

Abstract
Background and Purpose
Therapeutic options for treating glomerulopathies, the main cause of chronic kidney disease, are limited. Podocyte dedifferentiation is a major event in the pathogenesis of glomerulopathies. The goal of the present study was, therefore, to develop an assay to monitor podocyte differentiation suitable for compound screening.
Experimental Approach
We isolated and cultured glomeruli from transgenic mice, expressing cyan fluorescent protein (CFP) under the control of the promoter of nephrin, a marker of podocyte differentiation. Mean CFP fluorescence intensity per glomerulus (MFG) was determined by summation of all glomerular voxels from confocal z‐stacks in the absence and presence of pharmaceutical compounds.
Key Results
In untreated cultured glomeruli, MFG remained fairly stable during the first 5 days, when foot processes were already effaced, and the level of many podocyte‐specific proteins was only mildly affected, as revealed by proteomics. Between day 6 and 9, MFG decreased to almost zero. The decrease in MFG was paralleled by a decrease in CFP and nephrin expression, as determined by RT‐PCR, western blots and proteomics. Puromycin aminonucleoside (PAN), which damages podocytes, concentration‐dependently induced a complete loss of MFG. Dexamethasone (25 μM) and pioglitazone (10 μM) markedly attenuated the effect of 0.6 μg·mL−1 PAN on MFG.
Conclusion and Implications
In summary, we established a novel assay to assess the effect of pharmaceutical compounds on the differentiation of podocytes in situ. Our assay is suitable for compound screening to identify drugs for the treatment of glomerulopathies.
Abbreviations
- CFP
cyan fluorescent protein
- CKD
chronic kidney disease
- ESRD
end stage renal disease
- GBM
glomerular basement membrane
- MFG
mean CFP fluorescence intensity per glomerulus
- PAN
puromycin aminonucleoside
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptors a | Enzymes b |
| Itgb1 | Collagenase A |
| LIGANDS |
|---|
| Dexamethasone |
| Pioglitazone |
| Rac1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
In the past few years the number of patients suffering from chronic kidney disease (CKD) has increased worldwide and continues to rise (Couser et al., 2011). CKD is mainly caused by glomerulopathies, which are associated with a disturbance of the glomerular filtration barrier, and which eventually progress to end stage renal disease (ESRD). The glomerular filtration barrier consists of three parts that are essential for proper blood filtration: endothelial cells, the glomerular basement membrane (GBM) and podocytes. Podocytes are highly specialized postmitotic cells, which are attached to the GBM with their interdigitating foot processes (Endlich et al., 2001). The slit diaphragm is spanned between the interdigitating foot processes by the homophilic interaction of the transmembrane protein nephrin (Holzman et al., 1999; Ruotsalainen et al., 1999). This complex morphology depends on the expression of podocyte‐specific proteins like nephrin, podocin, actin‐associated proteins and others. Insufficient expression of proteins that are essential for podocyte function results in a disruption of the filtration barrier associated with proteinuria, the clinical hallmark of kidney disease, like minimal change nephropathy, focal segmental glomerulosclerosis, diabetic nephropathy and immunoglobulin A nephropathy (Doublier et al., 2001; Cooper et al., 2002; Kwoh et al., 2006). Since there are no specific drugs available to treat CKD, and since dialysis or kidney transplantation are the only therapies for the patients with ESRD to survive, it is of great interest to find new pharmacological compounds that could delay or even halt the progression of CKD. It was estimated that about two‐thirds of CKD are due to podocytopathies (Wiggins, 2007). Therefore, the search for novel drugs to treat CKD should focus on the podocyte.
Yamauchi et al. established the first assay to screen for compounds that act on podocytes (Yamauchi et al., 2006). For this purpose, they generated reporter cell lines derived from murine immortalized podocytes that were stably transfected with a gene encoding secreted alkaline phosphatase under the control of the nephrin promoter. By evaluation of the alkaline phosphatase activity, several endogenous substances were identified as regulators of nephrin expression. Another screening method was developed by Lee et al. (Lee et al., 2015). They studied the effect of small molecules on immortalized murine podocytes by phenotypical properties (e.g. cell morphology, cytoskeletal organization and focal adhesions). In this context, they discovered pyrintegrin as a novel podocyte‐protective agent. Since podocytes in cell culture change their morphological properties (sparse ‘foot processes’ if any, no slit diaphragms, no major processes) and functional properties (very low expression levels of the slit diaphragm proteins nephrin and podocin), it is worthwhile to study the effect of chemical compounds on fully differentiated podocytes in situ. Furthermore, in situ conditions preserve the crosstalk between the different glomerular cell types, such as podocytes, endothelial and mesangial cells.
Here, we describe a novel assay on cultured, isolated mouse glomeruli that enables screening of the effects of chemical compounds on the differentiation of podocytes in situ. The state of podocyte differentiation is measured non‐destructively over time through the fluorescence intensity of cyan fluorescent protein (CFP) that is expressed under the control of a nephrin promoter fragment.
Methods
Transgenic mice
We utilized transgenic nephrin:CFP‐mice that express CFP under control of a nephrin promoter fragment specifically in podocytes. This mouse strain was generated by subcloning the CFP cDNA into the EcoR1 site of the NPXRS nephrin construct (Wong et al., 2000; Cui et al., 2005). Mice were on an Institute of Cancer Research (ICR) background and were bred in separate colonies. Animals were housed in a room with controlled temperature (21°C) and humidity (60%), were exposed to a 12:12 h light–dark cycle and were given free access to water and standard chow. For the experiments, 100 mice at the age of 6 months were used. All procedures on mice were performed in accordance with national animal protection guidelines that conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local governmental authorities. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Glomeruli isolation
The different glomeruli isolations were done as described previously and will be explained in the following briefly.
- Sieving method
Kidney cortices were minced, and sieving steps were performed with filters of different mesh sizes (250, 150 and 70 μm) as described previously by Schiwek et al. (Schiwek et al., 2004).
- Collagenase method
Kidney cortices were minced, enzymatically digested (1 mg·mL−1 collagenase A for 45 min at 37°C, Roche Diagnostics Deutschland GmbH, Mannheim, Germany) and pushed through a 100 μm cell strainer (BD Biosciences, San Jose, CA, USA). The suspension was rinsed again through a 100 μm strainer, and glomeruli were collected with a sieve (70 μm; BD Bioscience). After the transfer into a falcon and centrifugation (10 min, 560 × g), the glomeruli were resuspended and cultured [related to (Helwig et al., 1974)].
- Dynabeads method
Glomeruli isolation with magnetic Dynabeads was performed as described by Takemoto et al. (Takemoto et al., 2002) with slight modifications. Mice were anaesthetized using ketamine/xylazine (both Selectavet Dr Otto Fischer GmbH, Weyarn‐Holzolling, Germany) and perfused through the left ventricle with Dynabeads M‐450 Epoxy (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, kidneys were minced, digested with collagenase A (1 mg·mL−1, 45 min at 37°C) and gently pressed through a 100 μm strainer. After centrifugation (230 × g, 7 min), the cell pellet was resuspended and diluted in PBS (Biochrom GmbH, Berlin, Germany) and finally collected by a magnetic particle concentrator.
Culturing glomeruli and live cell microscopy
After isolation, glomeruli were cultured on collagen IV‐coated μ‐plates (ibidi GmbH, Munich, Germany) in phenol red‐free RPMI‐1640 medium (Lonza Group Ltd., Basel, Switzerland) supplemented with 10% FBS (Invitrogen), 100 U·mL−1 penicillin and 0.1 mg·mL−1 streptomycin (Life Technologies, Thermo Fisher Scientific) at 37°C and 5% CO2. Glomeruli were visualized repeatedly over time by live cell imaging using a Leica TCS SP5 confocal laser‐scanning microscope (CLSM, Leica Microsystems, Wetzlar, Germany). Three‐dimensional animations were generated using Volocity 6.3 (Perkin Elmer, Waltham, MA, USA).
To examine the cellular viability of isolated glomeruli, the propidium iodide (PI) assay was performed; PI (Sigma‐Aldrich, Steinheim, Germany) was diluted in phenol red‐free RPMI medium to a final concentration of 3 μM. The intercalation of PI into the DNA of dead cells was imaged by CLSM.
Pharmacological treatment of glomeruli
Isolated glomeruli were treated with the following substances and concentrations: puromycin aminonucleoside (PAN; 0.1–200 μg·mL−1), dexamethasone (25 μM) and pioglitazone (10 μM, all from Sigma Aldrich). Each preparation of isolated glomeruli received all treatments, rendering group assignments unnecessary. The IC50 was calculated by fitting the data to a sigmoidal dose–response regression curve using Prism 5.01 (GraphPad Software, San Diego, USA).
Quantification of fluorescence intensity
To quantify fluorescence intensity, a z‐stack of isolated glomeruli was recorded by CLSM. In order to speed up the analysis, a programme was written in C++, which uses the OpenCV library. Glomeruli were identified by a combination of bright‐field and fluorescence images creating a binary image by Otsu's method. Then, edge detection was used to detect the outlines of the glomeruli, which were then masked by minimum bounding circles (MBC) and counted. A graphical user interface allowed manual corrections. Since we did not observe that the outgrowth of podocytes had any influence on the MFG, we included all glomeruli in our calculation. The single pictures of each z‐stack were summed by the programme, and the fluorescence intensity (FIGlom) as well as the area included by the MBC (AGlom) were measured. A glomeruli‐free region was selected to determine the mean background intensity (MBI). With this information, the corrected fluorescence intensity (FIcorr) was calculated using the following formula: FIcorr = FIGlom − (AGlom × MBI). FIcorr was related to the number of glomeruli determined before, and the mean CFP fluorescence intensity per glomerulus (MFG) was calculated. The procedure was repeated for three visual fields per well, which were selected randomly in the brightfield channel to exclude any bias for choosing specific fluorescence intensities. The MFGs of the three visual fields were averaged and related to an internal solvent control. All z‐stacks were generated using identical microscope settings. Blinding was achieved as automated image analysis for measuring MFG was performed by two independent scientists. MFG is proportional to the number of differentiated podocytes, depending on glomerular size (e.g. smaller cortical glomeruli vs. larger juxtamedullary glomeruli) and on the individual isolation. As a consequence, absolute changes in MFG in response to different conditions or treatments are not meaningful. Therefore, we normalized MFG to baseline values.
Immunocytochemistry
Cryosections of isolated glomeruli (thickness 20 μm) were fixed (2% PFA, 10 min) and blocked with blocking solution (PBS, 2% FBS, 2% bovine serum fraction V, 0.2% fish gelatin) for 1 h at room temperature (RT). Primary antibodies were incubated for 1 h at RT. The following polyclonal antibodies were used: rabbit anti‐WT‐1 (1:50, C‐19; Santa Cruz Biotechnology Inc., Dallas, TX, USA), guinea pig anti‐nephrin (1:100, GP‐N2; Progen Biotechnik GmbH, Heidelberg, Germany) and rabbit anti‐podocin (1:100, P0372; Sigma‐Aldrich). After being washed (3 × 5 min with PBS), cryosections were incubated with the following Cy3‐conjugated secondary antibodies for 1 h at RT: goat anti‐rabbit and donkey anti‐guinea pig (both from Jackson Immuno Research Laboratories Inc., West Grove, PA, USA). Nuclear staining was done using Hoechst 33342 (Sigma‐Aldrich). Finally, cryosections were embedded in mounting medium (40 mL PBS, 10 g Mowiol and 20 mL glycerol). Images were taken using the Leica TCS SP5.
Electron microscopy
For scanning electron microscopy (SEM), glomeruli were separated from the culture medium by filtration through a 0.2 μm pore size polycarbonate filter, which was then transferred into the fixation solution (2.5% glutaraldehyde in PBS) for 1 h at RT and 4°C overnight. Afterwards, glomeruli were postfixed in 1% osmium tetroxide in PBS for 60 min, dehydrated in a graded series of ethanol and critical point‐dried. Finally, samples were mounted on aluminium stubs, sputtered with gold/palladium and examined in an EVO LS10 scanning electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany).
For transmission electron microscopy (TEM), glomeruli were fixed in 2.5% glutaraldehyde in 0.1 M HEPES containing 0.1% MgCl2 and 0.05% CaCl2 for 1 h at RT and 4°C overnight. Subsequently, glomeruli were embedded in 2% low gelling agarose, postfixed in 2% osmium tetroxide for 2 h at 4°C and dehydrated through a graded ethanol series. Afterwards, the material was embedded in Epon (SERVA Electrophoresis GmbH, Heidelberg, Germany) and ultrathin sections were cut on an Ultracut UCT ultramicrotome (Leica Biosystems, Heidelberg, Germany). The sections were transferred onto a copper grid, contrasted with 5% uranyl acetate and lead citrate and analysed with a LIBRA 120 transmission electron microscope (Carl Zeiss Microscopy GmbH).
RT‐PCR
RNA was isolated with TRI reagent (Sigma‐Aldrich) according to the manufacturer's instructions, and reverse transcription of 1 μg denatured RNA was performed using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) following the corresponding protocol. Analysis of 1 μL cDNA was performed using the Phire Hot Start II DNA Polymerase (Thermo Fisher Scientific) and 500 nM specific sense and antisense primers. The following primers were purchased from Invitrogen: mouse nephrin (GenBank accession no. NM_019459.2), forward 5′‐GCC ACC ACC TTC ACA CTG AC‐3′, reverse 5′‐AGA CCA CCA ACC GCA AAG AG‐5′, 233 bp product size; CFP, forward 5′‐GGG CAC AAG CTG GAG TAC AA‐3′, reverse 5′‐ CTC AGG TAG TGG TTG TCG GG‐3′, 194 bp product size; mouse β‐Actin (GenBank accession no. NM_007393.5), forward 5′‐ GGC ACC ACA CCT TCT ACA ATG‐3′, reverse 5′‐GGA TGG CTA CGT ACA TGG C‐3′, 153 bp product size.
Generation of protein samples
Glomeruli were washed twice with PBS for 1 min, resuspended in 8 M urea /2 M thiourea and snap frozen in liquid nitrogen. Disruption was achieved by 3 cycles of rapid thawing at 30°C and subsequent freezing in liquid nitrogen, followed by sonication (3 × 3 s, 50% power) using a Sonoplus (Bandelin, Berlin, Germany) for nucleic acid fragmentation. The lysate was centrifuged (20 000 × g, 1 h at 4°C), and the protein‐containing supernatant was collected. The protein concentration was determined using a Pierce Bradford Assay kit (Thermo Fisher Scientific).
Western blots
Proteins (5 μg per lane) were separated using a Mini‐PROTEAN TGX Stain‐Free Precast Gel (Bio‐Rad Laboratories, München, Germany) and transferred to a nitrocellulose membrane. After a blocking step (5% skimmed milk powder in 0.05% TBST, 60 min), the membrane was incubated with primary antibody (1:10 000 guinea pig anti‐nephrin, GP‐N2, Progen; 1:20 000 mouse anti‐CFP, Living Colors JL‐8, Clontech Laboratories Inc., Mountain View, CA, USA; 1:4000 rabbit anti‐GAPDH, sc‐25 778, Santa Cruz Biotechnology Inc.), for detection of bound antibody, HRP‐coupled secondary antibodies (1:10 000 goat anti‐guinea pig, sc‐2438; goat anti‐mouse, sc‐2005; 1:17 500 goat anti‐rabbit, sc‐2030; all Santa Cruz Biotechnology Inc.) and the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) were used.
Proteomics analysis
Aliquots of 3 μg of total protein from each sample were reduced (2.5 mM DTT ultrapure for 1 h at 60°C, Invitrogen) and alkylated (10 mM iodoacetamide for 30 min at 37°C, Sigma‐Aldrich). Proteolysis was performed using LysC (1:100 for 3 h at 37°C) followed by tryptic digestion overnight at 37°C (both from Promega, Madison, WI, USA). The tryptic digestion was stopped by adding acetic acid at a final concentration of 1% followed by desalting using ZipTip‐μC18 tips (Merck Millipore, Darmstadt, Germany). Extracts were then concentrated by evaporation under vacuum and subsequently resolved in 0.1% acetic acid, 2% acetonitrile (ACN). Chromatographic separation of tryptic peptides was achieved on a reverse phase nano‐Acquity UPLC column (1.7, 100 μm i.d. × 100 mm, Waters GmbH, Eschborn, Germany) using a 90 min non‐linear gradient ranging from 2 to 60% ACN in 0.1% acetic acid at a flow rate of 0.3 μL·min−1.
The nano‐LC column was interfaced using electrospray ionization to an LTQ‐Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Precursor ions of m/z range 300–1500 (r = 30 000) were subjected to data dependent MS/MS fragmentation of top‐20 peaks in the ion trap at a collision‐induced energy of 35%. Repetitive MS/MS acquisition was avoided by setting dynamic exclusion of 60 s for already selected precursors.
The acquired MS/MS spectra were processed and searched using the Elucidator software (Ceiba Solutions, Boston, MA, USA). Data were searched against the UniProtKB/Swiss‐Prot mouse database version 2014_01 via the Sequest algorithm. The database search was performed using the following settings: trypsin as cleavage enzyme, no missed cleavages; oxidation on methionine was selected as a variable modification, and carbamidomethylation on cysteine residuals was selected as a static modification. Amino acid sequences identified at a false discovery rate <1% were annotated. Proteins with at least two unique peptides and a protein teller probability >0.9 were considered identified. For each sample, two technical replicates were analysed, and the results of three independent biological replicates were averaged.
Bioinformatics analysis
All proteins reaching the cut‐off criteria (fold change ≤0.5 and ≥1.5, q < 0.05) were used as input. The functional gene ontology analysis was performed utilizing Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources, and functional annotation charts as well as functional annotation clusters were created (Huang da et al., 2009). P‐values were determined by a modified Fisher's exact test with multiple test correction (Benjamini‐Hochberg). All charts/clusters with an enrichment score ≥ 1.5 and a P‐value <0.05 were considered for further evaluation.
Data and statistical analysis
All data are represented by mean ± SEM of biological replicates. Variance homogeneity was confirmed by Bartlett's test first. Then statistical significance was determined by parametric or non‐parametric one‐way ANOVA as appropriate. If F achieved a P‐value <0.05, post hoc tests were applied using the Bonferroni correction. For the proteome analysis, statistical analysis was performed on log10 transformed and central tendency median normalized data using Welch's t‐test followed by multiple test correction (Benjamini‐Hochberg). A P‐ or q‐value <0.05 was considered as statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
Isolation of intact glomeruli
Since three different methods for glomeruli isolation have been published (sieving, collagenase and Dynabeads method; Figure 1A), we analysed the quality of the isolated glomeruli obtained by these methods first. Utilization of nephrin::CFP mice, expressing CFP under a promoter fragment of the slit diaphragm protein nephrin (Cui et al., 2005), allowed direct visualization of the cyan fluorescent podocytes. As shown in Figure 1B, the fluorescence of the isolated glomeruli varied substantially depending on the isolation method.
Figure 1.
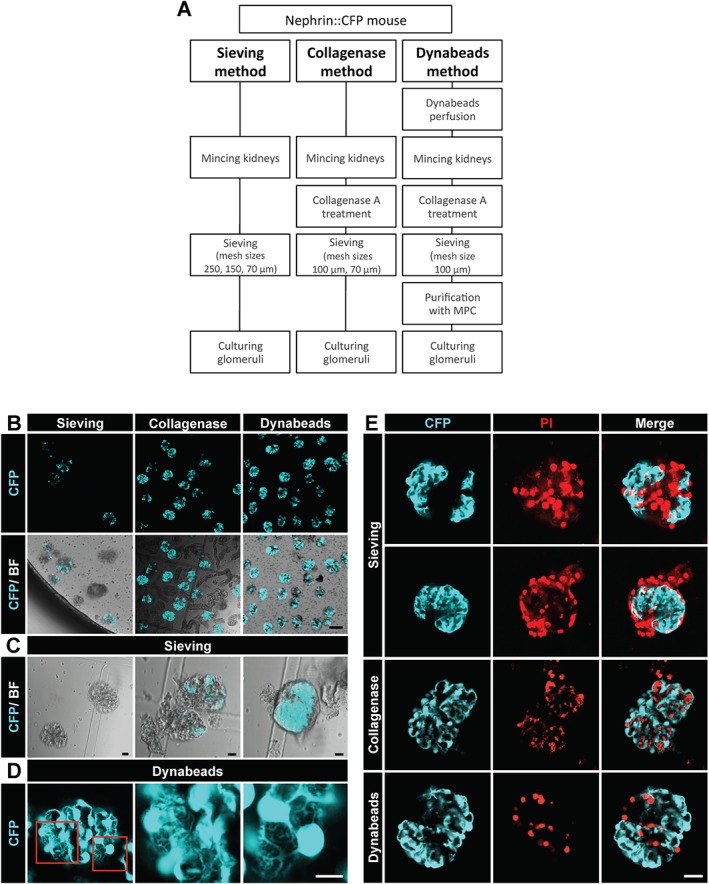
Methods for glomeruli isolation. (A) Schematic description of the three different methods for glomeruli isolation with their different steps to obtain glomeruli. MCP, magnetic particle concentrator. (B) CFP fluorescence alone and combined with bright‐field illumination (BF). Note the absence of CFP fluorescence in many glomeruli isolated by the sieving method and the tubular contamination using the collagenase method. (C) Glomeruli isolated by the sieving method were frequently decapsulated leading, probably, to mechanical damage of podocytes, which can be seen as a partial or complete loss of CFP fluorescence. The right image shows a capsulated glomerulus that has retained the CFP fluorescence in podocytes. (D) Isolation using the Dynabeads method yielded glomeruli with intact podocytes. The two enlargements of the boxed areas in the left image demonstrate the intact podocyte morphology (cf. Supp. Movie S1 and S2). (E) Isolated glomeruli were stained with the membrane impermeable DNA binding propidium iodide (PI). Areas lacking CFP fluorescence correspond to cells stained with PI. The Dynabeads method is associated with the least podocyte damage. Scale bars represent 100 μm (B), 10 μm (C–D) and 25 μm (E). Representative images of seven independent experiments are shown.
By the sieving method, mainly decapsulated glomeruli were isolated that had partly or completely lost their fluorescence (Figure 1B–C). Only capsulated glomeruli showed a strong fluorescence of podocytes (Figure 1C). Isolation by the use of collagenase resulted in a high amount of decapsulated glomeruli with fluorescent podocytes; however, it was associated with a significant contamination by tubule fragments (Figure 1B). The purest glomeruli isolation combined with a high amount of fluorescent podocytes was obtained by the use of Dynabeads (Figure 1B). As show in Figure 1D, podocytes of glomeruli isolated with Dynabeads retained their complex arborized morphology, further demonstrated in a 3D reconstruction from a z‐stack of a single glomerulus (Supp. Movies S1 and S2).
To assess the amount of cell damage by the different isolation methods, we stained the glomeruli with membrane impermeable, DNA binding PI. As it can be seen in Figure 1E, the sieving method resulted in damage of many podocytes and other glomerular cells. In contrast, little cell damage was observed in glomeruli isolated with the collagenase method and the Dynabeads method. Moreover, glomeruli isolated with the Dynabeads method showed an intensive staining for the podocyte‐specific proteins nephrin, podocin and WT‐1 (Figure 2). Since the Dynabeads method gave the best results, we used this method throughout the following experiments.
Figure 2.
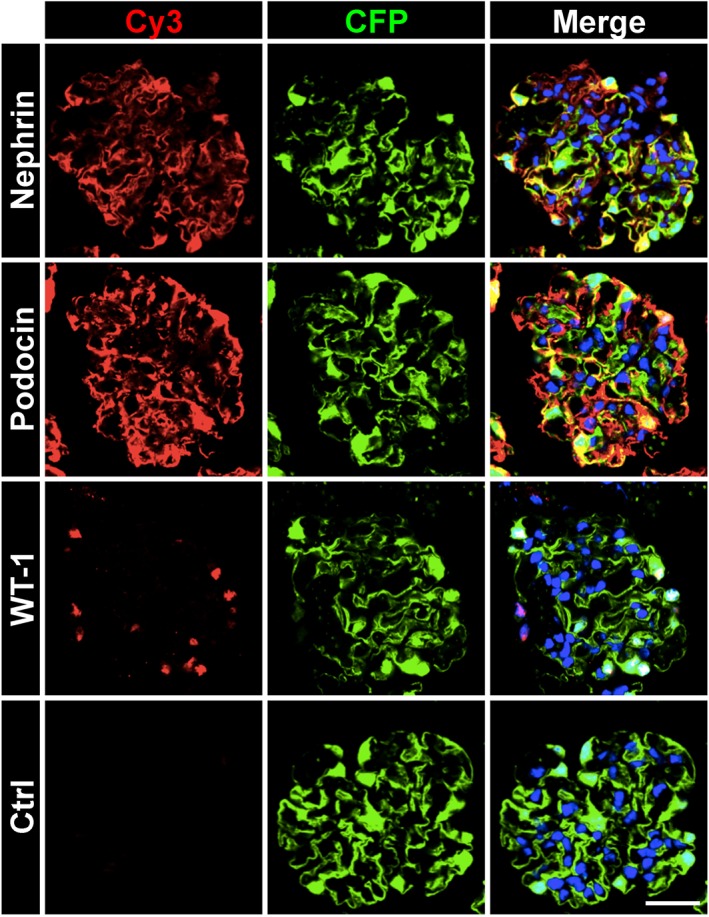
Localization of podocyte‐specific proteins in isolated glomeruli. Isolated glomeruli exhibit an intact staining pattern for the podocyte‐specific proteins nephrin, podocin and WT‐1. Freshly isolated glomeruli were stained for different podocyte markers using primary antibodies and Cy3‐conjugated secondary antibodies (red). CFP fluorescence is false coloured in green. Nuclei were stained with Hoechst 33 342 in blue. Scale bar represents 25 μm. Representative images of three independent experiments are shown.
Culturing glomeruli induces dedifferentiation of podocytes indicated by a decrease of CFP fluorescence
Since CFP is expressed under control of a nephrin promoter fragment, the CFP fluorescence indicates nephrin promoter activity and hence the state of podocyte differentiation in cultured glomeruli. CFP intensity of podocytes of cultured glomeruli remained rather stable during the first days and was progressively lost thereafter (Figure 3A). After 2 days in culture, we observed the first outgrowth of CFP expressing podocytes from single glomeruli (Supp. Figure S1). Podocyte outgrowth was observed for about 10% of the glomeruli. Interestingly, the outgrown podocytes exhibited a dramatic loss of CFP fluorescence and showed a heterogeneous morphology varying between arborized and cobblestone‐like phenotypes (Supp. Figure S1).
Figure 3.
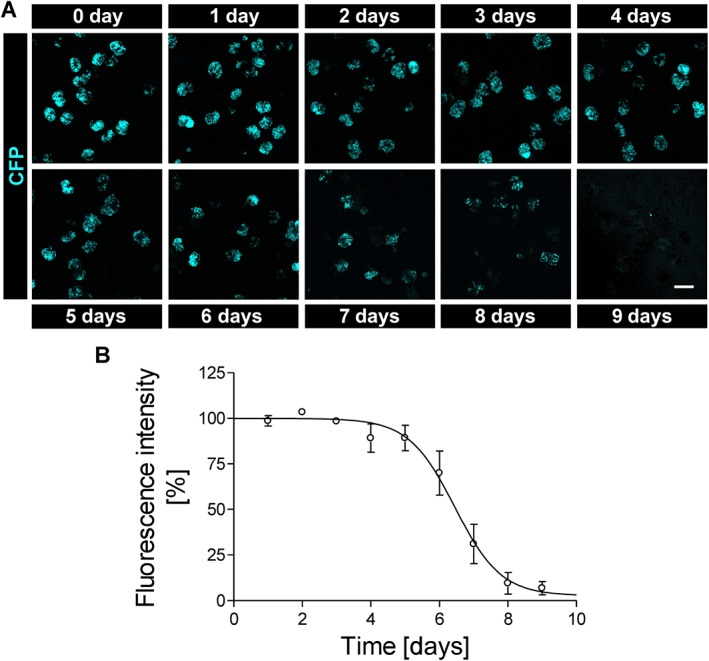
CFP fluorescence intensity of cultured glomeruli. (A) Representative images of cultured glomeruli after isolation (0 day) until day 9 in culture. Images are summed z‐stacks recorded by confocal laser‐scanning microscopy. Over time, the number of fluorescent glomeruli as well as the fluorescence intensity of individual glomeruli decreases, particularly after 6 days in culture. (B) CFP fluorescence was quantified and expressed as MFG. MFG remained fairly stable over the first 5 days and fell to almost zero after 9 days. Scale bar represents 100 μm. Data are means ± SEM of three independent experiments.
To quantify the time dependence of the CFP expression of glomerular podocytes, the MFG was measured every 24 h as an indicator of podocyte differentiation. MFG was calculated by summation of all glomerular voxels from z‐stacks recorded by confocal microscopy. MFG remained fairly constant for approximately 5 days in culture before it gradually decreased and practically vanished on day 9 (Figure 3B).
To confirm that CFP fluorescence indeed reflects nephrin expression, we analysed the expression of CFP and nephrin by RT‐PCR and Western Blot (Figure 4). Nephrin mRNA levels were strongly reduced in glomeruli after 9 days in culture, as it was the case for CFP mRNA. Nephrin and CFP expression decreased also on the protein level, albeit to a lesser extent as compared with the mRNA level.
Figure 4.
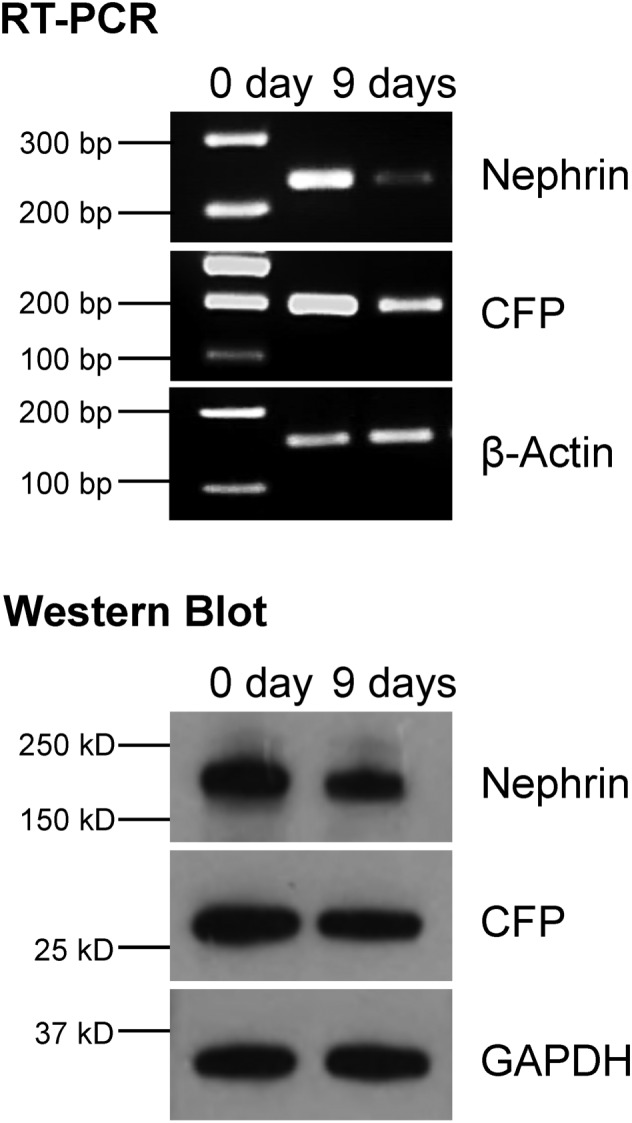
Expression of nephrin and CFP in cultured glomeruli. Nephrin and CFP expression were determined by RT‐PCR and western blots in freshly isolated glomeruli (0 day) and after 9 days in culture. Nephrin as well as CFP expression dropped at the mRNA and protein level. Representative images of three independent experiments are shown.
To find out whether the reduction of CFP and nephrin expression is associated with changes of podocyte morphology, we performed scanning electron microscopy (SEM) and TEM of glomeruli after 0, 4 and 9 days in culture. Foot processes of glomerular podocytes were effaced already after 4 days (Figure 5). In addition, we observed that the initially well‐preserved, fenestrated endothelium was nearly absent after 9 days in culture.
Figure 5.
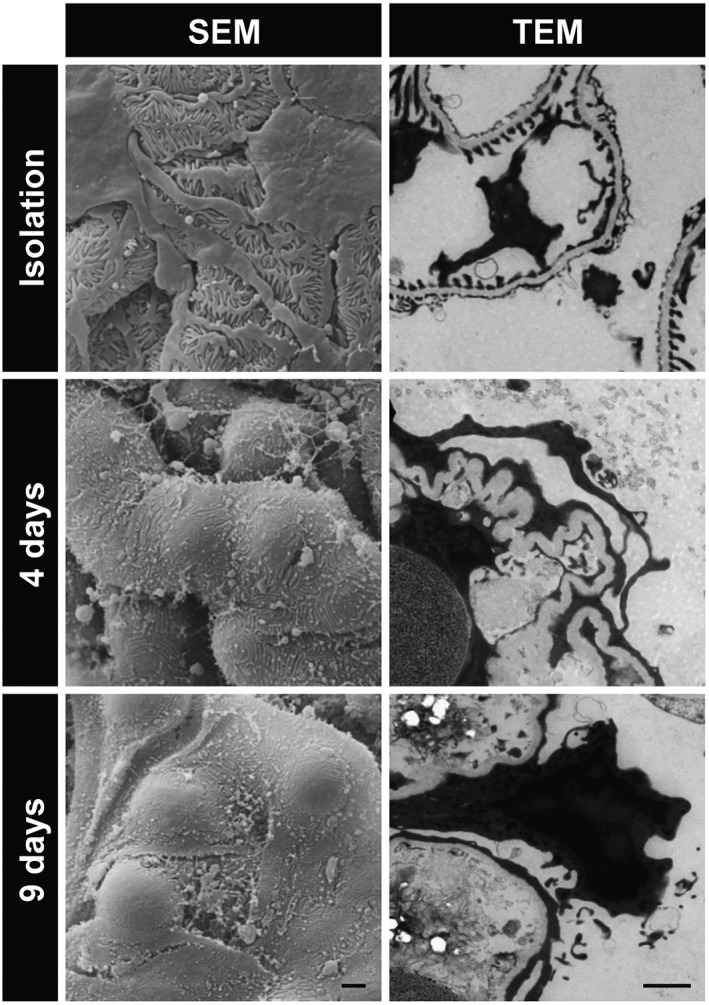
Ultrastructural analysis of cultured glomeruli. Scanning electron microscopy (SEM) and TEM were employed to analyse the ultrastructure of freshly isolated glomeruli and of glomeruli cultured for 4 and 9 days. Freshly isolated glomeruli exhibit interdigitating foot processes and fenestrated endothelium. In cultured glomeruli, foot processes of podocytes are effaced. Scale bars represent 2 μm. Representative images of three independent experiments are shown.
Proteomics analysis of cultured glomeruli at different time points
As quantified by the CFP fluorescence and as demonstrated by the expression and ultrastructural analysis above, podocytes of cultured glomeruli spontaneously dedifferentiate over time. To gain further insight into the process of glomerular dedifferentiation as well as to provide molecular details of the behaviour of our assay under control conditions, we collected glomeruli after 0, 3, 6 and 9 days in culture for protein isolation and subsequent proteomics analysis. Of the 2604 proteins identified in this study, between 6 and 14% were significantly regulated by at least 50% over time (Table 1).
Table 1.
Proteomics analysis of cultured glomeruli
| 3 days | 6 days | 9 days | |
|---|---|---|---|
| Significantly regulated proteins versus day 0 | 181 | 158 | 363 |
| Proteins mapped by DAVID | 173 | 153 | 351 |
| Functional annotation terms | 132 | 105 | 271 |
| Functional annotation clusters | 9 | 12 | 25 |
Proteomics were performed on cultured glomeruli (3, 6 and 9 days) and on freshly isolated glomeruli (day 0). Statistical significance and a change of at least 50% over time were defined as ‘significantly regulated’. DAVID was utilized to assign functional annotation terms (Supp. Table S1) to the significantly regulated proteins and to group these terms into functional annotation clusters (Supp. Table S2). Analysis was done on data obtained from three independent experiments.
After 9 days in culture, confirming the data obtained by Western blot analysis (Figure 4), CFP and nephrin (Nphs1) protein levels fell to 58 ± 10% and 44 ± 2% of control values (0 day), respectively, as determined by proteomics analysis. We further examined the regulation of several proteins (Figure 6) that are functionally relevant or specific for podocytes. Slit diaphragm proteins like nephrin (Nphs1) and podocin (Nphs2), which are exclusively expressed in podocytes, were most strongly down‐regulated over time. However, the expression of the slit diaphragm protein CD2AP remained unchanged. Except for Arhgap24, which strongly decreased, several cytoskeletal proteins were only mildly affected. Notably, the expression of the recently described podocyte‐specific actin cytoskeleton‐associated protein Schip1 (Perisic et al., 2015) increased by 76 ± 5% already after 3 days. The amount of podocyte‐specific matrix and cell matrix proteins remained stable or even slightly increased. Signalling proteins did not exhibit major changes, except for Rac1. The expression of Rac1, which has been implicated in foot process effacement (Yu et al., 2013), was up‐regulated throughout (Figure 6).
Figure 6.
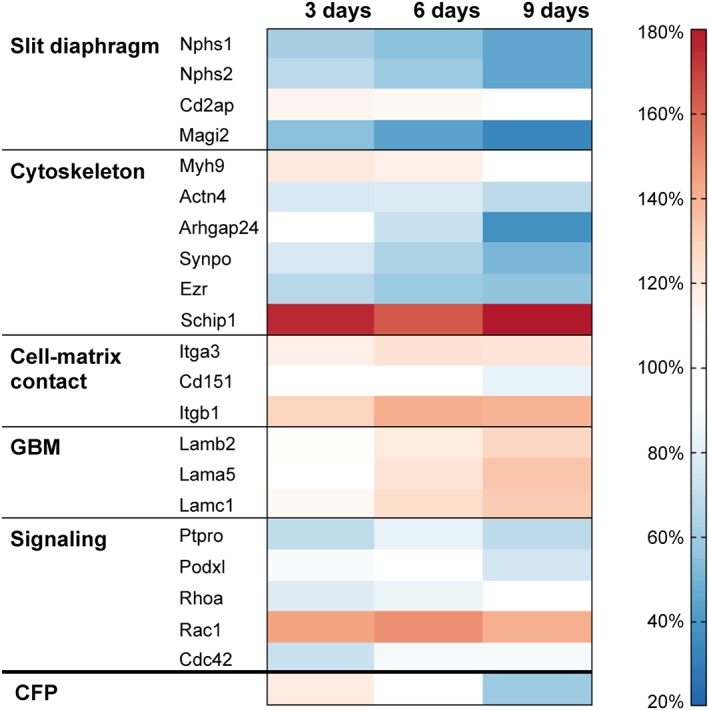
Proteomics analysis of cultured glomeruli. The expression levels of several proteins, which are functionally important for podocytes, are presented as a heat map of up‐ and down‐regulated proteins. Proteomics were performed on cultured glomeruli (3, 6 and 9 days) and on freshly isolated glomeruli. Protein levels of freshly isolated glomeruli were set to 100%. The strongest down‐regulation occurs for the slit diaphragm proteins nephrin (Nphs1), podocin (Nphs2) and Magi2, and for the actin‐associated protein Arhgap24. While a few proteins (Itgb1, Rhoa, Rac1 and Cdc42) are expressed in every glomerular cell type, the expression of all other shown proteins is podocyte‐specific. Data are means of three independent experiments.
For the further evaluation of proteomics data, we performed gene ontology analysis using the bioinformatics resource DAVID (Huang da et al., 2009). We created functional annotation charts to figure out the most overrepresented, statistically significant (fold enrichment ≥1.5, q < 0.05) biological terms at each time point (Supp. Table S1). These biological terms were further condensed into functionally related groups by functional annotation clustering (Supp. Table S2). At all times, annotation clusters associated with ‘ribosomes’, ‘tRNA’ and ‘metabolic cofactors’ achieved high enrichment scores (≥1.5). Furthermore, the ‘LIM domain’ related cluster was augmented after 6 and 9 days including proteins like Wtip, Fhl2 and Hic‐5. After 9 days, the annotation clusters ‘actin cytoskeleton’, ‘cell junctions’, ‘cell adhesion’ and ‘extracellular/basement membrane’ were identified.
Thus, the proteome data corroborated our choice to monitor the state of podocyte differentiation by utilizing nephrin promoter‐driven CFP expression of nephrin::CFP mice, as nephrin was one of the most strongly down‐regulated proteins. Consistent with the CFP fluorescence, massive reduction of slit diaphragm proteins occurred after 6 days in culture. Therefore, the CFP fluorescence of cultured glomeruli may serve as a reliable readout of podocyte differentiation for compound screening.
CFP fluorescence is a readout for podocyte damaging and protecting substances
To examine whether the CFP fluorescence of cultured glomeruli is indeed suited for compound screening, we measured CFP fluorescence in response to well‐characterized substances. Glomeruli were cultured in 15‐well plates containing about 150–200 glomeruli per well, and all substances were added to the medium right from the beginning. First, we applied PAN, an experimentally established inducer of podocyte effacement in rats and mice. As measured on day 6, PAN decreased MFG concentration‐dependently with an IC50 value of 0.31 μg·mL−1 (Figure 7A). Dexamethasone (25 μM), a member of the family of corticosteroids, and pioglitazone (10 μM), a PPAR‐γ activator used to regulate serum glucose levels in patients with diabetes mellitus, attenuated the effect of 0.6 μg·mL−1 PAN (Figure 7B). MFG on day 6 was 45 ± 14% and 70 ± 14% higher in the presence of dexamethasone and pioglitazone, respectively, as compared with the MFG of PAN treatment alone. When dexamethasone and pioglitazone were applied together, they acted additively in protecting against PAN‐induced podocyte dedifferentiation (Figure 7B). In the absence of PAN, neither dexamethasone nor pioglitazone enhanced the CFP expression (Figure 7C).
Figure 7.
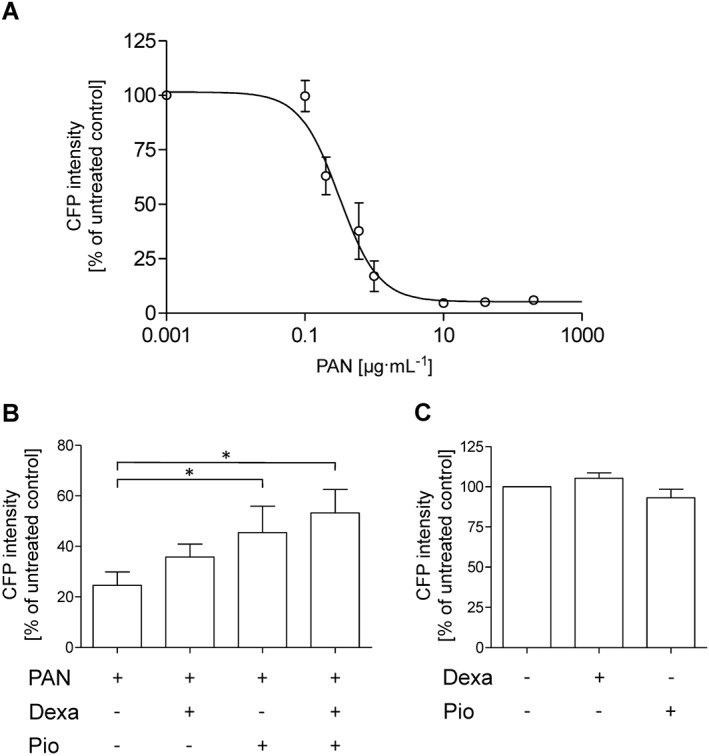
CFP fluorescence intensity in response to podocyte‐affecting substances. (A) Freshly isolated glomeruli were incubated with different concentrations of PAN, which is known to injure podocytes. After 6 days, MFG was measured. PAN concentration‐dependently decreased MFG to almost zero. (B) The decrease in MFG by 0.6 μg·mL‑1 PAN was attenuated by 25 μM dexamethasone (Dexa) or 10 μM pioglitazone (Pio) alone or in combination. (C) In the absence of PAN, Dexa (25 μM) or Pio (10 μM) did not affect MFG. MFG of untreated glomeruli was set to 100%. Data are means ± SEM of five independent experiments. *P < 0.05.
Discussion
Since dedifferentiation of podocytes plays a key role in the development of different types of kidney diseases like diabetic nephropathy, focal segmental glomerulosclerosis and other glomerulopathies, much effort has been applied to identify pathways and chemical compounds that could halt or reverse the dedifferentiation of this highly specific and postmitotic cell. In the past, immortalized podocyte cell lines were utilized to screen for drugs exerting differentiating or protective effects on podocytes (Yamauchi et al., 2006; Saito et al., 2010; Lee et al., 2015). However, podocytes, like many other cell types, change their mRNA and protein expression pattern in culture (Warsow et al., 2013). Therefore, the use of permanent podocyte cell lines to identify drugs to treat CKD is of limited use. Moreover, podocyte differentiation is affected by the crosstalk between glomerular endothelial cell, mesangial cells and podocytes. These limitations of podocyte cell lines can be overcome by studying podocytes in glomeruli in situ. For this purpose, we have developed a quick and easy to handle assay to assess the effect of chemical compounds on podocyte differentiation. This assay is also suitable for high‐throughput screening, for example in the search for drugs to treat CKD. In this study, we utilized one mouse for about 50 wells containing 150–200 glomeruli. Further optimization and miniaturization might allow to perform measurements on about 10 glomeruli per well. This implies that roughly 1000 measurements could be done with one animal.
The hallmark of podocyte differentiation is the expression of slit membrane proteins, in particular that of nephrin. It has been shown many times that glomerulopathies are associated with a decrease of nephrin expression (Kwoh et al., 2006; Patrakka and Tryggvason, 2007). Therefore, we hypothesized that a fluorescent reporter coupled to the nephrin promoter would be a well‐suited readout to follow easily and non‐destructively changes of the differentiation state of podocytes in cultured glomeruli in situ. Using the nephrin::CFP mice (Cui et al., 2005), we could demonstrate that the spontaneous dedifferentiation of podocytes in cultured glomeruli can be quantified by measuring the decrease of CFP fluorescence. As demonstrated by RT‐PCR, by Western blot and proteomics analysis, the decrease of CFP fluorescence reflects the decrease in nephrin expression. Furthermore, the well‐known podocyte‐damaging agent PAN diminished CFP fluorescence in a concentration‐dependent manner.
To assess how much the CFP fluorescence depends on the preparation method, we compared three isolation methods that are frequently used for the generation of primary and permanent podocyte cell cultures as well as for gene arrays and proteomics (Misra, 1972; Helwig et al., 1974; Takemoto et al., 2002; Schiwek et al., 2004). As observed in the present study, the sieving that was used in many laboratories in the past caused marked podocyte damage. In agreement with our findings, apoptosis in about 80% of the podocytes in glomeruli isolated by sieving was described by Ishikawa and Kitamura (Ishikawa and Kitamura, 1998). Podocyte damage was decreased by the collagenase method, but the isolate was significantly contaminated by tubular fragments. By the modified Dynabeads method, we obtained a high amount of pure glomeruli with mostly intact podocytes. These findings suggest that mechanical stress imposed by sieving on the glomeruli irreversibly damages most of the podocytes probably due to their exposed position on the outer capillary side. That mechanical stress could induce apoptosis and not only necrosis was already reported for myocytes and podocytes as well (Cheng et al., 1995; Dessapt et al., 2009).
Surprisingly, the CFP fluorescence remained fairly stable over the first 5 days in culture. Likewise, the levels of many podocyte‐specific proteins showed rather mild changes over the first 3–6 days in culture. However, foot processes were amply effaced after 4 days in culture. Our ultrastructural findings are consistent with those of Nørgaard (Norgaard, 1978) and Andrews (Andrews, 1981), who observed foot process effacement in cultured glomeruli within the first days. Since foot process effacement is associated with the disappearance of slit diaphragms, our proteomics results suggest that, at early time points, the ultrastructural changes are rather mediated by signalling and posttranslational protein modifications than by altered protein levels.
Proteomics data revealed significant expression changes in cultured glomeruli over time. It has to be kept in mind that podocytes, mesangial cells as well as endothelial cells may contribute to changes in the glomerular proteome. However, expression changes can be attributed to one cell type, as long as the expression of cell‐specific proteins is considered. Among the podocyte‐specific proteins, slit diaphragm proteins (nephrin, podocin, Magi2) were the most strongly down‐regulated group of proteins after 9 days in culture. As the slit diaphragm is responsible for the retention of plasma proteins, the spontaneous dedifferentiation of podocytes in cultured glomeruli allows for screening of chemical compounds that may preserve the expression of slit diaphragm proteins and thus alleviate proteinuria.
One of the earliest and most up‐regulated podocyte proteins was Schip1, a protein specifically expressed in foot processes (Perisic et al., 2015). Knockdown of Schip1 in zebrafish induces foot process disorganization and proteinuria. In cell culture, it was shown that Schip1 is localized at lamellipodia together with F‐actin, Nherf2 and ezrin. Interestingly, it was found by several groups that Schip1 is a PDGF‐B response gene inducing migration after stimulation with PDGF‐B (Chen et al., 2004; Schmahl et al., 2007; Perisic et al., 2015). Rac1, another migration‐regulating protein, was up‐regulated in culture throughout. Recently, it was demonstrated that podocyte‐specific expression of constitutively active Rac1 in mice results in foot process effacement, formation of dynamic membrane protrusions, detachment and proteinuria (Yu et al., 2013; Brahler et al., 2016). Together with the late but strong down‐regulation of Arhgap24, an inactivator of Rac1 (Akilesh et al., 2011), up‐regulation of Schip1 and Rac1 may lead to an enhanced motile phenotype of podocytes.
To prove whether the glomeruli assay might be useful to identify drugs that exert beneficial effects on podocytes, we applied dexamethasone and pioglitazone together with PAN treatment. Dexamethasone and pioglitazone alone or in combination were able to significantly counteract the effect of PAN. Dexamethasone belongs to the glucocorticoids, which are routinely used to treat many forms of CKD, including nephrotic syndrome. However, the use of the PPAR‐γ agonist pioglitazone, which is prescribed for diabetes mellitus type 2, has only recently been suggested for the treatment of proteinuria. Our results on pioglitazone are in agreement with those of Zuo et al. and Agrawal et al., who demonstrated a protective effect of pioglitazone in PAN‐treated rats and PAN‐treated cultured podocytes respectively (Agrawal et al., 2011; Zuo et al., 2012). Excitingly, Agrawal et al. have very recently reported that glucocorticoids and pioglitazone act additively in protecting rats from PAN‐induced glomerular injury (Agrawal et al., 2016), providing in vivo evidence for our assay results. In the absence of PAN, neither dexamethasone nor pioglitazone had any effect on CFP intensity. We therefore hypothesize that the dedifferentiation pathways evoked by PAN partially differ from the pathways involved in spontaneous dedifferentiation.
In summary, we have established a novel assay to assess the effect of pharmaceutical compounds on the differentiation of podocytes in situ via measurement of a fluorescent reporter. The handling of the assay is easy, and the non‐destructive measurements can be quickly and repeatedly performed over several days. Our assay is suited for compound screening with a markedly reduced number of animals to identify drugs for treating CKD or to assess renal toxicity.
Author contributions
F.K., K.E. and N.E. designed the study; F.K., E.H., A.B. and R.S. performed the experiments; F.K., S.K., P.K., S.E.Q., J.v.d.B., G.F., U.V., K.E. and N.E. contributed to the acquisition and analysis of the data; F.K., E.H., A.B., K.E. and N.E. interpreted the data; and F.K., K.E. and N.E. drafted the manuscript. All authors critically revised and approved the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Movie S1 The movie shows a z‐stack of a freshly isolated glomerulus with CFP‐expressing podocytes recorded by an inverted confocal laser‐scanning microscope. Scanning starts at the top of the glomerulus and continues down to the bottom.
Movie S2 The movie shows an animated 3D reconstruction of a z‐stack of a freshly isolated glomerulus with CFP‐expressing podocytes.
Table S1 Analysis of proteome data using DAVID: Lists of functional annotation terms.
Table S2 Analysis of proteome data using DAVID: Lists of functional annotation clusters.
Figure S1 Podocyte outgrowth from isolated glomeruli. Outgrowth of podocytes was observed in about 10% of the glomeruli after 2 days or longer in culture. Images were taken by confocal microscopy at the substrate level. CFP fluorescence is shown alone and combined with bright‐field illumination (BF). Outgrown podocytes exhibited arborized (arrowhead) or cobblestone‐like morphologies (arrow). Note the dramatic loss of CFP fluorescence in many outgrown podocytes that are attached to the substratum. Scale bar represents 25 μm. Representative images of five independent experiments are shown.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
{kind=link}
Acknowledgements
We are grateful to Bianca Haracska for assistance in fluorescence quantification and to Vishnu Mukund Dhople for assistance in mass spectrometry. This study was supported by grants of the German Federal Ministry of Education and Research (BMBF) to G.F. (VIP project, grant 03V0396), to N.E. (E‐Rare project ‘Rare‐G’, grant 01GM1208B and project ‘STOP‐FSGS’, grant 01GM1518B) and to U.V., K.E. and N.E. within the framework of the GANI_MED research project (grant 03IS2061A). The German Patent and Trade Mark Office (DPMA) issued a patent (no. 10.2015.102.445) that is related to this study.
Kindt, F. , Hammer, E. , Kemnitz, S. , Blumenthal, A. , Klemm, P. , Schlüter, R. , Quaggin, S. E. , van den Brandt, J. , Fuellen, G. , Völker, U. , Endlich, K. , and Endlich, N. (2017) A novel assay to assess the effect of pharmaceutical compounds on the differentiation of podocytes. British Journal of Pharmacology, 174: 163–176. doi: 10.1111/bph.13667.
References
- Agrawal S, Guess AJ, Benndorf R, Smoyer WE (2011). Comparison of direct action of thiazolidinediones and glucocorticoids on renal podocytes: protection from injury and molecular effects. Mol Pharmacol 80: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Chanley MA, Westbrook D, Nie X, Kitao T, Guess AJ et al. (2016). Pioglitazone enhances the beneficial effects of glucocorticoids in experimental nephrotic syndrome. Sci Rep 6: 24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R et al. (2011). Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PM (1981). Investigations of cytoplasmic contractile and cytoskeletal elements in the kidney glomerulus. Kidney Int 20: 549–562. [DOI] [PubMed] [Google Scholar]
- Brahler S, Yu H, Suleiman H, Krishnan GM, Saunders BT, Kopp JB et al. (2016). Intravital and kidney slice imaging of podocyte membrane dynamics. J Am Soc Nephrol 27: 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Delrow J, Corrin PD, Frazier JP, Soriano P (2004). Identification and validation of PDGF transcriptional targets by microarray‐coupled gene‐trap mutagenesis. Nat Genet 36: 304–312. [DOI] [PubMed] [Google Scholar]
- Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH et al. (1995). Stretch‐induced programmed myocyte cell death. J Clin Invest 96: 2247–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ME, Mundel P, Boner G (2002). Role of nephrin in renal disease including diabetic nephropathy. Semin Nephrol 22: 393–398. [DOI] [PubMed] [Google Scholar]
- Couser WG, Remuzzi G, Mendis S, Tonelli M (2011). The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int 80: 1258–1270. [DOI] [PubMed] [Google Scholar]
- Cui S, Li C, Ema M, Weinstein J, Quaggin SE (2005). Rapid isolation of glomeruli coupled with gene expression profiling identifies downstream targets in Pod1 knockout mice. J Am Soc Nephrol 16: 3247–3255. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessapt C, Baradez MO, Hayward A, Dei Cas A, Thomas SM, Viberti G et al. (2009). Mechanical forces and TGFbeta1 reduce podocyte adhesion through alpha3beta1 integrin downregulation. Nephrol Dial Transplant 24: 2645–2655. [DOI] [PubMed] [Google Scholar]
- Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG et al. (2001). Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol 158: 1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endlich K, Kriz W, Witzgall R (2001). Update in podocyte biology. Curr Opin Nephrol Hypertens 10: 331–340. [DOI] [PubMed] [Google Scholar]
- Helwig JJ, Zachary D, Bollack C (1974). Isolation of glomeruli and tubular fragments from rabbit kidney. Urol Res 2: 55–59. [DOI] [PubMed] [Google Scholar]
- Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR (1999). Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int 56: 1481–1491. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Kitamura M (1998). Spontaneous apoptosis of podocytes in explanted glomeruli. Technical note. Kidney Int 54: 2008–2013. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwoh C, Shannon MB, Miner JH, Shaw A (2006). Pathogenesis of nonimmune glomerulopathies. Annu Rev Pathol 1: 349–374. [DOI] [PubMed] [Google Scholar]
- Lee HW, Khan SQ, Faridi MH, Wei C, Tardi NJ, Altintas MM et al. (2015). A podocyte‐based automated screening assay identifies protective small molecules. J Am Soc Nephrol 26: 2741–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra RP (1972). Isolation of glomeruli from mammalian kidneys by graded sieving. Am J Clin Pathol 58: 135–139. [DOI] [PubMed] [Google Scholar]
- Norgaard JO (1978). Retraction of epithelial foot processes during culture of isolated glomeruli. Lab Invest 38: 320–329. [PubMed] [Google Scholar]
- Patrakka J, Tryggvason K (2007). Nephrin – a unique structural and signaling protein of the kidney filter. Trends Mol Med 13: 396–403. [DOI] [PubMed] [Google Scholar]
- Perisic L, Rodriguez PQ, Hultenby K, Sun Y, Lal M, Betsholtz C et al. (2015). Schip1 is a novel podocyte foot process protein that mediates actin cytoskeleton rearrangements and forms a complex with Nherf2 and ezrin. PLoS One 10: e0122067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestila M, Jalanko H et al. (1999). Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A 96: 7962–7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Okamura M, Nakajima S, Hayakawa K, Huang T, Yao J et al. (2010). Suppression of nephrin expression by TNF‐alpha via interfering with the cAMP‐retinoic acid receptor pathway. Am J Physiol Renal Physiol 298: F1436–F1444. [DOI] [PubMed] [Google Scholar]
- Schiwek D, Endlich N, Holzman L, Holthofer H, Kriz W, Endlich K (2004). Stable expression of nephrin and localization to cell–cell contacts in novel murine podocyte cell lines. Kidney Int 66: 91–101. [DOI] [PubMed] [Google Scholar]
- Schmahl J, Raymond CS, Soriano P (2007). PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet 39: 52–60. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y et al. (2002). A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 161: 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warsow G, Endlich N, Schordan E, Schordan S, Chilukoti RK, Homuth G et al. (2013). PodNet, a protein–protein interaction network of the podocyte. Kidney Int 84: 104–115. [DOI] [PubMed] [Google Scholar]
- Wiggins RC (2007). The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214. [DOI] [PubMed] [Google Scholar]
- Wong MA, Cui S, Quaggin SE (2000). Identification and characterization of a glomerular‐specific promoter from the human nephrin gene. Am J Physiol Renal Physiol 279: F1027–F1032. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Takano Y, Kasai A, Hayakawa K, Hiramatsu N, Enomoto N et al. (2006). Screening and identification of substances that regulate nephrin gene expression using engineered reporter podocytes. Kidney Int 70: 892–900. [DOI] [PubMed] [Google Scholar]
- Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS et al. (2013). Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol 33: 4755–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Yang HC, Potthoff SA, Najafian B, Kon V, Ma LJ et al. (2012). Protective effects of PPARgamma agonist in acute nephrotic syndrome. Nephrol Dial Transplant 27: 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 The movie shows a z‐stack of a freshly isolated glomerulus with CFP‐expressing podocytes recorded by an inverted confocal laser‐scanning microscope. Scanning starts at the top of the glomerulus and continues down to the bottom.
Movie S2 The movie shows an animated 3D reconstruction of a z‐stack of a freshly isolated glomerulus with CFP‐expressing podocytes.
Table S1 Analysis of proteome data using DAVID: Lists of functional annotation terms.
Table S2 Analysis of proteome data using DAVID: Lists of functional annotation clusters.
Figure S1 Podocyte outgrowth from isolated glomeruli. Outgrowth of podocytes was observed in about 10% of the glomeruli after 2 days or longer in culture. Images were taken by confocal microscopy at the substrate level. CFP fluorescence is shown alone and combined with bright‐field illumination (BF). Outgrown podocytes exhibited arborized (arrowhead) or cobblestone‐like morphologies (arrow). Note the dramatic loss of CFP fluorescence in many outgrown podocytes that are attached to the substratum. Scale bar represents 25 μm. Representative images of five independent experiments are shown.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item