Analysis was carried out using the GraphPad Prism (7.0) software (La Jolla, CA, USA). For biochemical analysis (of mRNA and protein expression) and stiffness comparisons, groups were compared using a Student's t‐test (two‐tailed) for parametric data. DPBA‐induced phosphorylation was compared using a two‐way anova. For grouped analyses, data were analysed with Sidak's multiple comparisons test. Significance was assumed at P < 0.05.
Results
In vivo aortic stiffness increases with age in the mouse
Pulse wave velocity is the gold standard for clinical assessment of aortic stiffness. Aortic stiffness is well‐known to increase with age in humans, beginning around age 50 55. However, to the best of our knowledge only a single paper has appeared demonstrating an age‐induced increase in PWV in the mouse, and this measurement was at a single age point of 29 months 56. The age of 29 months in the mouse is reported to be comparable to ~80 years of age in the human 57. The question arises as to whether aortic stiffness in the mouse model has a similar time course to that of the human. Here, before beginning in vitro studies, we determined the ageing time course of the increases in in vivo aortic stiffness in the mouse. As shown in Figure 1B, PWV was similar in 4‐ and 10‐month‐old mice (similar to 20–40 years of age in humans) 57, but increased in 18‐ and 24‐month‐old mice (comparable to 56–69 human years) 57. Thus, similar to humans, increased aortic stiffness begins in middle age and continues into old age in the mouse. In the present studies we used mice between 24 and 29 months of age where ageing‐induced aortic stiffness is well‐established.
Bioinformatic analysis identifies eight miRs that are high probability matches with the ageing phenotype
In order to determine whether aberrant expression of miRs could play a role in the previously described age‐related impairment of Src‐mediated mechanotransduction of vascular smooth muscle, we employed a reverse target prediction‐based bioinformatics method (Fig. 2A). First, we identified genes that encode multiple signalling molecules involved in the regulation of FAs and cytoskeletal plasticity in VSMCs from the literature 10, 11, 12, 16, 17, 18, 19, 46, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67. Then we used two distinct bioinformatic tools, TargetScan and miRbase, to predict miRs with a high probability of regulating the expression of these targets. Figure 2A highlights these miRs, predicted to target genes encoding FA proteins that we have previously associated with the regulation of cytoskeletal plasticity in vascular smooth muscle. From this analysis, miR‐34a, ‐137, ‐138, ‐145, ‐155, ‐203 and ‐222 were predicted to target the master regulator, Src, the FA proteins FAK, paxillin, vinculin, talin and Crk and, the ERK signalling proteins, mitogen‐activated protein kinase 1, protein kinase C‐α (PKC‐α) and Cav‐1. Next, we tested these putative associations by qRT‐PCR (Fig. 2B).
Ageing in the mouse aorta increases expression of four miRs that target members of the Src signalling network
The proximal aorta from young and aged mice was harvested for qRT‐PCR analysis and probed for the miRs predicted in Figure 2A. Ageing significantly increased the proximal aortic expression of miR‐34a, ‐137, ‐203 and ‐222 by 77%, 95%, 92% and 59%, respectively, whereas no significant differences were observed in the expression levels of miR‐21, ‐138, ‐145, and ‐155 (Fig. 2B).
miR‐203 mimic regulates expression levels of Src, Cav‐1 and Paxillin in A7r5 cells
As indicated in Figure 2A, miR‐203 is predicted to target multiple proteins of interest, namely Cav‐1, Crk, PKC‐α, paxillin, Src and Tln2, whereas the other increased miRs are predicted to target only one or 2 of these proteins. We therefore chose to focus the current study on determining whether the age‐associated increase in miR‐203 may cause changes in smooth muscle FA signalling.
To modify miR‐203 expression, we transfected with a miR‐203 mimic, which mimics the sequence of mature endogenous miR‐203, to enhance its activity in A7r5 cells (protocol diagrammed in Fig. 3A). Smooth muscle cell lysates were harvested at 24 hrs post‐transfection for gene expression analysis by qRT‐PCR. The miR‐203 mimic induced 64%, 64% and 25% decreases in Src, Cav‐1 and paxillin mRNA levels, respectively, but had no significant effect on PKC‐α or Tln2 expression (Fig. 3B). Surprisingly, overexpression of miR‐203 significantly increased Crk gene expression by 23%. Protein levels of these targets were then analysed by immunoblots of lysates of smooth muscle cells transfected with the miR‐203 mimic for 72 hrs. The miR‐203 mimic significantly decreased protein levels of Src, Cav‐1 and paxillin by 61%, 65% and 35%, respectively (Fig. 3C).
These findings indicate that miR‐203 regulates the expression of important FA proteins previously associated with signalling and remodelling of the cytoskeleton of aortic smooth muscle.
mRNA expression levels of miR‐203 targets are regulated by ageing in mouse aorta
We next returned to ageing studies and tested whether there is a correlation between ageing, endogenous miR‐203 and its validated targets (Fig. 4). Consistent with an increase in miR‐203 expression with ageing (Fig. 2), mRNA levels for all three miR‐203 targets are decreased in aged aorta (Fig. 4A). In addition, comparison of miR‐203 expression with the expression levels of its associated target mRNAs, from each individual sample, revealed a direct negative correlation between miR‐203 and Src, Cav‐1 and paxillin mRNA expression levels, albeit with high variability (i.e. low r 2 numbers) (Fig. 4B).
Src, Cav‐1 and paxillin protein levels are significantly changed with ageing
Protein levels were quantitated by immunoblot. A typical blot is shown in Figure 4C, left‐hand panel with mean values plotted in the right‐hand panel. Src, Cav‐1 and paxillin protein expression were decreased in aged aorta by 28%, 35% and 20%, respectively. Of note is the fact that the percentage change in protein expression is quantitatively smaller than the change in mRNA expression with ageing. This is likely due to the well‐known increased stability of many proteins compared to mRNA 68.
Taken together, these results suggest that miR‐203, which increases with ageing, may potentiate age‐induced downregulation of Src, Cav‐1 and paxillin protein expression levels in the mouse aorta.
miR‐203 impairs FA signalling in A7r5 cells in a cause‐and‐effect manner
Next, we examined the impact of an increase in miR‐203 expression on signalling events critical to FA dynamics and cytoskeletal plasticity 2, 17, 18, 69. To achieve this, we overexpressed miR‐203 in A7r5 cells for 72 hrs and quantitated phorbol ester (DPBA)‐induced tyrosine phosphorylation of the FA proteins CAS, FAK, and paxillin, and ERK activation (Fig. 5A–F). Overexpression of miR‐203 completely abolished agonist‐induced tyrosine phosphorylation of FAK pY‐925, CAS pY‐165, and paxillin pY‐118 in A7r5 cells (Fig. 5B–D). Furthermore, agonist‐induced ERK2 phosphorylation was also eliminated in miR‐203 transfected cells (Fig. 5F). However, ERK1 phosphorylation was not significantly affected by miR‐203 transfection in A7r5 cells (Fig. 5E).
Taken together, these findings suggest that increased miR‐203 expression is involved in the previously observed age‐related impairment of FA signalling, resulting in dysfunction of vascular smooth muscle cytoskeletal dynamics.
miR‐203 increases ex vivo active smooth muscle stiffness in young moue aorta
We next determined the effect of overexpressing miR‐203 on ex vivo stiffness of young mouse aorta. Similar to the method for transfecting smooth muscle cells, we transfected excised aortic rings for 72 hrs and then assessed PE‐induced aortic stiffness increase. We have previously experienced difficulty in transfecting vascular tissue from larger animals. However, as shown in Figure 6B, miR‐203 was significantly overexpressed in mouse aortic rings transfected with the miR‐203 mimic compared to scrambled miR mimic control. Interestingly, miR‐203 overexpression enhanced PE‐induced stiffness increases in young aorta (stiffness increase for control: 182.0 ± 19.4 kPa and miR‐203: 262.3 ± 12.0 kPa) to a similar extent as observed with ageing (stiffness increase for young: 151.1 ± 16.6 kPa and aged: 249.0 ± 27.6 kPa) (Fig. 6C).
Figure 6.
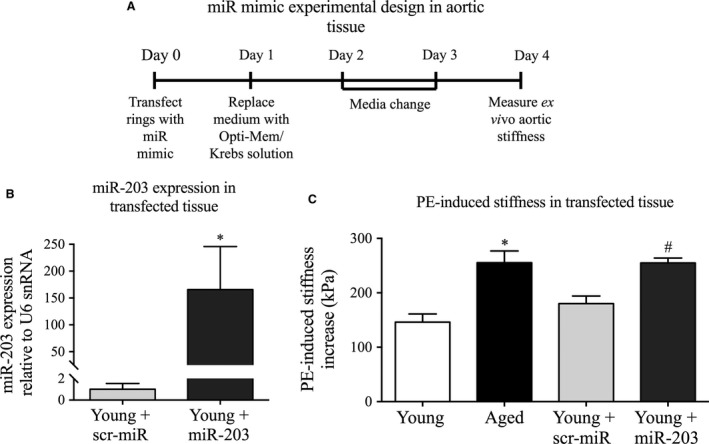
Overexpression of miR‐203 in young aorta reproduces the ageing phenotype of increased aortic stiffness. (A) Schematic of the protocol for transfection of aortic tissue with miR‐203 mimic or control mimic. (B) miR‐203 expression levels in tissue transfected with miR‐203 mimic or control (n = 4 experiments). (C) Agonist‐induced stiffness increase in young (white, n = 10) versus aged (black, n = 6) aorta and young aorta transfected with scrambled miR mimic control (light grey, n = 10) versus miR‐203 mimic (pattern grey, n = 9). *P < 0.05 from young aorta or # scrambled mimic control (two‐tailed Student's t‐test).
These findings are consistent with increased miR‐203 expression in aged aorta playing a role in the age‐related increase in active smooth muscle stiffness.
Ageing negatively regulates mediators of DNA methylation
The question arises as to the molecular mechanism by which miR‐203 levels are regulated by ageing. Previous studies, in other systems, have shown that the expression of miR‐34a, ‐137 and ‐203 are regulated by epigenetic methylation events in their respective promoter regions 82. We therefore hypothesized that aberrant expression of DNMTs played a role in the age‐related regulation of miR‐203 expression in vascular smooth muscle. As illustrated in Figure 7A, the mRNA levels of the three main DNMTs, DNMT1, 3a, and 3b, are significantly decreased in the aorta from aged mice by 68%, 65% and 67%, respectively. In addition, treatment of A7r5 cells with the DNMT inhibitor, 5‐Aza, increased miR‐203 expression several‐fold (Fig. 7B). These findings support the premise that age‐related hypomethylation of the miR‐203 promoter may cause the observed increase in its expression in aged aorta.
Figure 7.
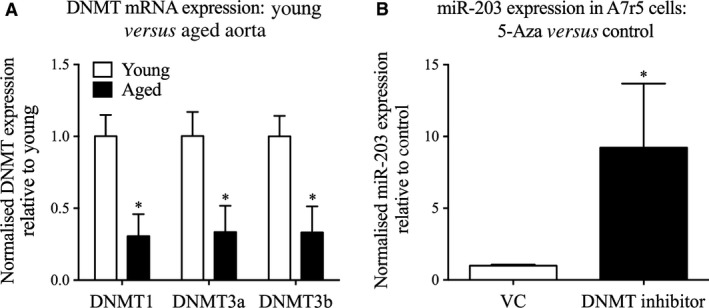
DNA methyltransferases (DNMTs) influence miR‐203 expression. (A) Gene expression of DNMT1, 3a and 3b in young (white) and aged (black) proximal mouse aorta (n = 10 mice). *Significant difference from young mice (two‐tailed Student's t‐test). (B) miR‐203 expression in cells treated with the DNMT inhibitor 5‐Aza for 72 hrs and harvested for quantification of miR‐203 expression (n = 6 experiments). *P < 0.05 (two‐tailed Student's t‐test).
Discussion
In this study we demonstrate, for the first time, that age‐related increases in miR‐203 expression in mouse aorta are associated with downregulation of critical FA signalling proteins in the VSMC, providing a mechanistic basis (summarized in Fig. 8) for the previously described defect in FA signalling with aortic ageing 2. In addition, in vivo data confirm that ageing is also associated with increased aortic stiffness in this mouse model, as measured by the gold standard for clinical assessment of stiffness, PWV. Interestingly, ageing and miR‐203 overexpression in young aorta similarly increased ex vivo measurements of activated smooth muscle stiffness. It appears, therefore, that miR‐203 contributes to the impairment of FA signalling in aged aorta, which is thought to be critical in the normal functioning of the proximal aorta as a buffer against the high systolic pressures generated by the heart.
Figure 8.
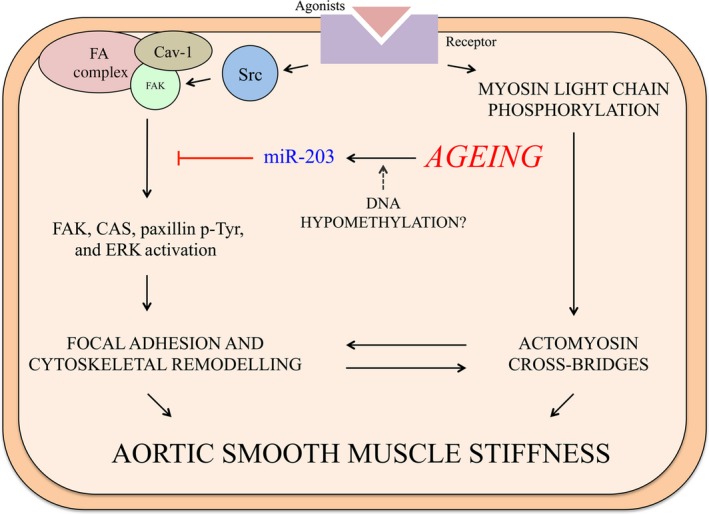
Proposed mechanism. Ageing triggers an increase in miR‐203 expression, leading to a reduction in Src‐mediated signalling. The age‐related increase in miR‐203 expression, at least partially caused by hypomethylation of the miR‐203 promoter region, negatively influences the expression of Src, Cav‐1 and paxillin. Consequently; downstream tyrosine phosphorylation (p‐Tyr) of FAK, paxillin and CAS and ERK activation are impaired, contributing to the age‐related defect in FA remodelling and impaired regulation of stiffness.
In this study, we found that miR‐203 expression is increased in aged aorta, and since it is predicted to target many interesting FA‐related proteins of interest, we further examined its role in aortic smooth muscle cells. We also demonstrated that, consistent with the observed increase in miR‐203 expression, ageing was associated with a decrease in expression of its confirmed targets Src, Cav‐1 and paxillin at both the message and protein levels. These proteins are known to play an essential role in vascular smooth muscle function 10, 17, 19, 70, 71, 72, 73, 74, 75. Unexpectedly, Crk message levels increase following miR‐203 transfection. This is an intriguing finding, since miRs ordinarily negatively regulate target expression. Notwithstanding the possibility that miR‐203 downregulates an unknown repressor of Crk expression, miRs can also directly increase gene expression by interacting with promoter sequences 76. Although this is thought to be a rare occurrence, the influence of miR‐203 on Crk expression warrants further investigation.
It has recently been shown that the expression of miR‐203 is under the influence of methylation events in diverse animals and tissues 52, 53, 77, 78, 79, 80, 81, 82, 83. It is therefore reasonable to speculate that an age‐induced alteration in DNA methylation of the miR‐203 promoter might contribute to the increase in its expression. Indeed, we show here that the expression of key regulators of DNA promoter methylation, DNMTs, are decreased with ageing. Furthermore, we show that cells treated with a DNMT inhibitor display increased expression of miR‐203. Hence, hypomethylation, which is known to occur with ageing 84, could be the cause of this aberrant miR expression in aged aorta. Future experiments will be needed, however, to confirm this observation by specifically examining miR‐203 promoter methylation.
We report here that miR‐203 plays a role in the regulation of VSMC stiffness with increased ageing. Aortic stiffness, as measured in vivo by PWV, was increased at or before 18 months of age, which is consistent with the well‐established observation of increased aortic stiffness in humans between 50 and 60 years of age. Changes in aortic stiffness have often assumed to be dominated by alterations in the ECM. However, we and others have found that the activated smooth muscle contributes to overall aortic stiffness, challenging this dogma 2, 22, 23, 24, 25, 26, 27. Further, we find here that ageing increases activated (PE‐induced) smooth muscle stiffness of aortic rings, supporting the notion that changes at the level of the VSMC could lead to increased aortic stiffness. Of particular importance was the further finding that miR‐203 overexpression in young aorta also increased active aortic stiffness, therefore mimicking the aged phenotype. It should be noted that the approach used in the current study, miR mimic transfection, exaggerates the overexpression of miR‐203, although much of the transfected miR may not actually become incorporated into the RNA‐induced silencing complex 85. While it would be beneficial to study the chronic effects of smaller increases in miR‐203, it is not possible to keep tissue alive ex vivo for the required length of time. Further work will be required to determine the long‐term consequence of miR‐203 overexpression through genetic approaches 86. Despite this caveat, the data presented here point to the increase in miR‐203 expression playing a significant role in increased aortic stiffness with ageing.
The finding that an impairment of Src‐mediated signalling is associated with increased aortic stiffness is interesting. In contrast with our finding, FAK/Src‐mediated signalling has been found to directly increase ECM stiffness in fibroblasts 87, 88, and changes in FAK and CAS protein levels were found to increase cell stiffness 88, 89. However, it is important to point out that in the current study we did not actually see a change in FAK/CAS protein levels, but rather a change in Src‐dependent FAK/CAS signalling. Thus, for the ageing smooth muscle cell we propose an alternative mechanism. Our data are consistent with a model where an inactive FA signalling pathway, primarily caused by age‐ and miR‐203‐related reduction in Src expression, leads to impaired plasticity of VSMCs (Fig. 8). We have previously demonstrated that Src tyrosine kinase is a key player in the regulation of VSMC stiffness, through its effects on a program of FA and actin cytoskeletal remodelling pathways. The tyrosine phosphorylation of the FA proteins CAS pY‐165, FAK pY‐925, and paxillin pY‐118 causes their redistribution, which we previously found to occur through an endocytic recycling pathway 28, allowing cyclic remodelling of the FAs 10, 17, 69, 71. Interestingly, knockout of the Src gene supresses tyrosine phosphorylation, which is associated with their defective turnover and cyclic remodelling 90. We suggest that this cyclic remodelling process, which is lost with ageing, is critical for the proximal aorta to function as a shock absorber against the pulsatile forces from the heart 2.
The regulation of VSMC stiffness has also recently been shown to involve the ERK activation pathway 24. We show here that miR‐203 overexpression disrupts the activation of ERK2 in aortic smooth muscle cells, supporting a recent finding in airway smooth muscle 91. In this study, miR‐203 was found to downregulate the expression of Abl, a Src‐dependent kinase, which modulates ERK activation and, in turn, inhibits cell proliferation 91. In contractile smooth muscle, however, ERK activation promotes phosphorylation and inactivation of the actin‐binding protein caldesmon, alleviating caldesmon‐induced inhibition of myosin‐actin interactions, thus promoting VSMC contraction 92, 93, 94. Furthermore, a pathway linking Src‐dependent FA signalling with an ERK scaffolding function of FAs has recently been implicated in the regulation of VSMC stiffness 24. The molecular mechanism leading to miR‐203‐induced impairment of ERK activation are likely complex, but could be caused by the downregulation of the ERK scaffold Cav‐1, which facilitates the assembly of signalling cascades 46, 72. Further to its role as an ERK scaffold, Cav‐1 has recently been suggested to directly interact with Src to mediate FA signalling 95, 96. In this scenario, Src is scaffolded to lipid rafts by directly binding to Cav‐1, which leads to Cav‐1 phosphorylation at Y14. FA signalling promotes FAK autophosphorylation, which induces Src/Cav‐1 to relocalize to FAK‐containing FA complexes. Subsequent phosphorylation of FAK at Y925, which was abolished by miR‐203 in the present study, and Cav‐1 dephosphorylation are then important events in FA turnover and cell motility 95, 96. In a separate study, the interaction between Cav‐1 and Src was found to be essential for the accumulation of Src in FAs 97. It was interesting, therefore, that Cav‐1 protein was downregulated in the current study, which likely contributes to the defective FA signalling we have reported previously 2. When taken together, these results suggest that miR‐203 regulates the expression of Src, Cav‐1 and paxillin, thereby disrupting the dynamic nature of FA remodelling pathways, which leads to dysregulation of VSMC stiffness in aged aorta (Fig. 8). Focal adhesion signalling is highly complex, however, and it is likely that changes to both inside‐out and outside‐in signalling, involving critical relationships between the ECM, integrins, and the actin cytoskeleton, are responsible for changes in VSMC stiffness. Furthermore, the development of aortic stiffness is clearly multifaceted and involves changes in ECM composition as well as to the intrinsic properties of endothelial and smooth muscle cells.
Although we have focussed our detailed analysis on miR‐203 in the present study, we did find that miR‐34a, ‐137 and ‐222 were also significantly upregulated in aged mouse aorta. Thus, ageing could alter an entire program of VSMC signalling. This also supports previous studies in aged mouse aorta, which have reported an association between miRs that trigger increased inflammatory signalling 98, 99. We confirmed the previously reported increase in miR‐34a expression in aged mouse aorta, which is associated with decreases in the well‐studied longevity gene Sirtuin 1 98. Interestingly, miR‐34a is also known to target vinculin 100, which links the transmembrane integrins to the actin filaments via talin interactions, and could therefore also be an important player in the regulation of VSMC stiffness and contractility 10, 58, 63. This is the first study to find an association between vascular ageing and miR‐137 expression. It is important to note that both miR‐34a and ‐137 are also predicted to target Src and paxillin. Furthermore, several studies have suggested a role for miR‐222 in VSMC function 101, 102, 103, 104, including an association with vascular smooth muscle contractility changes in diabetic rats 102. Further studies will be required to address the functional implications in multiple disease‐associated settings of the age‐related increase in miR‐34a, ‐137 and ‐222 expressions in aortic smooth muscle.
In summary, we show here that male C57BL/6 mice develop aortic stiffness at or before 18 months of age, which is consistent with the well‐established observation of increased aortic stiffness in humans between 50 and 60 years of age. Ageing increases the expression of miR‐203, possibly through promoter hypomethylation, which leads to a decrease in Src, Cav‐1 and paxillin expression. Overexpression of miR‐203 impairs agonist‐induced FA signalling in aortic smooth muscle cells, and increases VSMC stiffness of aortic tissue from young mice. We have previously shown that the impairment of Src‐mediated FA signalling disrupts a VSMC stiffness regulatory pathway in aortas from aged mice 2. Hence, miR‐203 is an upstream mediator of this defective regulation, which interferes with the ability of the aorta to modulate pulsatile pressures coming out of the heart. In conclusion, this study provides novel insights into the molecular mechanisms of age‐related impairment of VSMC function and points to potential therapeutic targets for the modification of aortic stiffness development with ageing.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Author contributions
C.J.N. conceived, designed and performed the experiments, analysed and interpreted the data, and co‐wrote the manuscript. F.S. and S.L. designed and performed the experiments and helped interpret the results. K.G.M. conceived and designed the experiments and interpreted the data, and co‐wrote the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
The authors thank Dr. Kuldeep Singh for critical input regarding the text of the manuscript. This work was supported by NIA AGO50599 to K.G.M. and the Aortic Stiffness Affinity Research Collaborative of the Boston University Evans Center.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, et al Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015; 131: e29–322. [DOI] [PubMed] [Google Scholar]
- 2. Gao YZ, Saphirstein RJ, Yamin R, et al Aging impairs smooth muscle‐mediated regulation of aortic stiffness: a defect in shock absorption function? Am J Physiol Heart Circ Physiol. 2014; 307: H1252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luchsinger PC, Sachs M, Patel DJ. Pressure‐radius relationship in large blood vessels of man. Circ Res. 1962; 11: 885–8. [DOI] [PubMed] [Google Scholar]
- 4. Mitchell GF, Hwang SJ, Vasan RS, et al Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010; 121: 505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Michener KH, Mitchell GF, Noubary F, et al Aortic stiffness and kidney disease in an elderly population. Am J Nephrol. 2015; 41: 320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsao CW, Lyass A, Larson MG, et al Relation of central arterial stiffness to incident heart failure in the community. J Am Heart Assoc. 2015; 4: e002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pase MP, Himali JJ, Mitchell GF, et al Association of aortic stiffness with cognition and brain aging in young and middle‐aged adults: the framingham third generation cohort study. Hypertension. 2016; 67: 513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012; 110: 1097–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol. 2012; 590: 4145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen S, Wang R, Li QF, et al Abl knockout differentially affects p130 Crk‐associated substrate, vinculin, and paxillin in blood vessels of mice. Am J Physiol Heart Circ Physiol. 2009; 297: H533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El‐Yazbi AF, Abd‐Elrahman KS, Moreno‐Dominguez A. PKC‐mediated cerebral vasoconstriction: role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem Pharmacol. 2015; 95: 263–78. [DOI] [PubMed] [Google Scholar]
- 12. Ohanian V, Gatfield K, Ohanian J. Role of the actin cytoskeleton in G‐protein‐coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension. 2005; 46: 93–9. [DOI] [PubMed] [Google Scholar]
- 13. Retailleau K, Toutain B, Galmiche G, et al Selective involvement of serum response factor in pressure‐induced myogenic tone in resistance arteries. Arterioscler Thromb Vasc Biol. 2013; 33: 339–46. [DOI] [PubMed] [Google Scholar]
- 14. Shaw L, Ahmed S, Austin C, et al Inhibitors of actin filament polymerisation attenuate force but not global intracellular calcium in isolated pressurised resistance arteries. J Vasc Res. 2003; 40: 1–10; discussion. [DOI] [PubMed] [Google Scholar]
- 15. Sun Z, Parrish AR, Hill MA, et al N‐cadherin, a vascular smooth muscle cell‐cell adhesion molecule: function and signaling for vasomotor control. Microcirculation. 2014; 21: 208–18. [DOI] [PubMed] [Google Scholar]
- 16. Mills RD, Mita M, Nakagawa J, et al A role for the tyrosine kinase Pyk2 in depolarization‐induced contraction of vascular smooth muscle. J Biol Chem. 2015; 290: 8677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Min J, Reznichenko M, Poythress RH, et al Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol. 2012; 227: 3585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saphirstein RJ, Gao YZ, Jensen MH, et al The focal adhesion: a regulated component of aortic stiffness. PLoS ONE. 2013; 8: e62461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Srinivasan R, Forman S, Quinlan RA, et al Regulation of contractility by Hsp27 and Hic‐5 in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol. 2008; 294: H961–9. [DOI] [PubMed] [Google Scholar]
- 20. Kim HR, Gallant C, Leavis PC, et al Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008; 295: C768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. North AJ, Gimona M, Lando Z, et al Actin isoform compartments in chicken gizzard smooth muscle cells. J Cell Sci. 1994; 107(Pt 3): 445–55. [DOI] [PubMed] [Google Scholar]
- 22. Hong Z, Sun Z, Li M, et al Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J Physiol. 2014; 592: 1249–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qiu H, Zhu Y, Sun Z, et al Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010; 107: 615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saphirstein RJ, Gao YZ, Lin QQ, et al Cortical actin regulation modulates vascular contractility and compliance in veins. J Physiol. 2015; 593: 3929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sehgel NL, Sun Z, Hong Z, et al Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension. 2015; 65: 370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sehgel NL, Vatner SF, Meininger GA. “Smooth muscle cell stiffness syndrome”‐revisiting the structural basis of arterial stiffness. Front Physiol. 2015; 6: 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sehgel NL, Zhu Y, Sun Z, et al Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol. 2013; 305: H1281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poythress RH, Gallant C, Vetterkind S, et al Vasoconstrictor‐induced endocytic recycling regulates focal adhesion protein localization and function in vascular smooth muscle. Am J Physiol Cell Physiol. 2013; 305: C215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Albinsson S, Bhattachariya A, Hellstrand P. Stretch‐dependent smooth muscle differentiation in the portal vein‐role of actin polymerization, calcium signaling, and microRNAs. Microcirculation. 2014; 21: 230–8. [DOI] [PubMed] [Google Scholar]
- 30. Brozovich FV, Nicholson CJ, Degen CV, et al Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. 2016; 68: 476–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Albinsson S, Skoura A, Yu J, et al Smooth muscle miRNAs are critical for post‐natal regulation of blood pressure and vascular function. PLoS ONE. 2011; 6: e18869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Albinsson S, Suarez Y, Skoura A, et al MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol. 2010; 30: 1118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhattachariya A, Dahan D, Ekman M, et al Spontaneous activity and stretch‐induced contractile differentiation are reduced in vascular smooth muscle of miR‐143/145 knockout mice. Acta Physiol (Oxf). 2015; 215: 133–43. [DOI] [PubMed] [Google Scholar]
- 34. Bhattachariya A, Dahan D, Turczynska KM, et al Expression of microRNAs is essential for arterial myogenic tone and pressure‐induced activation of the PI3‐kinase/Akt pathway. Cardiovasc Res. 2014; 101: 288–96. [DOI] [PubMed] [Google Scholar]
- 35. Dahan D, Ekman M, Larsson‐Callerfelt AK, et al Induction of angiotensin‐converting enzyme after miR‐143/145 deletion is critical for impaired smooth muscle contractility. Am J Physiol Cell Physiol. 2014; 307: C1093–101. [DOI] [PubMed] [Google Scholar]
- 36. Turczynska KM, Bhattachariya A, Sall J, et al Stretch‐sensitive down‐regulation of the miR‐144/451 cluster in vascular smooth muscle and its role in AMP‐activated protein kinase signaling. PLoS ONE. 2013; 8: e65135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Turczynska KM, Hellstrand P, Sward K, et al Regulation of vascular smooth muscle mechanotransduction by microRNAs and L‐type calcium channels. Commun Integr Biol. 2013; 6: e22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turczynska KM, Sadegh MK, Hellstrand P, et al MicroRNAs are essential for stretch‐induced vascular smooth muscle contractile differentiation via microRNA (miR)‐145‐dependent expression of L‐type calcium channels. J Biol Chem. 2012; 287: 19199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013; 14: 475–88. [DOI] [PubMed] [Google Scholar]
- 40. Albinsson S, Sward K. Targeting smooth muscle microRNAs for therapeutic benefit in vascular disease. Pharmacol Res. 2013; 75: 28–36. [DOI] [PubMed] [Google Scholar]
- 41. Weisbrod RM, Shiang T, AlSayah L, et al Arterial stiffening precedes systolic hypertension in diet‐induced obesity. Hypertension. 2013; 62: 1105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Corcoran JJ, Nicholson C, Sweeney M, et al Human uterine and placental arteries exhibit tissue‐specific acute responses to 17beta‐estradiol and estrogen‐receptor‐specific agonists. Mol Hum Reprod. 2014; 20: 433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marganski WA, Gangopadhyay SS, Je HD, et al Targeting of a novel Ca2+/calmodulin‐dependent protein kinase II is essential for extracellular signal‐regulated kinase‐mediated signaling in differentiated smooth muscle cells. Circ Res. 2005; 97: 541–9. [DOI] [PubMed] [Google Scholar]
- 44. Firulli AB, Han D, Kelly‐Roloff L, et al A comparative molecular analysis of four rat smooth muscle cell lines. In Vitro Cell Dev Biol Anim. 1998; 34: 217–26. [DOI] [PubMed] [Google Scholar]
- 45. Kimes BW, Brandt BL. Characterization of two putative smooth muscle cell lines from rat thoracic aorta. Exp Cell Res. 1976; 98: 349–66. [DOI] [PubMed] [Google Scholar]
- 46. Vetterkind S, Poythress RH, Lin QQ, et al Hierarchical scaffolding of an ERK1/2 activation pathway. Cell Commun Signal. 2013; 11: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Camici GG, Steffel J, Akhmedov A, et al Dimethyl sulfoxide inhibits tissue factor expression, thrombus formation, and vascular smooth muscle cell activation: a potential treatment strategy for drug‐eluting stents. Circulation. 2006; 114: 1512–21. [DOI] [PubMed] [Google Scholar]
- 48. Seo HJ, Park HJ, Choi HS, et al BMI‐1026 treatment can induce SAHF formation by activation of Erk1/2. BMB Rep. 2008; 41: 523–8. [DOI] [PubMed] [Google Scholar]
- 49. Yu HN, Lee YR, Noh EM, et al Tumor necrosis factor‐alpha enhances DMSO‐induced differentiation of HL‐60 cells through the activation of ERK/MAPK pathway. Int J Hematol. 2008; 87: 189–94. [DOI] [PubMed] [Google Scholar]
- 50. Brozovich FV, Morgan KG. Stimulus‐specific changes in mechanical properties of vascular smooth muscle. Am J Physiol. 1989; 257: H1573–80. [DOI] [PubMed] [Google Scholar]
- 51. Rhee AY, Brozovich FV. The smooth muscle cross‐bridge cycle studied using sinusoidal length perturbations. Biophys J. 2000; 79: 1511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chim CS, Wan TS, Wong KY, et al Methylation of miR‐34a, miR‐34b/c, miR‐124‐1 and miR‐203 in Ph‐negative myeloproliferative neoplasms. J Transl Med. 2011; 9: 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Noguchi S, Mori T, Nakagawa T, et al DNA methylation contributes toward silencing of antioncogenic microRNA‐203 in human and canine melanoma cells. Melanoma Res. 2015; 25: 390–8. [DOI] [PubMed] [Google Scholar]
- 54. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 55. Mitchell GF, Guo CY, Benjamin EJ, et al Cross‐sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation. 2007; 115: 2628–36. [DOI] [PubMed] [Google Scholar]
- 56. Reddy AK, Li YH, Pham TT, et al Measurement of aortic input impedance in mice: effects of age on aortic stiffness. Am J Physiol Heart Circ Physiol. 2003; 285: H1464–70. [DOI] [PubMed] [Google Scholar]
- 57. Flurkey K, Harrison DE. Mouse models in aging research In: Fox JG, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The mouse in biomedical research. 2nd ed Elsevier, Amsterdam, The Netherlands; 2007. pp. 637–72. [Google Scholar]
- 58. Huang Y, Day RN, Gunst SJ. Vinculin phosphorylation at Tyr 1065 regulates vinculin conformation and tension development in airway smooth muscle tissues. J Biol Chem. 2014; 289: 3677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang Y, Zhang W, Gunst SJ. Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J Biol Chem. 2011; 286: 3630–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim HR, Graceffa P, Ferron F, et al Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator‐stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010; 298: C559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mills RD, Mita M, Walsh MP. A role for the Ca‐dependent tyrosine kinase Pyk2 in tonic depolarization‐induced vascular smooth muscle contraction. J Muscle Res Cell Motil. 2015; 36: 479–89. [DOI] [PubMed] [Google Scholar]
- 62. Ohanian J, Pieri M, Ohanian V. Non‐receptor tyrosine kinases and the actin cytoskeleton in contractile vascular smooth muscle. J Physiol. 2014; 593: 3807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Opazo Saez A, Zhang W, Wu Y, et al Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol. 2004; 286: C433–47. [DOI] [PubMed] [Google Scholar]
- 64. Pavalko FM, Adam LP, Wu MF, et al Phosphorylation of dense‐plaque proteins talin and paxillin during tracheal smooth muscle contraction. Am J Physiol. 1995; 268: C563–71. [DOI] [PubMed] [Google Scholar]
- 65. Tang DD, Tan J. Role of Crk‐associated substrate in the regulation of vascular smooth muscle contraction. Hypertension. 2003; 42: 858–63. [DOI] [PubMed] [Google Scholar]
- 66. Tang DD, Zhang W, Gunst SJ. The adapter protein CrkII regulates neuronal Wiskott‐Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J Biol Chem. 2005; 280: 23380–9. [DOI] [PubMed] [Google Scholar]
- 67. Wang T, Cleary RA, Wang R, et al Role of the adapter protein Abi1 in actin‐associated signaling and smooth muscle contraction. J Biol Chem. 2013; 288: 20713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012; 13: 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anfinogenova Y, Wang R, Li QF, et al Abl silencing inhibits CAS‐mediated process and constriction in resistance arteries. Circ Res. 2007; 101: 420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Drab M, Verkade P, Elger M, et al Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin‐1 gene‐disrupted mice. Science. 2001; 293: 2449–52. [DOI] [PubMed] [Google Scholar]
- 71. Zhang W, Huang Y, Wu Y, et al A novel role for RhoA GTPase in the regulation of airway smooth muscle contraction. Can J Physiol Pharmacol. 2015; 93: 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Je HD, Gallant C, Leavis PC, et al Caveolin‐1 regulates contractility in differentiated vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2004; 286: H91–8. [DOI] [PubMed] [Google Scholar]
- 73. Murata T, Lin MI, Huang Y, et al Reexpression of caveolin‐1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin‐1 knockout mice. J Exp Med. 2007; 204: 2373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pojoga LH, Yao TM, Opsasnick LA, et al Dissociation of hyperglycemia from altered vascular contraction and relaxation mechanisms in caveolin‐1 null mice. J Pharmacol Exp Ther. 2014; 348: 260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sung DJ, Noh HJ, Kim JG, et al Serotonin contracts the rat mesenteric artery by inhibiting 4‐aminopyridine‐sensitive Kv channels via the 5‐HT2A receptor and Src tyrosine kinase. Exp Mol Med. 2013; 45: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Place RF, Li LC, Pookot D, et al MicroRNA‐373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008; 105: 1608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bueno MJ, Perez de Castro I, Gomez de Cedron M, et al Genetic and epigenetic silencing of microRNA‐203 enhances ABL1 and BCR‐ABL1 oncogene expression. Cancer Cell. 2008; 13: 496–506. [DOI] [PubMed] [Google Scholar]
- 78. Craig VJ, Cogliatti SB, Rehrauer H, et al Epigenetic silencing of microRNA‐203 dysregulates ABL1 expression and drives Helicobacter‐associated gastric lymphomagenesis. Cancer Res. 2011; 71: 3616–24. [DOI] [PubMed] [Google Scholar]
- 79. Du Y, Liu Z, Gu L, et al Characterization of human gastric carcinoma‐related methylation of 9 miR CpG islands and repression of their expressions in vitro and in vivo . BMC Cancer. 2012; 12: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Saini S, Arora S, Majid S, et al Curcumin modulates microRNA‐203‐mediated regulation of the Src‐Akt axis in bladder cancer. Cancer Prev Res (Phila). 2011; 4: 1698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sandhu R, Rivenbark AG, Coleman WB. Loss of post‐transcriptional regulation of DNMT3b by microRNAs: a possible molecular mechanism for the hypermethylation defect observed in a subset of breast cancer cell lines. Int J Oncol. 2012; 41: 721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sato F, Tsuchiya S, Meltzer SJ, et al MicroRNAs and epigenetics. FEBS J. 2011; 278: 1598–609. [DOI] [PubMed] [Google Scholar]
- 83. Tiwari P, Gupta KP. Modulation of miR‐203 and its regulators as a function of time during the development of 7, 12 dimethylbenz [a] anthracene induced mouse skin tumors in presence or absence of the antitumor agents. Toxicol Appl Pharmacol. 2014; 278: 148–58. [DOI] [PubMed] [Google Scholar]
- 84. Zampieri M, Ciccarone F, Calabrese R, et al Reconfiguration of DNA methylation in aging. Mech Ageing Dev. 2015; 151: 60–70. [DOI] [PubMed] [Google Scholar]
- 85. Thomson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011; 39: 6845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jin HY, Gonzalez‐Martin A, Miletic AV, et al Transfection of microRNA mimics should be used with caution. Front Genet. 2015; 6: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bae YH, Mui KL, Hsu BY, et al A FAK‐Cas‐Rac‐lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal. 2014; 7: ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fabry B, Klemm AH, Kienle S, et al Focal adhesion kinase stabilizes the cytoskeleton. Biophys J. 2011; 101: 2131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Janostiak R, Brabek J, Auernheimer V, et al CAS directly interacts with vinculin to control mechanosensing and focal adhesion dynamics. Cell Mol Life Sci. 2014; 71: 727–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Volberg T, Romer L, Zamir E, et al pp60(c‐src) and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. J Cell Sci. 2001; 114: 2279–89. [DOI] [PubMed] [Google Scholar]
- 91. Liao G, Panettieri RA, Tang DD. MicroRNA‐203 negatively regulates c‐Abl, ERK1/2 phosphorylation, and proliferation in smooth muscle cells. Physiol Rep. 2015; 3: e12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li Y, Je HD, Malek S, et al ERK1/2‐mediated phosphorylation of myometrial caldesmon during pregnancy and labor. Am J Physiol Regul Integr Comp Physiol. 2003; 284: R192–9. [DOI] [PubMed] [Google Scholar]
- 93. Li Y, Je HD, Malek S, et al Role of ERK1/2 in uterine contractility and preterm labor in rats. Am J Physiol Regul Integr Comp Physiol. 2004; 287: R328–35. [DOI] [PubMed] [Google Scholar]
- 94. Morgan KG. The importance of the smooth muscle cytoskeleton to preterm labour. Exp Physiol. 2014; 99: 525–9. [DOI] [PubMed] [Google Scholar]
- 95. Nethe M, Hordijk PL. A model for phospho‐caveolin‐1‐driven turnover of focal adhesions. Cell Adh Migr. 2011; 5: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Su B, Gao L, Meng F, et al Adhesion‐mediated cytoskeletal remodeling is controlled by the direct scaffolding of Src from FAK complexes to lipid rafts by SSeCKS/AKAP12. Oncogene. 2013; 32: 2016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gottlieb‐Abraham E, Shvartsman DE, Donaldson JC, et al Src‐mediated caveolin‐1 phosphorylation affects the targeting of active Src to specific membrane sites. Mol Biol Cell. 2013; 24: 3881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Badi I, Burba I, Ruggeri C, et al MicroRNA‐34a induces vascular smooth muscle cells senescence by SIRT1 downregulation and promotes the expression of age‐associated pro‐inflammatory secretory factors. J Gerontol A Biol Sci Med Sci. 2014; 70: 1304–11. [DOI] [PubMed] [Google Scholar]
- 99. Boon RA, Seeger T, Heydt S, et al MicroRNA‐29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011; 109: 1115–9. [DOI] [PubMed] [Google Scholar]
- 100. Bernardo BC, Gao XM, Winbanks CE, et al Therapeutic inhibition of the miR‐34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci USA. 2012; 109: 17615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leung A, Trac C, Jin W, et al Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res. 2013; 113: 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li T, Yang GM, Zhu Y, et al Diabetes and hyperlipidemia induce dysfunction of VSMCs: contribution of the metabolic inflammation/miRNA pathway. Am J Physiol Endocrinol Metab. 2015; 308: E257–69. [DOI] [PubMed] [Google Scholar]
- 103. Liu X, Cheng Y, Yang J, et al Cell‐specific effects of miR‐221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol. 2012; 52: 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu X, Cheng Y, Zhang S, et al A necessary role of miR‐221 and miR‐222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009; 104: 476–87. [DOI] [PMC free article] [PubMed] [Google Scholar]