

Abstract
Background and Purpose
KV7.1 voltage‐gated potassium channels are expressed in vascular smooth muscle cells (VSMC) of diverse arteries, including mesenteric arteries. Based on pharmacological evidence using R‐L3 (KV7.1 channel opener), HMR1556, chromanol 293B (KV7.1 channel blockers), stimulation of these channels has been suggested to evoke profound relaxation in various vascular beds of rats. However, the specificity of these drugs in vivo is uncertain.
Experimental Approach
We used Kcnq1 −/− mice and pharmacological tools to determine whether KV7.1 channels play a role in the regulation of arterial tone.
Key Results
R‐L3 produced similar concentration‐dependent relaxations (EC50 ~ 1.4 μM) of arteries from wild‐type (Kcnq1 +/+) and Kcnq1 −/− mice, pre‐contracted with either phenylephrine or 60 mM KCl. This relaxation was not affected by 10 μM chromanol 293B, 10 μM HMR1556 or 30 μM XE991 (pan‐KV7 channel blocker). The anti‐contractile effects of the perivascular adipose tissue (PVAT) were normal in Kcnq1 −/− arteries. Chromanol 293B and HMR1556 did not affect the anti‐contractile effects of (PVAT). Isolated VSMCs from Kcnq1 −/− mice exhibited normal peak KV currents. The KV7.2–5 channel opener retigabine caused similar relaxations in Kcnq1 −/− and wild‐type vessels.
Conclusion and Implications
We conclude that KV7.1 channels were apparently not involved in the control of arterial tone by α1‐adrenoceptor agonists and PVAT. In addition, R‐L3 is an inappropriate pharmacological tool for studying the function of native vascular KV7.1 channels in mice.
Abbreviations
- 4‐AP
4‐aminopyridine
- ADRF
adipocyte‐derived relaxing factor
- HMR1556
N‐(6‐cyano‐3‐hydroxy‐2,2‐dimethyl‐3,4‐dihydrochromen‐4‐yl)‐N‐methylethanesulfonamide
- ML277
(2R)‐N‐[4‐(4‐methoxyphenyl)‐1,3‐thiazol‐2‐yl]‐1‐(4‐methylphenyl)sulfonylpiperidine‐2‐carboxamide
- PVAT
perivascular adipose tissue
- VSMC
vascular smooth muscle cells
Tables of Links
| TARGETS |
|---|
| Voltage‐gated ion channels a |
| KV7.1 potassium channels |
| KV7.2 potassium channels |
| KV7.3 potassium channels |
| KV7.4 potassium channels |
| KV7.5 potassium channels |
| L type CaV1.2 calcium channels |
| GPCRs b |
| α1A‐adrenoceptor |
| α1B‐adrenoceptor |
| α1D‐adrenoceptor |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Recent data suggest that the KCNQ family of voltage‐gated K+ (KV7) channels represents a new therapeutic target in cardiovascular disease (Mackie and Byron, 2008; Greenwood and Ohya, 2009; Gurney et al., 2010; Tano et al., 2014). The KV7 channel family is composed of five different isoforms, namely KV7.1–5. Among them, KV7.1 channels are highly expressed at the mRNA and protein level in different types of vessels in humans and animals (Yeung et al., 2007; Ng et al., 2011; Chadha et al., 2012). Based on the effects of pharmacological drugs, stimulation of KV7.1 channels has been suggested to cause profound relaxation in various vascular tissues of rats, including the aorta, mesenteric and pulmonary arteries (Chadha et al., 2012). Immunohistochemical studies using anti‐KCNQ1 antibodies indicated Kcnq1 expression in murine arteries (aortic artery), including the endothelial and smooth muscle layers, at late embryonic and fetal stages (E14.5 to E16.5) (de Castro et al., 2006) and in adult arteries (Yeung et al., 2007). Of note, blood pressure is enhanced in Kcnq1 −/− mice (Takagi et al., 2007). In humans, mutations in the KCNQ1 gene cause Jervell and Lange–Nielsen syndrome (Wang et al., 1996; Goldenberg et al., 2008). A recent meta‐analysis suggests a possible causative role of the KCNQ1 gene in type 2 diabetes (Liu et al., 2013). However, it is unknown whether KCNQ1 gene products are related to vascular pathologies and/or blood pressure regulation in humans.
KV7.1 channels are expressed in vascular smooth muscle cells (VSMCs) of thoracic aorta, carotid, femoral and mesenteric arteries in mice (Yeung et al., 2007; Schleifenbaum et al., 2014) and rats (Chadha et al., 2012). Although these channels presumably do not contribute to resting vascular tone, KV7.1 channel activators, such as R‐L3 (L‐364373), were shown to be effective vasorelaxants (Chadha et al., 2012). R‐L3 responses were inhibited by blockers of KV7.1 channels, such as HMR1556 or chromanol 293B, which suggests that KCNQ1 activation is an important mechanism underlying vascular relaxation (Chadha et al., 2012). However, the specificity of the drugs used to target native KV7.1 channels is unknown, since the pore‐forming KV7.1 subunit can interact with several accessory KCNE subunits, which are known to modulate the biophysical properties of KV7 channels in vivo (Jespersen et al., 2005). Second, these accessory subunits may influence the pharmacological properties of native, complex, multimeric KV7.1 channels (MacVinish et al., 2001). Therefore, the aim of this study was to test the contribution of KV7.1 channels in the regulation of arterial vascular tone by using Kcnq1 −/− mice and pharmacological tools, such as R‐L3, chromanol 293B and HMR1556. We also used a novel KV7 channel opener, ML277, which has recently been shown to activate KV7.1 channels in cardiomyocytes with an EC50 of 260 nM (Mattmann et al., 2012).
Methods
Mouse model
All animal care and experimental procedures followed American Physiological Society guidelines and were approved by the local authorities (Landesamt für Gesundheit und Soziales Berlin, LAGeSo). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Animals were housed in individually ventilated cages under standardized conditions with an artificial 12 h dark–light cycle with free access to water and food.
We used Kcnq1 −/− mice (C57BL/6 background) with targeted disruption of exon 2 of the Kcnq1 gene (Casimiro et al., 2001). Heterozygous mice were used for breeding to obtain homozygous knockout (Kcnq1 −/−) mice. Littermate (12–16 weeks old) male wild‐type mice (+/+) were used as controls (Figures 1C,D; 4C; 5 and Figures S4A; S5C,D; S6; S7); as everywhere else age‐matched (12–16 weeks old) male C57BL/6 mice purchased from Charles River, Sulzfeld, Germany. For experiments involving rat mesenteric arteries, male Sprague Dawley rats (Charles River, Sulzfeld, Germany, age 10 weeks) were used. Animals were randomly assigned to the experimental procedures in accordance with the German legislation on protection of animals.
Figure 1.
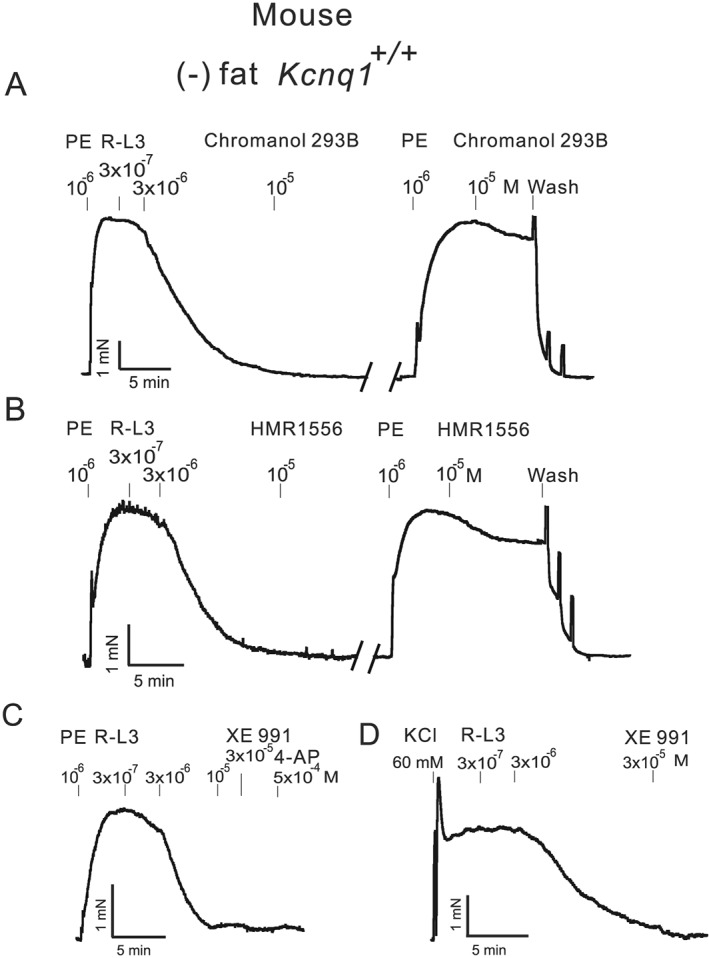
Original recordings showing the effects of 0.3‐3 μM R‐L3, 10 μM chromanol 293B, 10 μM HMR1556, 0.5 mM 4 aminopyridine (4‐AP) and 10–30 μM XE991 on arterial tone of isolated mesenteric artery rings without PVAT (−) fat. Mesenteric arteries were either precontracted with 1 μM phenylephrine (PE) (A,B,C) or with 60 mM KCl (D). Effects of chromanol 293B (A) and HMR1556 (B) on arterial tone induced by PE. All vessels were from wild‐type (Kcnq1 +/+) mice.
Genotyping of Kcnq1−/− and Kcnq1+/+ mice
Tail samples from 4‐week‐old mice were digested in 150 μL tail lysis buffer (75 μL 25 mM NaOH; 75 μL 40 mM TrisHCl, pH 5.0) for 30 min at 95°C. Genotyping PCR was carried out on 3.5 μL tail lysate with Go Taq ® G2 Green Master mix according to manufacturer's specifications in 15 μL reactions. The following PCR programme was used: 94°C 6 min, repeat 40 times 94°C for 45 s, 58°C for 45 s, 72°C for 50 s followed by 72°C for 8 min then holding at 4°C. Following primers were used: forward primer 5′‐ CCAGGAGTGGGTGGTTCTAC‐3′, reverse primer 5′‐GCCAGCACTAAAGATCTTGC‐3′, Neo forward primer 5′‐CGCTTCCTCGTGCTTTACG‐3′, as previously described (Casimiro et al., 2001). PCR reactions were analysed on 2% agarose gels containing ethidium bromide, visualized by exposure to ultraviolet light with DNA ladder 100 bp manufactured by PeqLab.
Quantitative real‐time PCR
Experiments were performed according to MIQE guidelines (Bustin et al., 2009). Briefly, total RNA was isolated from either Kcnq1 +/+ or Kcnq1 −/− mesenteric arteries (first branches) by using the RNeasy RNA isolation kit (Qiagen, Hamburg, Germany) according to the manufacturer's instruction. Isolated RNA concentration was measured and RNA quality was tested by NanoDrop‐1000 spectrophotometer (PeqLab, Erlangen, Germany). For the synthesis of cDNA, equivalent amounts of RNA (2 μg) were used and processed by a TaqMan® Reverse Transcription Reagents (Life Technologies GmbH, Darmstadt, Germany, catalogue number: N8080234). Real‐time PCR were done using 2.0 μL of cDNA in a total volume of 25 μL using the Faststart Universal SYBR green Master Mix (Roche, catalogue number: 04913850001). Experiments were run on an Applied Biosystems 7500 Fast Real‐Time PCR System (Life Technologies Corporation, Carlsbad, CA, USA). Primers were designed using Primer 3 software on different exons to exclude any DNA contamination. Specificity of amplified products was validated in silico (blast) and empirically with gel electrophoresis and analysis of melt curves. Primers were synthesized by BioTez (Berlin, Germany); the sequences are provided below. The cycling conditions were the following: initial activation at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Samples and negative controls were run in parallel. The expression level of the target genes was normalized by the expression of 18 s. Under our experimental conditions, expression of 18 s as a reference gene did not differ between Kcnq1 +/+ and Kcnq1 −/− tissues. The fold change in gene expression between Kcnq1 +/+ and Kcnq1 −/− was calculated using 2 ΔΔCt method (Livak and Schmittgen, 2001). The following primers were used:
18s: F: 5′‐ACATCCAAGGAAGGCAGCAG‐3′; R: 5′‐XTTTTCGTCACTACCTCCCCG‐3′
Kcnq3: F: 5′‐CAGTATTCGGCCGGACATCT‐3′; R: 5′‐GAGACTGCTGGGATGGGTAG‐3′
Kcnq4: F: 5′‐CACTTTGAGAAGCGCAGGAT‐3′; R: 5′‐CCAGGTGGCTGTCAAATAGG‐3′
Kcnq5: F: 5′‐CCTCACTACGGCTCAAGAGT‐3′; R: 5′‐TTAAGTGGTGGGGTGAGGTC‐3′
Wire myography
Mesenteric or renal arteries were removed immediately after killing the mice with isoflurane anaesthesia, quickly transferred to cold (4°C), oxygenated (95% O2/5% CO2) physiological salt solution (PSS) containing (in mmol L−1) 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose, 1.6 CaCl2 and dissected into 2 mm rings. The perivascular fat (PVAT) and connective tissue was either intact “(+) fat” or removed “(−) fat” from each ring without damaging adventitia. For experiments involving endothelium‐denuded arteries, a 1 mL air bubble was used to disrupt the endothelium and removal of a functional endothelium was confirmed by absence of a vasodilatory response to 10 μM ACh. Each ring was positioned between two stainless steel wires (diameter 0.0394 mm) in a 5 mL organ bath of a Mulvany Small Vessel Myograph (DMT 610 M; Danish Myo Technology, Denmark). The organ bath was filled with PSS. The bath solution was continuously oxygenated with a gas mixture of 95% O2 and 5% CO2 and kept at 37°C (pH 7.4) (Fésüs et al., 2007). The mesenteric and renal rings were placed under a tension equivalent to that generated at 0.9 times the diameter of the vessel at 100 mm Hg by stepwise distending the vessel using LabChart DMT Normalization module. This normalization procedure was performed to obtain the passive diameter of the vessel at 100 mm Hg (Fésüs et al., 2007). The software Chart5 (AD Instruments Ltd. Spechbach, Germany) was used for data acquisition and display. After 60 min, incubation arteries were pre‐contracted either with isotonic external 60 mM KCl or 1–3 μM phenylephrine until a stable resting tension was acquired. The composition of 60 mM KCl (in mmol L−1) was 63.7 NaCl, 60 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose and 1.6 CaCl2. Drugs were added to the bath solution if not indicated otherwise. Tension is expressed as a percentage of the steady‐state tension (100%) obtained with isotonic external 60 mM KCl.
Isolation of arterial VSMCs
VSMCs from mesenteric arteries were isolated as described (Gollasch et al., 1998; Plüger et al., 2000; Schleifenbaum et al., 2014). Briefly, the first order of mesenteric or main renal arteries was removed and quickly transferred to cold (4°C) oxygenated (95% O2–5% CO2) PSS. The arteries were cleaned, cut into pieces and placed into a Ca2 +‐free Hank's solution (mM): 55 NaCl, 80 sodium glutamate, 5.6 KCl, 2 MgCl2, 1 mg·mL−1 BSA (Sigma, Taufkirchen), 10 glucose and 10 HEPES (pH 7.4 with NaOH) containing 0.5 mg·mL−1 papain (Sigma) and 1.0 mg·mL−1 DTT for 50 min at 37°C. The segments then were placed in Hank's solution containing 1 mg·mL−1 collagenase (Sigma, type F an H, ratio 30 and 70%) and 0.1 mM CaCl2 for 10 min at 37°C. Following several washes in Ca2 +‐free Hank's solution (containing 1 mg·mL−1 BSA), single cells were dispersed from artery segments by gentle triturating. Cells were then stored in the same solution at 4°C.
Electrophysiology
Voltage dependent potassium (KV) currents were measured in the conventional whole‐cell configuration of the patch‐clamp technique at room temperature as previously described (Gollasch et al., 1996; Essin et al., 2007; Schleifenbaum et al., 2014). Patch pipettes (resistance, 3–5 MΩ) were filled with a solution containing (in mM): 130 KCl, 1 MgCl2, 3 Na2‐ATP, 0.1 Na3‐GTP, 10 HEPES and 5 EGTA (pH 7.2; Yeung and Greenwood, 2005). The external bath solution contained (in mM): 126 NaCl, 5 KCl, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 11 Glucose (pH 7.2; Yeung and Greenwood, 2005). Holding potential was −60 mV. Whole cell currents were recorded using an Axopatch 200B amplifier (Axon Instruments/Molecular Devices, Sunnyvale, CA, USA) or an EPC 7 amplifier (List, Darmstadt, Germany) and digitized at 5 kHz, using a Digidata 1440A digitizer (Axon CNS, Molecular Devices) and pClamp software versions 10.1 and 10.2.
Data and statistical analysis
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM. EC50 values were calculated using a Hill equation: T = (B0−Be)/(1 + ([D]/EC50)n) + Be, where T is the tension in response to the drug (D); Be is the maximum response induced by the drug; B0 is a constant; EC50 is the concentration of the drug that elicits a half‐maximal response (Bychkov et al., 1998). Curve fittings and data analysis were done by GraphPad6 (Software, La Jolla California USA) using nonlinear regression. Statistical significance was determined by t‐test or ANOVA with multiple comparison by Holm–Sidak method. The post hoc tests were run only if F achieved P < 0.05, and there was no statistical significant variance in homogeneity. Extra sum‐of‐squares F test was performed for comparison of concentration–response curves. P values <0.05 were considered statistically significant; n represents the number of animals used; data from multiple rings, multiple cells from the same animal were averaged and treated as a single n. The data analyst was blinded, whereas blinding for the operator was not possible due to the shaker‐waltzer phenotype of mice (Video S8). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
All salts and other chemicals were obtained from Sigma‐Aldrich (Germany) or Merck (Germany). All drugs were freshly dissolved in the day of each experiment accordingly to the material sheet. When DMSO was used as solvent, maximal DMSO concentration after application did not exceed 0.5%. The following concentration of drugs were used: phenylephrine (Sigma Aldrich) ranged from 0.1 to 100 μM, retigabine (Valeant Research North America) from 0.01 to 100 μM, R‐L3 (Merck) from 0.3 to 10 μM, ML277 (Sigma Aldrich) from 0.1 to 10 μM, 30 μM XE991 (Tocris), 10 μM chromanol 293B (Sigma Aldrich), 0.5 mM 4‐aminopyridine (4‐AP; Sigma Aldrich). KCNQ1/KCNE1 subunits are blocked by chromanol 293B with EC50 ~ 7 μM; whereas homomeric KCNQ2, KCNQ3, KCNQ4 and heteromeric KCNQ2/KCNQ3 channels are only slightly blocked by 100 μM (Lerche et al., 2007). Chromanol 293B is also known as (3R,4S)‐293B or 3S,4R‐293B (Seebohm et al., 2001). XE991 was found to inhibit either KV7.1 homomeric or KV7.1/KCNE channels with IC50 ~ 0.8 μM and 11.1 μM respectively (Wang et al., 2000). As a pan KCNQ inhibitor, XE991 also inhibits KCNQ2/3 channels (EC50 ~ 1 μM) (Wang et al., 1998), KCNQ4 (EC50 ~ 5.5 μM) (Søgaard et al., 2001) and KCNQ5 (EC50 ~ 65 μM) (Schroeder et al., 2000). 10 μM HMR1556 is known to inhibit 97% of IKs current (mainly composed of KV7.1/KCNE channels) (Bosch et al., 2003). ML277 is a potent activator of KCNQ1 with EC50 260 nM and >100‐fold selective versus KCNQ2 and KCNQ4 channels (Mattmann et al., 2012). Retigabine is an activator of the KCNQ2/3 (EC50 5.2 μM) (Main et al., 2000) and KCNQ3/KCNQ5 channels (EC50 1.4 μM) (Wickenden et al., 2001).
Results
Failure of KV7.1 channel blockers to reverse arterial relaxation induced by R‐L3
First, we tested the vasodilatory effects of R‐L3, which supposedly acts through activation of KV7.1 channels. R‐L3 produced concentration‐dependent relaxation of murine mesenteric arteries pre‐contracted with 1 μM phenylephrine (Figures 1A,B,C and 2A,B). Both chromanol 293B and HMR1556 (KV7.1 channel blockers) on the contrary did not affect contractions induced by 1 μM phenylephrine (Figure 1A,B) (increase by 8.4 ± 4.3%, n = 5, P > 0.05, paired t‐test; increase by −7.3 ± 3.8%, n = 5, P > 0.05, paired t‐test, respectively). Neither 10 μM chromanol 293B (Figure 1A), 10 μM HMR1556 (Figure 1B), 30 μM XE991 nor 0.5 mM 4‐AP (Figure 1C) reversed R‐L3‐induced relaxations of the murine mesenteric rings (but see Figure 3 for rat mesenteric arteries). Pretreatment of the murine mesenteric arteries with 10 μM chromanol 293B did not prevent relaxation by R‐L3 (Figure S1A). Similar results were obtained in rat mesenteric arteries (Figure S1B). Interestingly, the resting tone of murine mesenteric arteries was not affected by KV7.1 channel blockade, that is neither by 10 μM chromanol 293B (increase by −0.4 ± 0.2%, n = 5, P > 0.05, paired t‐test) nor by 10 μM HMR1556 (increase by −0.2 ± 0.1%, n = 5, P > 0.05, paired t‐test). Additionally and similarly to the mice, the resting tone of rat mesenteric arteries was not affected by KV7.1 channel blockade with 10 μM chromanol 293B (increase by −0.5 ± 0.3%, n = 5, P > 0.05, paired t‐test).
Figure 2.
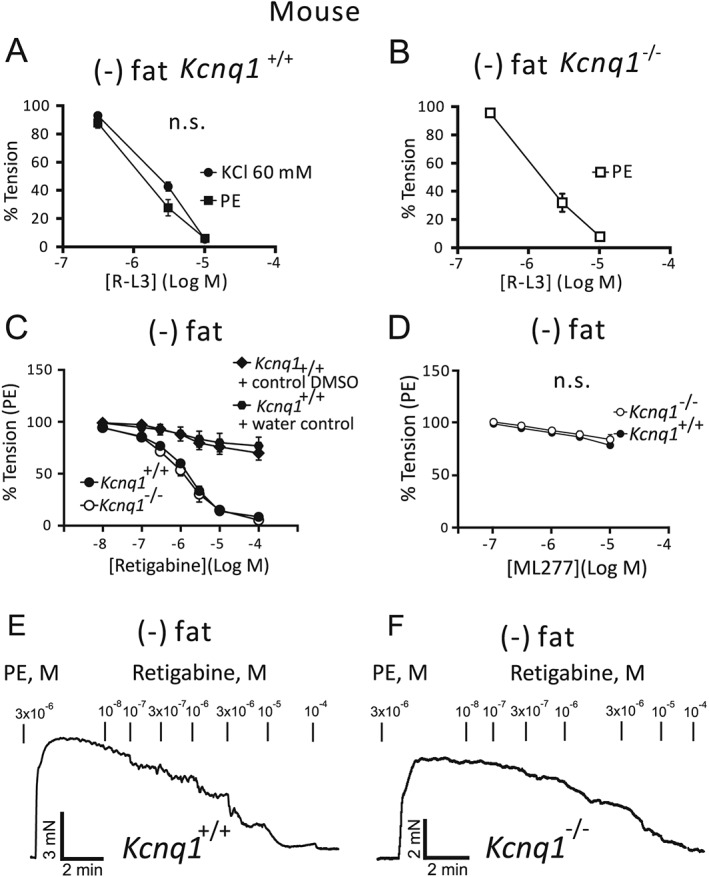
Relaxation of (−) fat mesenteric artery rings by 0.3‐3 μM R‐L3, 0.1‐10 μM ML 277 or 0.01‐10 μM retigabine. Vessels were isolated from wild‐type (Kcnq1 +/+) mice (A) or Kcnq1 −/− mice (B). Mesenteric arteries were precontracted by phenylephrine (PE) 1 μM (A,B) or KCl 60 mM (A). Representative traces showing relaxation induced by retigabine in rings from Kcnq1 +/+ (E) and Kcnq1 −/− mice (F) and average values compared with vehicle (DMSO) and water controls (C). ML277 effects on rings from Kcnq1 −/− and Kcnq1 +/+ mice (D). Tension is expressed as a percentage of KCl or PE‐induced contractions. n = 5 per group.
Figure 3.
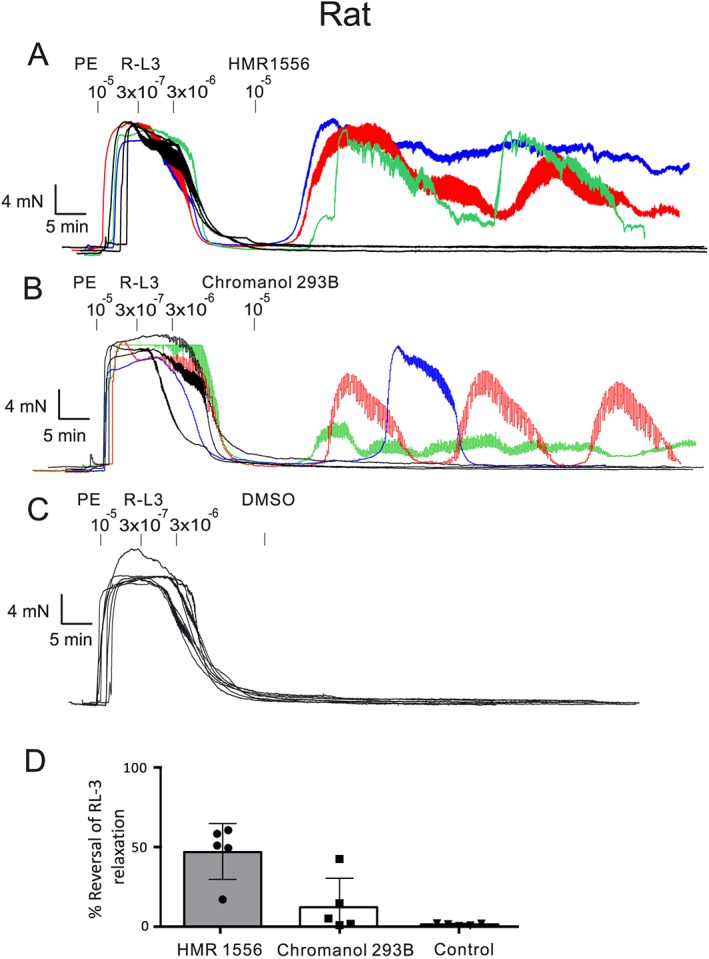
R‐L3‐induced relaxation of (−) fat mesenteric arteries from rats and subsequent exposure to KV7.1 channel blockers 10 μM HMR 1556 (A), 10 μM chromanol 293B (B), vehicle control DMSO (C). Percentage reversal of R‐L3‐induced relaxation by HMR 1556, chromanol 293B (D). The reversal was caused by inducing long‐lasting oscillatory contractions in ~50% of vessel rings. n = 5 per group.
Next, we aimed to determine whether the R‐L3 effects rely on K+ channel activation through experiments using 60 mM KCl‐induced contractions, which are largely determined by Ca2 + influx through L‐type Cav1.2 channels (Moosmang et al., 2005; Essin et al., 2007). Importantly, R‐L3 produced similar concentration‐dependent relaxation in mesenteric rings regardless of whether they were pre‐contracted with KCl or PE (Figures 1D and 2A). Similar results were also obtained in renal arteries (Figure S2). A total of 30 μM XE991 was unable to reverse R‐L3‐induced relaxations of rings pre‐contracted with KCl (Figure 1D). R‐L3 produced normal relaxations in arterial rings, from Kcnq1 −/− mice and the EC50 values were ~1.4 μM in rings from both Kcnq1 +/+ and Kcnq1 −/− mice (Figure 2A,B). The relaxant effects of R‐L3 were independent of the endothelium (Figure S3A) (see also Figure S3B for similar results in rat mesenteric arteries). Our data suggests that R‐L3, a supposed KV7.1 channel activator, did not induce relaxations of murine mesenteric arteries through activation of KV7.1 channels.
Effects of ML277, another activator of KV7.1 channels
In order to better understand the possible contribution of KV7.1 channels in vasoregulation, we studied the vasodilatory effects of ML277, a potent activator of KV7.1 channels EC50, (260 nM) (Mattmann et al., 2012). Studies suggest that it changes the conformational dynamics of the KV7.1 pore and/or global motions in the channel, including regions critical for KV7 gating transitions (Xu et al., 2015). In our experiments, ML277 induced relaxation of mesenteric arteries only at high concentrations, EC50 ~100 μM (Figure 2D, Figure S4). These effects were not different between arterial rings from wild‐type and Kcnq1 −/− mice and may have occurred due to activation of other KV7 channels (Mattmann et al., 2012).
Anti‐contractile effects of PVAT
The PVAT produces endothelium‐independent relaxation by opening VSMC KV channels, which can be blocked by the pan KV7 channel blocker XE991 and depends on the release of adipocyte‐derived relaxing factor (ADRF) (Gollasch, 2012; Tano et al., 2014). To test whether KV7.1 channels play a role in these effects, we performed a series of experiments using KV7.1 channel inhibitors and arteries from Kcnq1 +/+ and Kcnq1 −/− mice (Figure 4). Mesenteric arteries were prepared either (+) fat or (−) fat (with or without PVAT). Chromanol 293B (10 μM) did not diminish the anti‐contractile effects of PVAT (Figure 4A). Also, HMR1556 (10 μM) had no effect on phenylephrine‐induced contractions of both (−) fat rings and (+) fat rings isolated from Kcnq1 +/+ mice. Moreover, HMR1556 did not diminish the anti‐contractile effects of PVAT (Figure 4B, see also Figure S5A,B for data analysis by curve fitting using nonlinear regression). Similarly, the anti‐contractile effects of PVAT were normal in arteries isolated from Kcnq1 −/− mice (Figure 3C, see also Figure S5C,D for data analysis by curve fitting using non‐linear regression). Interestingly, the activator of KV7.2–5 channels, retigabine, induced normal concentration‐dependent relaxations of Kcnq1 −/− arteries (EC50 ~1 μM) (Figure 2C,E,F). Taken together, our data indicate that KV7.1 channels did not contribute to the anti‐contractile effects of PVAT in our arterial rings.
Figure 4.
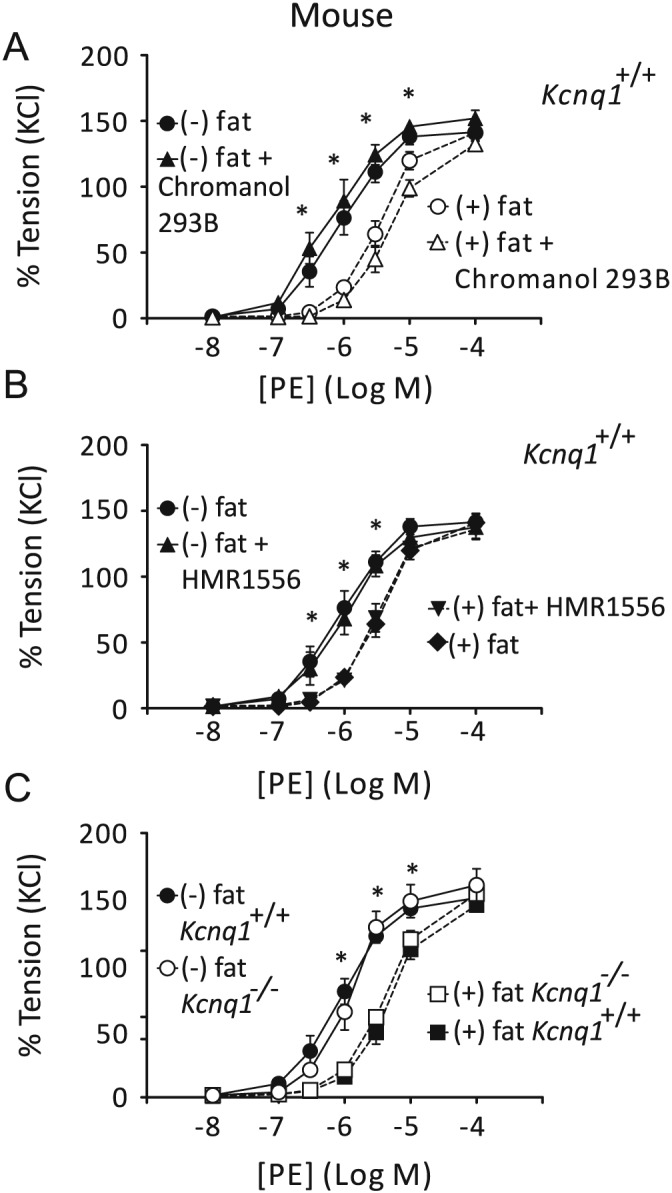
Effects of 10 μM chromanol 293B or 10 μM HMR1556 on dose–response relationships for phenylephrine (PE)‐induced contractions of arterial mesenteric rings with [(+) fat] (n = 5 per group) and without (−) PVAT [(−) fat] (n = 5 per group) (A,B). Dose–response relationships for PE‐induced contractions of (+) fat and (−) fat rings isolated from wild‐type (Kcnq1 +/+) [(+) fat; (−) fat, n = 7 per group] and Kcnq1 −/− mice [(+) fat; (−) fat, n = 7 per group] (C). Tension is expressed as a percentage of the steady‐state tension (100%) obtained with isotonic external 60 mM KCl. *P < 0.05, significant difference between (−) fat and (+) fat rings.
Electrophysiology
Consistent with the above data indicating that KV7.1 channels do not contribute to resting vascular tone, we found that outward voltage‐dependent KV currents were normal in VSMCs isolated from Kcnq1 −/− mesenteric arteries (Figure 5). There were no differences in normalized peak KV currents (Kcnq1 +/+, 11.3 ± 1.5 pA/pF; Kcnq1 −/−, 13.5 ± 1.8 pA/pF, panel B), cell capacities (Kcnq1 +/+, 23.5 ± 1.8 pF; Kcnq1 −/−, 23.3 ± 1.5 pF, panel C) and block of KV currents by XE991 (30 μM) (Kcnq1 +/+, 56.8 ± 12%; Kcnq1 −/−, 62.5 ± 9% panels D,E,F) between Kcnq1 +/+ and Kcnq1 −/− cells. Moreover, we did not observe statistically significant differences in steady state current, fast and slow time constants of current activation at +20 and +40 mV [steady state current +20 mV: Kcnq1 +/+: 13.4 ± 1.7 pA/pF (n = 5); Kcnq1 −/−: 15.9 ± 1.6 pA/pF (n = 5); +40 mV: Kcnq1 +/+ 23.5 ± 3.1 pA/pF (n = 5); Kcnq1 −/−: 24.7 ± 3.3 pA/pF (n = 5); slow time constant +20 mV: Kcnq1 +/+: 70.7 ± 19.3 ms (n = 5); Kcnq1 −/−: 138.9 ± 55.6 ms (n = 5); +40 mV: Kcnq1 +/+: 51.2 ± 25.1. ms (n = 5); Kcnq1 −/−: 49.5 ± 15.9 ms (n = 5); fast time constant +20 mV: Kcnq1 +/+: 45.1 ± 4.6 ms (n = 5); Kcnq1 −/−: 56.3 ± 13.0 ms (n = 5); +40 mV: Kcnq1 +/+: 23.3 ± 3.2 ms (n = 5); Kcnq1 −/−: 34.7 ± 5.2 ms (n = 5)].
Figure 5.
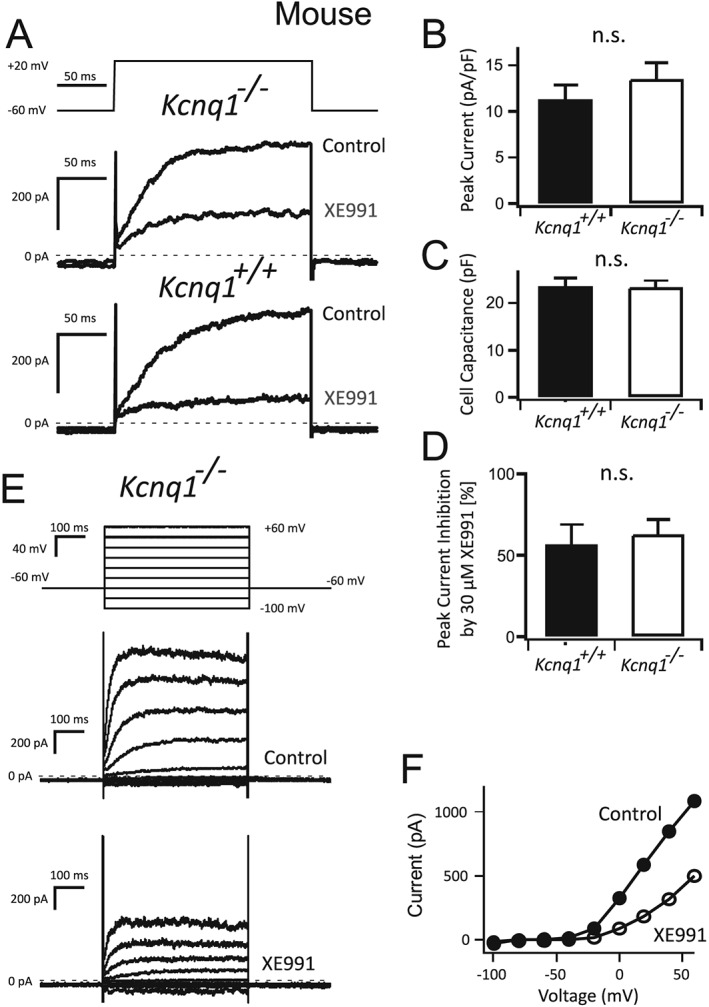
Voltage‐dependent potassium KV currents in freshly isolated Kcnq1 −/− VSMCs. Original recordings of Kv currents in VSMCs from Kcnq1 −/− and wild‐type (WT) (Kcnq1 +/+) mice (A). Normalized peak Kv currents (B), cell capacitance (C) and relative inhibition of KV currents by 30 μM XE991 (D). Original recordings of whole‐cell KV currents in a Kcnq1 −/− VSMC in the absence (Control) and presence of 30 μM XE991 (E). Voltage clamp protocol included command voltage steps (500 ms) ranging from −100 to +60 mV in 20 mV increments. Holding potential was −60 mV; test pulse frequency was 1·20 s−1. Current–voltage (I–V) relationship of the currents from (E) (F). n = 5 per group.
Discussion
Our findings provide compelling evidence that KV7.1 channels do not contribute to vascular contraction and to the anti‐contractile effects of PVAT in mouse mesenteric arteries. We observed no functional role for KV7.1 channels in R‐L3‐induced relaxations. R‐L3‐induced relaxations fully persisted after genomic deletion of Kcnq1 and after pharmacological blockade of KV7.1 channels by two compounds, namely HMR1556 or chromanol 293B. These conclusions were supported by findings obtained using another potent KV7.1 channel opener, ML277. Our experiments rule out a major role for anticipated downstream targets of ADRF signalling in mesenteric vessels, namely the Kcnq1 channel gene family. Instead, our genetic mouse model revealed an unappreciated role of R‐L3 in antagonizing high KCl‐induced L‐type CaV1.2 channel‐dependent vascular contractions. This relaxation occurred independent of the endothelium and opening of K+ channels, including KV7.1 channels. We thus conclude that R‐L3 is an inappropriate pharmacological tool for studying native vascular KV7.1 channels in mice.
KV7.1 channels and vascular tone
According to our experimental data obtained using chromanol 293B, HMR1556 and genomic deletion, the KV7.1 isoform is unlikely involved in the control of vascular tone of murine mesenteric arteries. Based on the effects of putative KV7.1 channel blockers (HMR1556 and L‐7) in reversing R‐L3‐induced relaxation, Chadha et al. proposed that KV7.1 channels can be functionally relevant in rat mesenteric arteries (Chadha et al., 2012). These results were reproduced in this study (Figure 3), as the relaxant effect of R‐L3 was reversed by the KV7.1 channel blockers chromanol 293B and HMR1556 in rings from rat mesenteric arteries. However, since the observation periods in our experiments were longer than in Chadha et al., we noticed that HMR1556 produced a sustained reversal of R‐L3‐induced relaxations only in one out of six vascular rings. Instead, HMR1556 and chromanol 293B produced rather long‐lasting oscillations. This phenomenon was observed only in ~50% of the vessels. In extension to the studies of Chadha et al., we explored whether blockade of KV7.1 channels (by HMR1556) could prevent R‐L3 relaxations. Our results demonstrate that KV7.1 channel blockade (chromanol 293B) failed to prevent R‐L3 relaxations (Figure S1), raising doubts that R‐L3 vasodilation in rat mesenteric arteries is indeed primarily caused by KV7.1 channel openings. Finally, the KV7.1 channel activator ML277 (EC50 260 nM in cardiomyocytes) even at 10 μM, failed to induce relaxation in murine mesenteric arteries. Together, the data support the notion that stimulation of KV7.1 channels cannot evoke profound relaxation in mesenteric arteries of mice.
Recent data suggest a major role of KV family of K+ channels as putative downstream targets of ADRF and possibly other relaxing factors released by PVAT (Schleifenbaum et al., 2010; Tano et al., 2014). This hypothesis is supported by the blockade of the anti‐contractile effects of PVAT by XE991 in rat aortas (Köhn et al., 2012). Similar to the aorta, smaller visceral and skeletal muscle arteries, which are important in the regulation of peripheral vascular resistance, rely on opening of XE991‐sensitive KV channels to mediate the paracrine effect of ADRF (Schleifenbaum et al., 2010). In this study, we found that both chromanol 293B and HMR1556 did not affect the anti‐contractile effects of PVAT. The anti‐contractile effects of PVAT were normal in Kcnq1 −/− vessels. We therefore conclude that KV7.1 channels are not the putative downstream K+ channel targets of ADRF that we seek. It is likely that the ADRF effects are mediated by opening of other Kv channels, for example KV7.2–5 (Figure 6).
Figure 6.
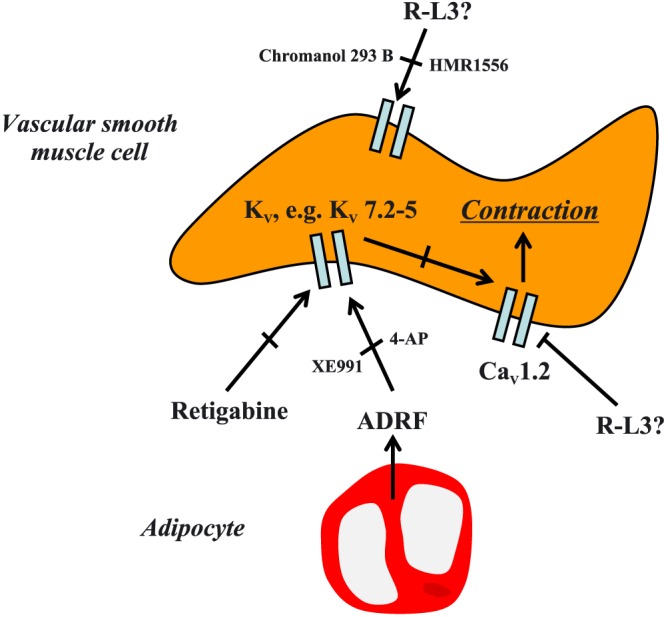
Schematic representation of voltage‐dependent potassium (KV) channels involved in vasoconstriction and ADRF‐dependent relaxation. K+ efflux through KV7.1 channels does not seem to be involved in either process and the compound R‐L3 is not specific for these channels in mice. L‐type Cav1.2 channels play a dominant role in depolarization‐induced contraction. The focus for future studies should be other KV channels and the use of selective pharmacological agents to identify the channels involved in this process. 4‐AP, 4‐aminopyridine.
A limitation of our study is that we cannot unequivocally exclude protein overexpression of other KV channels in Kcnq1 −/− arteries to functionally compensate for the loss of Kcnq1. However, we found that the pan KV7.2–5 channel activator retigabine (Yeung et al., 2007) produced normal relaxations in Kcnq1 −/− arteries. In addition, we also found similar expression of Kcnq3, Kcnq4 and Kcnq5 at the mRNA levels between Kcnq1 −/− and Kcnq1 +/+ mesenteric arteries (Figure S6). Kcnq2 mRNA is not detectable in this preparation, either in Kcnq1 +/+ (Schleifenbaum et al., 2014) or in Kcnq1 −/− (our data, not shown). These results argue against a significant up‐regulation of KV7.2–5 channels in these arteries. Furthermore, we found that KV currents and their block by XE991 were normal in Kcnq1 −/− VSMCs, which also argues against a contribution of functionally up‐regulated KV7.2–5 channels in the vessels of this mouse model. Moreover, another KV7.1 channel inhibitor (L‐735821) has been tested for inhibition of IKs current in the Kcnq1 −/− mice used in our study (Knollmann et al., 2004). Notably, L‐735821 inhibited IKs and prolonged action potential duration in Kcnq1 +/+ but not Kcnq1 −/− hearts. These data are also consistent with the idea that KV7.2–5 channels are not up‐regulated in mice deficient in KV7.1 channels.
Specificity of R‐L3
R‐L3 has been used to study the functional role of KV7.1 channels in various vascular beds and species (Seebohm et al., 2003; Chadha et al., 2012). The results indicate that KV7.1 channels may play a functional role in mesenteric (Chadha et al., 2012), but not coronary arteries of rats (Khanamiri et al., 2013). However, it is worth stressing that results with R‐L3 are difficult to interpret because the enantiomeric ratio of the drug can, at least in heart (Corici et al., 2013), affect activation kinetics of native KV7.1 channels. Furthermore, R‐L3 has been suggested to be a potent inhibitor of native vascular L‐type CaV1.2 channels in mouse aorta (Yeung et al., 2007), but not in rat arteries (Chadha et al., 2012). Our genetic model supports this idea for being particularly relevant in murine mesenteric arteries (Figure 6). Our conclusion is also based on similar cumulative response curves for R‐L3 in both Kcnq1 −/− and Kcnq1 +/+ arteries. Moreover, the relaxant effects of R‐L3 were inhibited neither by chromanol 293B, HMR1556, XE991 nor by 4‐AP. Furthermore, R‐L3 produced similar concentration‐dependent relaxation in mesenteric rings regardless of whether they were pre‐contracted with phenylephrine or KCl. Of note, tamoxifen‐induced smooth muscle‐specific inactivation of the L‐type CaV1.2 Ca2 + channel gene revealed a dominant role of these channels in KCl contractions (Moosmang et al., 2005; Essin et al., 2007). R‐L3 is thus not suitable for the study of native vascular KV7.1 channels in mice.
R‐L3 may represent a more appropriate tool to study KV7.1 channels in the rat vasculature (Figure 3). However, it is not clear why we observed R‐L3 effects, which were inhibited by HMR1556 or chromanol 293B only in 50% of the arteries. One possibility is that the enantiomeric ratio of the drugs used could play an important role. It is also well known that KCNE subunits influence the pharmacology of KV7.1 channels (Melman et al., 2002, 2004), which may explain drug, tissue and species differences in vivo. As R‐L3 enantiomers have opposing effects on cardiac IKs (KCNQ1) currents (Corici et al., 2013), KV7.x channel subunits can hetero‐oligomerize, and KCNE subunits can influence the pharmacology of KCNQ channels (Melman et al., 2002, 2004). Further studies should clarify the putative role of KCNE subunits/isoforms in enantiomeric enrichment of R‐L3 enabling the stimulatory effects of R‐L3 on native KV7.1 channels in vivo. New studies are needed to clarify a possible contribution of KV7.1 channels in producing oscillatory contractions in the rat vasculature and the specificity of KCNQ1 modulators in these effects.
Clinical relevance
One of the most dangerous potential side effects of a drug is its pro‐arrhythmic action due to cardiac QTc prolongation, in particular by KCNQ1 channel inhibition. Recently, arterial KCNQ1 (KV7.1) channels have attracted particular attention as putative novel drug targets for regulating arterial vascular tone and systemic blood pressure. Our genetic mouse model revealed novel insights into the specificity of pharmacological drugs commonly used to characterize KCNQ1 channel function in vivo. Our data indicate that KV7.1 channels are apparently not involved in the control of mesenteric and renal arterial tone in mice. Our studies highlight the importance of KCNQ1 channels in playing a critical role in cardiac arrhythmias, such as long QT syndrome, with negligible impact on KCNQ1 in the peripheral arteries, at least in mice.
In conclusion, our study demonstrated that KV7.1 channels were not required for the control of arterial tone by α1‐adrenoceptor agonists and PVAT in mesenteric and renal arteries of mice. Furthermore, R‐L3, recently described as a specific activator of KV7.1 channels, did not meet this criterion in our hands in the murine vasculature. R‐L3‐induced relaxations persisted after genomic deletion of Kcnq1 and following pharmacological blockade by two KV7.1 channel blockers, HMR1556 or chromanol 293B.
Author contributions
All authors planned and designed experimental studies. D.T. and C.L. performed the wire myography experiments. M.K. and J.Y.T. performed the electrophysiological experiments. D.T. and M.G. drafted the article, and all authors contributed to its completion.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Pretreatment of (−) fat murine (panel A) and (−) fat rat (panel B) mesenteric arteries with 10 μM chromanol 293B [(+) chromanol 293B] and subsequent exposure to R‐L3. (−) Chromanol 293B; control, non‐treated vessels. n.s., P > 0.05. n = 5 per group.
Figure S2 Relaxation of (−) fat murine renal artery rings by R‐L3. Vessels were isolated from wild‐type (Kcnq1 +/+) mice. Arteries were precontracted by 1 μM phenylephrine (PE) or 60 mM KCl. Tension is expressed as a percentage of KCl or PE induced contractions. *, P < 0.05. n = 5 per group.
Figure S3 Relaxation of endothelium‐denuded murine (panel A) and rat (panel B) mesenteric artery rings by R‐L3. (−) Endothelium: (−) fat, endothelium‐denuded vessels; (+) Endothelium: (−) fat, endothelium‐intact vessels. Arteries were precontracted by 1 μM phenylephrine (PE). Murine mesenteric arteries were isolated from Kcnq1 +/+ mice. Tension is expressed as a percentage of PE‐induced contractions. n.s., P > 0.05. n = 5 per group.
Figure S4 Original traces showing relaxation induced by the Kv7.1 channel opener ML277 in (−) fat murine mesenteric artery rings isolated from Kcnq1 +/+ (panel A) and Kcnq1 −/− mice (panel B) preconstricted with 1 μM phenylephrine.
Figure S5 Dose response curves and EC50 values for phenylephrine (PE)‐induced contractions of (−) fat and (+) fat murine mesenteric artery rings isolated from Kcnq1 +/+ and Kcnq1 −/− mice. Nonlinear regression model of dose–response curves to PE in the presence or absence of HMR1556 with or without fat (n = 5 per group) (panel A) and their corresponding EC50 values (panel B). Nonlinear regression model of dose–response curves to PE in Kcnq1 +/+ ((+) fat; (−) fat, n = 7 per group) and Kcnq1 −/− arteries ((+) fat; (−) fat, n = 7 per group) (panel C) and their corresponding EC50 values (panel D). *, P < 0.05.
Figure S6 Relative expression of Kcnq3–5 channels at RNA levels in (−) fat mesenteric arteries from Kcnq1 +/+ and Kcnq1 −/− mice normalized to 18 s. Relative mRNA levels for Kcnq3 (panel A) (n = 6 for Kcnq1 +/+; n = 5 for Kcnq1 −/−), Kcnq4 (panel B) (n = 6 for Kcnq1 +/+; n = 6 for Kcnq1−/−) and Kcnq5 expression (panel C) (n = 5 for Kcnq1 +/+; n = 5 for Kcnq1 −/−). n.s., P > 0.05, unpaired t‐test.
Figure S7 PCR genotyping of Kcnq1 +/+ and Kcnq1 −/− mice. Amplification by using the forward and reverse primers gives a 240‐bp product specific to the wild‐type (+/+) allele. Amplification by using the Neo forward and reverse primers gives a 370‐bp product specific to the null allele (−/−) (Casimiro et al., 2001).
Video S8 Demonstration of shaker‐waltzer phenotype (hyperactivity, head shaking, and/or circling), due to abnormality of the vestibular apparatus in Kcnq1 −/− mice. The video shows the typical behaviour of lack of KV7.1 channel function in the mouse.
Supporting info item
Supporting info item
Acknowledgements
This study was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to M.G. and the Deutsche Akademische Austauschdienst (DAAD) to M.G. and D.T. We thank Dr Iain A. Greenwood for providing HMR1556. D.T. is recipient of the ERA/EDTA Fellowship and J.Y.T. is an Alexander von Humboldt Fellow. We thank Atakan Aydin and Lajos Markó for help and advice in RT‐qPCR and Kornelia Buttke for expert technical assistance.
Tsvetkov, D. , Kaßmann, M. , Tano, J. ‐Y. , Chen, L. , Schleifenbaum, J. , Voelkl, J. , Lang, F. , Huang, Y. , and Gollasch, M. (2017) Do KV7.1 channels contribute to control of arterial vascular tone?. British Journal of Pharmacology, 174: 150–162. doi: 10.1111/bph.13665.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch RF, Schneck AC, Csillag S, Eigenberger B, Gerlach U, Brendel J et al. (2003). Effects of the chromanol HMR 1556 on potassium currents in atrial myocytes. Naunyn Schmiedebergs Arch Pharmacol 367: 281–288. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55: 611–622. [DOI] [PubMed] [Google Scholar]
- Bychkov R, Gollasch M, Steinke T, Ried C, Luft FC, Haller H (1998). Calcium‐activated potassium channels and nitrate‐induced vasodilation in human coronary arteries. J Pharmacol Exp Ther 285: 293–298. [PubMed] [Google Scholar]
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Greene AE, Franz MR et al. (2001). Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange‐Nielsen Syndrome. Proc Natl Acad Sci U S A 98: 2526–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Zunke F, Davis AJ, Jepps TA, Linders JTM, Schwake M et al. (2012). Pharmacological dissection of K(v)7.1 channels in systemic and pulmonary arteries. Br J Pharmacol 166: 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corici C, Kohajda Z, Kristóf A, Horváth A, Virág L, Szél T et al. (2013). L‐364373 (R‐L3) enantiomers have opposite modulating effects on IKs in mammalian ventricular myocytes. Can J Physiol Pharmacol 91: 586–592. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro MP, Aránega A, Franco D (2006). Protein distribution of Kcnq1, Kcnh2, and Kcne3 potassium channel subunits during mouse embryonic development. Anat Rec A Discov Mol Cell Evol Biol 288: 304–315. [DOI] [PubMed] [Google Scholar]
- Essin K, Welling A, Hofmann F, Luft FC, Gollasch M, Moosmang S (2007). Indirect coupling between Cav1.2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J Physiol 584: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fésüs G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC et al. (2007). Adiponectin is a novel humoral vasodilator. Cardiovasc Res 75: 719–727. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Zareba W, Moss AJ (2008). Long QT Syndrome. Curr Probl Cardiol 33: 629–694. [DOI] [PubMed] [Google Scholar]
- Gollasch M (2012). Vasodilator signals from perivascular adipose tissue. Br J Pharmacol 165: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollasch M, Ried C, Bychkov R, Luft FC, Haller H (1996). K+ currents in human coronary artery vascular smooth muscle cells. Circ Res 78: 676–688. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Wellman GC, Knot HJ, Jaggar JH, Damon DH, Bonev AD et al. (1998). Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circ Res 83p: 1104–1114. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Ohya S (2009). New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol 156: 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney AM, Joshi S, Manoury B (2010). KCNQ potassium channels: new targets for pulmonary vasodilator drugs? Adv Exp Med Biol 661: 405–417. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Olesen S‐P (2005). The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 20: 408–416. [DOI] [PubMed] [Google Scholar]
- Khanamiri S, Soltysinska E, Jepps TA, Bentzen BH, Chadha PS, Schmitt N et al. (2013). Contribution of Kv7 channels to basal coronary flow and active response to ischemia. Hypertension 62: 1090–1097. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann BC, Casimiro MC, Katchman AN, Sirenko SG, Schober T, Rong Q et al. (2004). Isoproterenol exacerbates a long QT phenotype in Kcnq1‐deficient neonatal mice: possible roles for human‐like Kcnq1 isoform 1 and slow delayed rectifier K+ current. J Pharmacol Exp Ther 310: 311–318. [DOI] [PubMed] [Google Scholar]
- Köhn C, Schleifenbaum J, Szijártó IA, Markó L, Dubrovska G, Huang Y et al. (2012). Differential effects of cystathionine‐γ‐lyase‐dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One 7: e41951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Bruhova I, Lerche H, Steinmeyer K, Wei AD, Strutz‐Seebohm N et al. (2007). Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol Pharmacol 71: 1503–1511. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang F, Wu Y, Huang X, Sheng L, Xu J et al. (2013). Meta‐analysis of the effect of KCNQ1 gene polymorphism on the risk of type 2 diabetes. Mol Biol Rep 40: 3557–3567. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Byron KL (2008). Cardiovascular KCNQ (Kv7) potassium channels: physiological regulators and new targets for therapeutic intervention. Mol Pharmacol 74: 1171–1179. [DOI] [PubMed] [Google Scholar]
- MacVinish LJ, Guo Y, Dixon AK, Murrell‐Lagnado RD, Cuthbert AW (2001). Xe991 reveals differences in K(+) channels regulating chloride secretion in murine airway and colonic epithelium. Mol Pharmacol 60: 753–760. [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA (2000). Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol 58: 253–262. [DOI] [PubMed] [Google Scholar]
- Mattmann ME, Yu H, Lin Z, Xu K, Huang X, Long S et al. (2012). Identification of (R)‐N‐(4‐(4‐methoxyphenyl)thiazol‐2‐yl)‐1‐tosylpiperidine‐2‐carboxamide, ML277, as a novel, potent and selective Kv7.1 (KCNQ1) potassium channel activator. Bioorg Med Chem Lett 22: 5936–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman YF, Krummerman A, McDonald TV (2002). KCNE regulation of KvLQT1 channels: structure–function correlates. Trends Cardiovasc Med 12: 182–187. [DOI] [PubMed] [Google Scholar]
- Melman YF, Um SY, Krumerman A, Kagan A, McDonald TV (2004). KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 42: 927–937. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Lenhardt P, Haider N, Hofmann F, Wegener JW (2005). Mouse models to study L‐type calcium channel function. Pharmacol Ther 106: 347–355. [DOI] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A et al. (2011). Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol 162: 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plüger S, Faulhaber J, Fürstenau M, Löhn M, Waldschütz R, Gollasch M et al. (2000). Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res 87: E53–E60. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Köhn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T et al. (2010). Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Kassmann M, Szijártó IA, Hercule HC, Tano J‐Y, Weinert S et al. (2014). Stretch‐activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 115: 263–272. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates m‐type currents. J Biol Chem 275: 24089–24095. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Lerche C, Pusch M, Steinmeyer K, Brüggemann A, Busch AE (2001). A kinetic study on the stereospecific inhibition of KCNQ1 and I(Ks) by the chromanol 293B. Br J Pharmacol 134: 1647–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Pusch M, Chen J, Sanguinetti MC (2003). Pharmacological activation of normal and arrhythmia‐associated mutant KCNQ1 potassium channels. Circ Res 93: 941–947. [DOI] [PubMed] [Google Scholar]
- Søgaard R, Ljungstrøm T, Pedersen KA, Olesen SP, Jensen BS (2001). KCNQ4 channels expressed in mammalian cells: functional characteristics and pharmacology. Am J Physiol Cell Physiol 280: C859–C866. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi T, Nishio H, Yagi T, Kuwahara M, Tsubone H, Tanigawa N et al. (2007). Phenotypic analysis of vertigo 2 Jackson mice with a Kcnq1 potassium channel mutation. Exp Anim 56: 295–300. [DOI] [PubMed] [Google Scholar]
- Tano J‐Y, Schleifenbaum J, Gollasch M (2014). Perivascular adipose tissue, potassium channels, and vascular dysfunction. Arterioscler Thromb Vasc Biol 34: 1827–1830. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ et al. (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17–23. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M‐channel. Science 282: 1890–1893. [DOI] [PubMed] [Google Scholar]
- Wang HS, Brown BS, McKinnon D, Cohen IS (2000). Molecular basis for differential sensitivity of KCNQ and I(Ks) channels to the cognitive enhancer XE991. Mol Pharmacol 57: 1218–1223. [PubMed] [Google Scholar]
- Wickenden AD, Zou A, Wagoner PK, Jegla T (2001). Characterization of KCNQ5/Q3 potassium channels expressed in mammalian cells. Br J Pharmacol 132: 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Zhang M, Jiang M, Rosenhouse‐Dantsker A, Wassenaar T et al. (2015). Probing binding sites and mechanisms of action of an I(Ks) activator by computations and experiments. Biophys J 108: 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Greenwood IA (2005). Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol 146: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Pucovský V, Moffatt JD, Saldanha L, Schwake M, Ohya S et al. (2007). Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol 151: 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Pretreatment of (−) fat murine (panel A) and (−) fat rat (panel B) mesenteric arteries with 10 μM chromanol 293B [(+) chromanol 293B] and subsequent exposure to R‐L3. (−) Chromanol 293B; control, non‐treated vessels. n.s., P > 0.05. n = 5 per group.
Figure S2 Relaxation of (−) fat murine renal artery rings by R‐L3. Vessels were isolated from wild‐type (Kcnq1 +/+) mice. Arteries were precontracted by 1 μM phenylephrine (PE) or 60 mM KCl. Tension is expressed as a percentage of KCl or PE induced contractions. *, P < 0.05. n = 5 per group.
Figure S3 Relaxation of endothelium‐denuded murine (panel A) and rat (panel B) mesenteric artery rings by R‐L3. (−) Endothelium: (−) fat, endothelium‐denuded vessels; (+) Endothelium: (−) fat, endothelium‐intact vessels. Arteries were precontracted by 1 μM phenylephrine (PE). Murine mesenteric arteries were isolated from Kcnq1 +/+ mice. Tension is expressed as a percentage of PE‐induced contractions. n.s., P > 0.05. n = 5 per group.
Figure S4 Original traces showing relaxation induced by the Kv7.1 channel opener ML277 in (−) fat murine mesenteric artery rings isolated from Kcnq1 +/+ (panel A) and Kcnq1 −/− mice (panel B) preconstricted with 1 μM phenylephrine.
Figure S5 Dose response curves and EC50 values for phenylephrine (PE)‐induced contractions of (−) fat and (+) fat murine mesenteric artery rings isolated from Kcnq1 +/+ and Kcnq1 −/− mice. Nonlinear regression model of dose–response curves to PE in the presence or absence of HMR1556 with or without fat (n = 5 per group) (panel A) and their corresponding EC50 values (panel B). Nonlinear regression model of dose–response curves to PE in Kcnq1 +/+ ((+) fat; (−) fat, n = 7 per group) and Kcnq1 −/− arteries ((+) fat; (−) fat, n = 7 per group) (panel C) and their corresponding EC50 values (panel D). *, P < 0.05.
Figure S6 Relative expression of Kcnq3–5 channels at RNA levels in (−) fat mesenteric arteries from Kcnq1 +/+ and Kcnq1 −/− mice normalized to 18 s. Relative mRNA levels for Kcnq3 (panel A) (n = 6 for Kcnq1 +/+; n = 5 for Kcnq1 −/−), Kcnq4 (panel B) (n = 6 for Kcnq1 +/+; n = 6 for Kcnq1−/−) and Kcnq5 expression (panel C) (n = 5 for Kcnq1 +/+; n = 5 for Kcnq1 −/−). n.s., P > 0.05, unpaired t‐test.
Figure S7 PCR genotyping of Kcnq1 +/+ and Kcnq1 −/− mice. Amplification by using the forward and reverse primers gives a 240‐bp product specific to the wild‐type (+/+) allele. Amplification by using the Neo forward and reverse primers gives a 370‐bp product specific to the null allele (−/−) (Casimiro et al., 2001).
Video S8 Demonstration of shaker‐waltzer phenotype (hyperactivity, head shaking, and/or circling), due to abnormality of the vestibular apparatus in Kcnq1 −/− mice. The video shows the typical behaviour of lack of KV7.1 channel function in the mouse.
Supporting info item
Supporting info item