

Abstract
Background and Purpose
Propranolol is a vasoactive drug that shows antiangiogenic and antitumour activities in melanoma. However, it is unknown whether these activities are dose‐dependent and whether there is a relationship between systemic vascular effects of propranolol and anti‐melanoma activity.
Experimental Approach
Effects of increasing doses of propranolol (10, 20, 30 and 40 mg·kg−1·day−1) on tumour growth were studied in B16F10 melanoma‐bearing mice. Histological and biochemical analyses were used to assess propranolol effects on angiogenesis and cancer cell proliferation. Systemic vascular resistance (SVR) was evaluated by measuring cardiac output and arterial BP.
Key Results
In vitro analyses revealed that B16F10 cells expressed β‐adrenoceptors, but neither isoprenaline, a β‐adrenoceptor agonist, nor the β‐blocker propranolol affected cancer cell proliferation. In vivo studies showed that the antitumour efficacy of propranolol follows a U‐shaped biphasic dose–response curve. Low doses (10 and 20 mg·kg−1·day−1) significantly inhibit tumour growth, whereas higher doses are progressively less effective. We also found that high‐dose propranolol stimulates tumour arteriogenesis whereas no effect on angiogenesis was observed at any dose. Based on these data and considering that propranolol is a vasoactive drug, we hypothesized that it causes systemic vasoconstriction or vasodilation depending on the dose and thus alters tumour perfusion and growth. Consistent with this hypothesis, we found that propranolol has a biphasic effect on SVR with low and high doses producing vasoconstriction and vasodilation respectively.
Conclusions and Implications
Propranolol inhibits melanoma growth in a U‐shaped biphasic manner. A direct relationship exists between SVR and anti‐melanoma activity.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- CO
cardiac output
- GAPDH
Glyceraldehyde‐3‐phosphate dehydrogenase
- HR
heart rate
- IB4
isolectin B4
- SV
stroke volume
- SVR
systemic vascular resistance
- vWF
Von Willebrand Factor
Tables of Links
| TARGETS |
|---|
| GPCRs |
| β1‐adrenoceptors |
| β2‐adrenoceptors |
| LIGANDS |
|---|
| Angiopoietin 1 |
| Isoprenaline |
| Propranolol |
| VEGF |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
β‐Adrenoceptors are a family of G‐protein coupled receptors comprising three subtypes, β1, β2 and β3, which act by activating a Gs protein. These receptors play a role in the regulation of peripheral vascular resistance, heart function and airway reactivity, as well as a variety of metabolic and CNS functions. β‐Adrenoceptors also regulate cellular processes involved in cancer and angiogenesis and are the molecular target of β‐blockers, a class of drugs which inhibits the interaction of catecholamines with β‐adrenoceptors.
In vitro studies indicate that propranolol, the prototype of β‐blockers, reduces melanoma cell proliferation in human and murine melanoma cell lines (Dal Monte et al., 2013; Moretti et al., 2013; Calvani et al., 2015; Wrobel and Le Gal, 2015). Furthermore, the systemic administration of propranolol slows down tumour development in immunodeficient mice transplanted with human melanoma cells (Wrobel and Le Gal, 2015) and inhibits the effects of stress on the growth and metastasis in the B16F10 melanoma mouse model (Hasegawa and Saiki, 2002; Glasner et al., 2010; Barbieri et al., 2012). Together, these data suggest that propranolol has potential anticancer activity against melanoma. However, it is unknown whether propranolol dose is linearly related to inhibition of tumour growth. Knowledge of this relationship is critical for the safe and effective use of this drug in melanoma treatment.
β‐Adrenoceptors are expressed not only in cancer cells but also in cells of tumour stroma such as endothelial cells (Flacco et al., 2013). Propranolol inhibits the proliferation, migration and differentiation of human endothelial cells (Lamy et al., 2010). Furthermore, systemic administration of propranolol down‐regulates pro‐angiogenic factors and reduces the formation and growth of new blood vessels in animal models of choroidal and retinal neovascularization (Ristori et al., 2011; Lavine et al., 2013) as well as melanoma angiogenesis in immunodeficient mice (Wrobel and Le Gal, 2015). Additionally, other preclinical studies have shown that activation of β‐adrenoceptors promotes angiogenesis (Iaccarino et al., 2005; Thaker et al., 2006; Xu et al., 2015). Collectively, these results indicate that β‐AR blockade by propranolol has the potential to inhibit angiogenesis which is a crucial process for tumour growth. However, whether the anticancer activity of propranolol against melanoma in immunocompetent mice is due to inhibition of angiogenesis remains to be ascertained.
Limiting tumour blood flow using antiangiogenic drugs is an established therapeutic strategy for cancer. Modulating tumour perfusion with agents that can indirectly regulate tumour blood flow through vasoconstriction or vasodilation of normal tissues can represent an alternative strategy. β‐blockers are vasoactive drugs and their major therapeutic effects are on the cardiovascular system. Administration of propranolol decreases cardiac output (CO), but peripheral vascular resistance can increase to maintain BP as a result of blockade of vascular β2‐adrenoceptors and reflex activation of vascular α‐adrenoceptors. This pharmacological activity, particularly peripheral vasoconstriction, is important. It seems to be one of the mechanisms underlying the successful use of propranolol in the management of infantile hemangiomas, the most common benign tumour in children (Bingham et al., 2012). However, it is unknown whether the doses of propranolol that inhibit melanoma growth would also cause peripheral vasoconstriction.
To answer these questions, the effects of increasing doses of propranolol were evaluated on tumour growth, vessel density, expression of growth factors related to angiogenesis and cytokines in tumour specimens from B16F10 melanoma‐bearing mice. In addition, cultures of B16F10 melanoma cells were used to evaluate direct antiproliferative effects of propranolol on tumour cells. Given that the major pharmacological effects of propranolol are on the cardiovascular system, we then examined whether propranolol causes systemic haemodynamic changes such as peripheral vasoconstriction or vasodilation that can affect tumour perfusion and growth (Zlotecki et al., 1995; Isenberg et al., 2008; Wright et al., 2011). Here, we report that propranolol inhibits tumour growth with a U‐shaped biphasic effect, with low doses producing inhibition and higher doses being less effective or even ineffective. We also found that propranolol had no effect on tumour angiogenesis, tumour inflammation and in vitro cancer cell proliferation, indicating that these are not the cause of reduced tumour growth. Finally, we found that low‐dose propranolol increased systemic vascular resistance (SVR), which returned to basal values at higher dosage. This implies that vasoconstrictor doses of propranolol have anti‐melanoma activity whereas vasodilating doses do not. In conclusion, propranolol inhibits tumour growth in a biphasic manner and a direct relationship exists between its anti‐melanoma activity and the SVR. The present results provide guidance for the clinical application of propranolol to melanoma treatment.
Methods
Animals
All animal care and experimental procedures were in compliance with the Italian guidelines for animal care (DL 116/92), the European directives (2010/63/EU) and the standards required by the UKCCCR guidelines on the welfare and use of animals in cancer research (Workman et al., 2010). Animal studies were performed in compliance with the ARRIVE and BJP guidelines (Kilkenny et al., 2010; Curtis et al., 2015; McGrath and Lilley, 2015). Animals were deeply anaesthetised with isoflurane before being killed by cervical dislocation. All efforts were made to minimize animal suffering.
C57BL/6 male mice of 14–16 weeks of age were used in all experiments and were purchased from Charles River Laboratories (Calco, Como, Italy). Mice were housed in groups of three adults per cage and maintained in standardized conditions in our animal facility at 20 ± 2°C room temperature, 40 ± 5% relative humidity and a 12 h light/dark cycle with dawn/dusk effect, water and standard pathogen‐free chow diet provided ad libitum.
Blinding and data normalization
All the data were analysed by two observers who were blinded to the group assignment of animals. Cell viability measurements and quantitative analysis of gene expression were normalized. Moreover, quantitative RT‐PCR (q‐PCR) data were adjusted by using corresponding reference genes (such as GAPDH or β‐actin). We calculated the control mean, and then all values in all groups respectively to the mean value of the control group so the control group value is 1 or 100%, and all the individual values expressed as fold of control mean (Curtis et al., 2015). The Y‐axis was labelled as fold of control mean values.
Cell cultures and reagents
The B16F10 cell line was cultured at 37°C in a humidified atmosphere containing 5% CO2. Low passage B16F10 murine melanoma cell line, obtained from ATCC, was maintained in DMEM high glucose medium (EuroClone, West York, UK) supplemented with 10% heat inactivated FBS (EuroClone, West York, UK) in the presence of penicillin and streptomycin.
Cell viability assay
CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) was used to determine the effects of β‐ adrenoceptor agonists and antagonists on cancer cell proliferation, as previously reported (Buoncervello et al., 2012). The B16F10 cell line was plated at a density of 1.5 × 103 cell per well, in 96‐well plates, and then treated with propranolol or isoprenaline (Sigma‐Aldrich, Milan, Italy) over a concentration range from 0.01 to 10 μM for 24, 48 and 72 h. At the end of treatments, 20 μL of 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H–tetrazolium (MTS) reagent were added to each well. The plates were then incubated for 2 h at 37°C in the dark. All values were normalized with respect to the viability of untreated cells (see data normalization).
Melanoma model and treatment
The B16F10 melanoma model, the most frequently used syngeneic murine melanoma, was established , as previously described (Kwong et al., 2013). B16F10 mouse melanoma cells (8 × 105 cells) in 200 μL PBS were injected s.c. into the back of C57BL/6 male mice. Animals were randomly divided into five groups of six animals each: one control group and four treatment groups. The control group was treated with vehicle (sterile saline) whereas the other groups received varying doses of propranolol hydrochloride. Propranolol was dissolved in sterile saline and administered i.p. in a volume of 5 mL·kg−1 and at 10, 20, 30 and 40 mg·kg−1·day−1 for 15 consecutive days. Treatments were started immediately after the injection of the tumour cells and continued until day 15, when mice were killed. Tumour length (L) and width (W) were measured using a calliper, and the tumour volume was calculated as L × W2 × 0.5. At the end of the experiment, tumour tissues were collected, weighed, frozen in liquid nitrogen and stored at −80°C until use or formalin fixed and paraffin embedded for later sectioning. Drug efficacy was expressed as the percentage tumour growth inhibition, calculated using the equation 100 − (T/C*100), where T is the mean volume of the treated tumour and C is the mean volume in the control group at the end of the experiment kill.
Measurements of cardiac output and systemic vascular resistance
To quantify the effects of propranolol on SVR, aortic BP and CO were measured by aortic catheterisation (1.4‐Fr micromanometer‐tipped catheter, mod. SPR 839, Millar Instruments, Houston, TX, USA) and echocardiography (SONOLINE G50, Siemens AG, Erlangen, German) respectively. Mice were anaesthetised with isoflurane (2.0% in 100% of oxygen) and placed in a supine position on a heated operating table to maintain core body temperature at 37°C. The conductance catheter was inserted into the ascending aorta to measure BP via the right common carotid artery. Echocardiography was performed as previously described (Patrizio et al., 2007). Briefly, the chest was shaved using a chemical hair remover and warmed ultrasound gel was applied to the surface of the thorax to optimize the visibility of the cardiac chambers. Left ventricular end‐diastolic (LVEDD) and end‐systolic (LVESD) diameters were measured from a M Mode image of a parasternal short axis view at the level of the papillary muscles and left ventricular end‐diastolic and end‐systolic volumes were calculated by the ‘D3’ formula. Stroke volume (SV) was derived from the difference of LV end‐diastolic and end‐systolic volumes calculated respectively as LVEDD3 and LVESD3. CO was calculated as the product of SV and heart rate (HR) (Tournoux et al., 2011). SVR was calculated as mean arterial pressure (MAP) divided by CO. MAP was calculated as diastolic BP plus a third of pulse pressure.
Following 10 min of stabilization after surgical preparation, haemodynamic and echocardiographic parameters were obtained before and 20 min after propranolol administration. Propranolol was administered i.p. in two groups of healthy C57BL/6 mice at 20 or 40 mg·kg−1. Haemodynamic signals were sampled at 1 KHz, stored and analysed with a software package for cardiovascular analysis (IOX 1.7; EMKA Technologies, Paris, France).
Quantification of tumour vessel density
Tumour samples were fixed in 10% neutral‐buffered formalin, dehydrated and embedded in paraffin before microtome sectioning. Paraffin‐embedded sections of 2.5 μm were prepared for histological analysis. The sections were dewaxed by heating at 65°C for 30 min, rehydrated with descending alcohols and subjected to antigen retrieval procedure using citrate buffer pH 6.0 before staining. Slides were washed with PBS and then incubated for 30 min at room temperature with a blocking solution (10% donkey serum in PBS). After blocking, the sections were incubated overnight at 4°C with the following primary antibodies: 1:100 mouse anti‐smooth muscle actin (α‐SMA, Sigma‐Aldrich, Milan, Italy), anti‐Von Willebrand Factor (vWF, Abcam, Milan, Italy) and isolectin B4 (GS‐IB4, Alexa Fluor 568 conjugated, Thermo Fisher Scientific, Milan, Italy). After washing with PBS, the slides were incubated with fluorescent‐conjugated secondary antibodies, 1:500, for 1 h at room temperature (donkey anti‐mouse). Negative controls were performed using the secondary antibody only. After washing, the sections were mounted with Vectashield mounting medium with DAPI. Assessment of tumour vascularity was performed according to Koitabashi et al. (2011). Specifically, intratumoural capillaries and arterioles were visualized by immunofluorescent staining with GS‐IB4 or with vWF plus α‐SMA, respectively, and counted in representative high power fields at 40X magnification using iVision software. Capillary and arteriolar densities were expressed as number of capillaries or arterioles mm−2.
RNA isolation and quantification
Total RNA was extracted from tumour tissue and spleen by using TRIzol (Invitrogen, Milan, Italy) and purified by using RNA purelink mini kit (Invitrogen, Milan, Italy). The concentration and purity of the RNA solution were determined by using a NanoDrop spectrophotometer (Fisher Scientific, Milan, Italy), whereas its overall quality was analysed using the Agilent 2100 bioanalyser with an RNA LabChip (RNA 6000 Nano kit, Agilent, Milan, Italy). cDNA was obtained by random primers and ThermoScript reverse transcriptase (Thermo Fisher, Milan, Italy) or by using the High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). mRNA expression levels for cytokines and vascular growth factors were quantified by using SensiMix SYBR Kit (Quantace, Watford, United Kingdom) and TaqMan gene expression assays respectively. qRT‐PCR analysis was performed using 7500 Real‐Time PCR system (Applied Biosystems, Foster City, CA, USA). The ΔCt was used for statistical analysis, and the treated group values were presented as fold of control mean value (see data normalization).
Cytokine array
Cytokine and chemokine expression in the sera and tumour tissues from untreated and propranolol‐treated mice were determined by Dot‐blot‐based mouse cytokine antibody array (ARY006, R&D Systems, Minneapolis, MN, USA) as recommended by the manufacturer. Protein concentration was normalized before performing the assay. Images were acquired by ChemiDoc System. Spots were averaged, background subtracted by Image Lab Software and average values were reported for each cytokine.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as the mean ± SEM of at least five determinations per experimental group and analysed using GraphPad Prism version 5.03 software program (GraphPad Software Inc., San Diego, CA, USA). Statistical significance between different groups was determined by unpaired t‐test for two groups or one‐way ANOVA with Bonferroni's post hoc test to compare all pairs of columns for more than two groups. A value of P < 0.05 was considered statistically significant.
Results
Propranolol does not affect B16F10 cell proliferation in vitro
Given that propranolol is a β1‐ and β2‐adrenoceptor antagonist, we first evaluated whether B16F10 tumour cells express the β1‐ and/or β2‐adrenoceptor subtypes. Heart tissue samples obtained from β‐adrenoceptor knockout and wild‐type mice were used as negative and positive controls respectively. As shown in Figure 1A, q‐PCR analysis revealed that B16F10 cells express a moderate level of β2‐adrenoceptors but lack β1‐adrenoceptors. Next, the effects of the β‐blocker propranolol or isoprenaline, a β‐adrenoceptors agonist, on cancer cell proliferation were assessed by MTS assay. Despite the presence of β2‐adrenoceptors, neither isoprenaline nor propranolol, at concentrations up to 10 μM, had any effect on cancer cell proliferation (Figure 1B). Together, these results indicate that propranolol has no direct antiproliferative effects on tumour cells and that the β2‐adrenoceptor signalling is not required for B16F10 melanoma cell proliferation in vitro.
Figure 1.
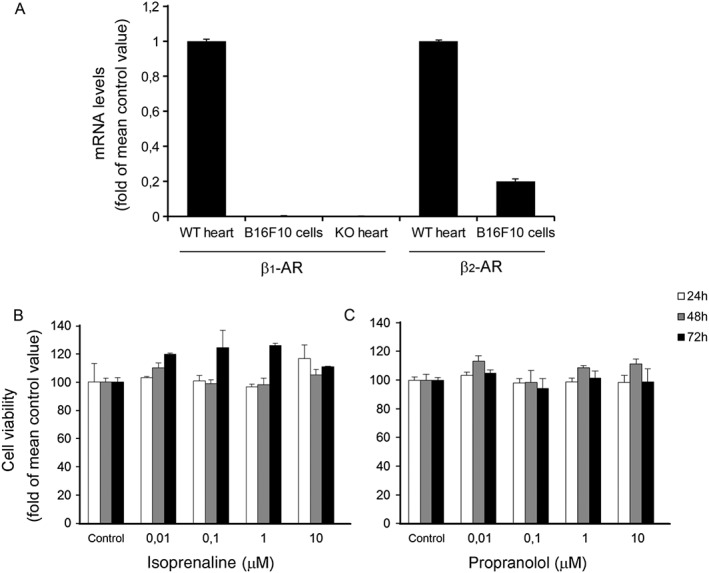
β‐adrenoceptor (β‐AR) ligands have no direct antitumour effects. (A) β1‐ and β2‐adrenoceptor mRNA levels in cultured cancer B16F10 cells. mRNA level was assessed by q‐PCR after being normalized by the mRNA level of GAPDH. Heart tissue samples from β1‐adrenoceptor knockout (KO) and wild‐type (WT) mice were used as negative and positive controls respectively. B16F10 cells expressed a moderate level of β2‐adrenoceptors whereas β1‐adrenoceptors were lacking (n = 10 per group). (B) Tumour cell proliferation. A MTS proliferation assay was used for in vitro assessment of propranolol, a β‐adrenoceptor antagonist, and isoprenaline, a β‐adrenoceptor agonist, effects on B16F10 cancer cell proliferation. Neither isoprenaline nor propranolol had any effect on cancer cell proliferation (n = 10 per group). Data are expressed as percentage of cell viability after treatment with β‐adrenoceptor ligands at different incubation times (24, 48 and 72 h).
Anti‐melanoma activity of propranolol in vivo follows a biphasic dose–response curve
To evaluate the anti‐melanoma activity of propranolol, we used the B16F10 melanoma model, a well‐established and widely used preclinical model of melanoma. Mice were treated i.p. with vehicle (sterile saline) or propranolol at different doses (10, 20, 30 and 40 mg·kg−1·day−1) for 15 consecutive days, starting immediately after tumour cell inoculation. The experiment had to be terminated after 15 days due to the large tumour sizes in the control group. At this time, maximal inhibition was obtained at 20 mg·kg−1·day−1 beyond which a further dose increase resulted in less tumour‐growth inhibitory activity (Figure 2A). The effects of propranolol treatment on tumour volume at day 15, for each dose, are shown in Figure 2B. Together, these results indicate that the relationship between propranolol dose and inhibition of tumour growth is not linear but follows a biphasic dose–response curve with a U‐shaped configuration.
Figure 2.
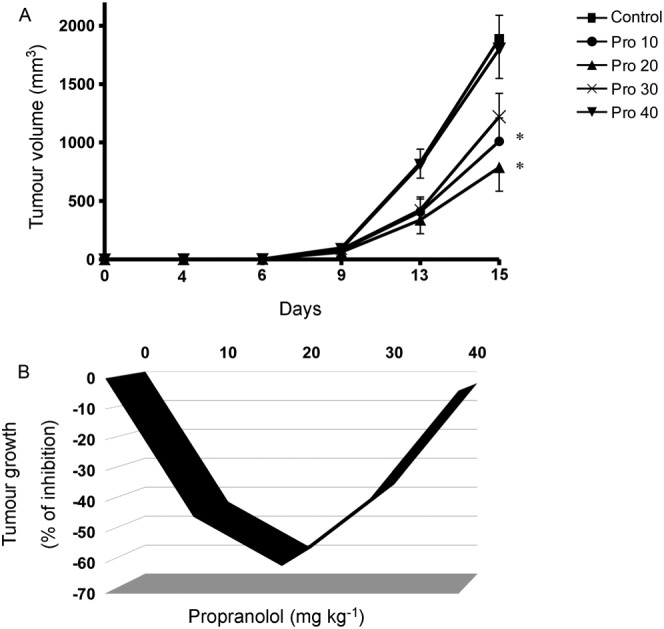
Biphasic effect of propranolol on melanoma growth. (A) Tumour volume. B16F10 tumour‐bearing mice were treated with vehicle (Control) or with increasing doses of propranolol (10, 20, 30 and 40 mg·kg−1·day−1). Propranolol was administered via i.p. route once daily for 15 days. Maximal inhibition of tumour growth was obtained at the dose of 20 mg·kg−1·day−1. Data represent mean tumour volume ± SEM (n = 6 per group). * P < 0.05, significantly different from the control group). (B) Tumour inhibition rate. Data were calculated by using the equation 100 − (T/C*100), where T is the mean volume of the treated tumour and C is the mean volume in the control group at the day 15 after tumour cell inoculation. Propranolol inhibits tumour growth in mice in a U‐shaped biphasic manner. Control, saline‐treated mice; Pro 10, mice treated with 10 mg·kg−1·day−1; Pro 20, mice treated with 20 mg·kg−1·day−1; Pro 30, mice treated with 30 mg·kg−1·day−1; Pro 40, mice treated with 40 mg·kg−1·day−1.
Propranolol affects tumour arteriogenesis but not angiogenesis
In order to investigate whether the effects of propranolol on tumour growth were due to inhibition of angiogenesis, capillary density and the expression of genes involved in angiogenesis were evaluated in tumour samples. Immunostaining of endothelial cells with IB4 revealed that there was no significant effect of propranolol treatment on tumour capillary density at any dose (Figure 3A,B). Furthermore, q‐PCR showed that vascular growth factors such as VEGF and angiopoietin‐1 were expressed in tumour samples, but propranolol treatment did not change their expression (Figure 3C). Collectively, these results indicate that tumour angiogenesis is not the primary target of the anti‐melanoma activity of propranolol in vivo.
Figure 3.
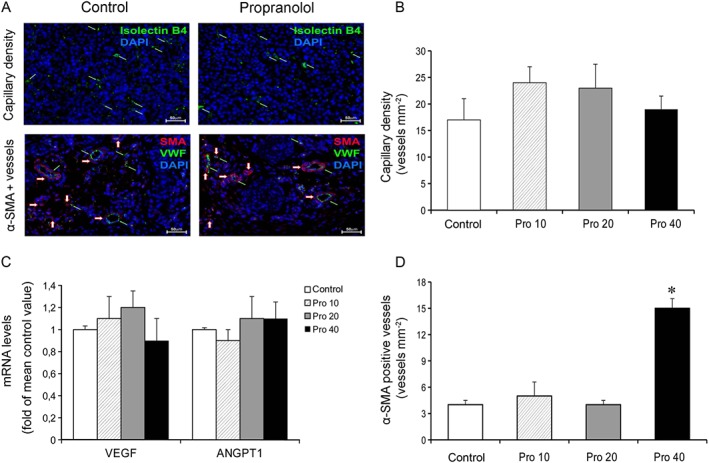
Propranolol affects arteriogenesis but not angiogenesis. (A) Representative images of capillary density and α‐SMA positive vessels in B16F10 tumours from mice treated with saline (Control) or 20 mg·kg−1·day−1 of propranolol. Tumour sections (40X magnification) were stained with IB4 or with anti‐vWF and anti‐α‐SMA antibodies for assessment of capillary density (green arrow) or pericyte‐positive vessels (red arrow) respectively. (B) Tumour capillary density. Ten sections from each tumour were counted and averaged. Tumour capillary density was similar in saline‐ and propranol‐treated mice (n = 5 per group). (C) mRNA levels of VEGF‐A (primer/probe set Mm00437306_m1) and angiopoietin 1 (ANGPT1, primer/probe set Mm00456503_m1) in tumours. Propranolol treatment did not change mRNA expression of vascular growth factors (n = 10 per group). (D) Tumour α‐SMA positive vessels. B16F10 tumour vessels were also characterized with respect to pericyte coverage. Administration of propranolol at the dose of 40 mg·kg−1·day−1 significantly increased the α‐SMA positive vessel density. Lower doses were ineffective (n = 5 per group). * P < 0.05, significantly different from the control group).
As arteriogenesis, one of the two forms of vessel growth after birth, represents an important adaptive response to haemodynamic changes or chronic hypoxia (Boroujerdi et al., 2012; Vandersmissen et al., 2015), we evaluated whether propranolol impairs arteriogenic remodelling in tumours, by performing immunostaining of endothelial cells for vWF and pericytes for α‐SMA. Pericyte‐positive vessels were defined as vWF‐positive structures associated with at least one α‐SMA‐positive cell. We found that propranolol at the dose of 40 mg·kg−1·day−1 significantly increased the α‐SMA positive vessel density whereas 10 and 20 mg·kg−1·day−1 had no effect (Figure 3D). These results indicate that high‐dose propranolol, while lacking antitumour activity, promotes tumour arteriogenic remodelling.
Propranolol does not affect the expression of key inflammatory cytokines
The interactions between cancer and stromal cells, such as endothelial and inflammatory cells, are regulated through a complex network of cytokines and growth factors which evoke important inflammatory and immunological responses by controlling multiple signalling pathways, including angiogenesis. Likewise, experimental and clinical evidence supports an effect of inflammatory cells in cancer growth. To date, it is unknown whether and how propranolol affects inflammatory cytokine and chemokine expression in tumours. To this end, we first evaluated the effects of propranolol on cytokine mRNA levels in B16F10 tumours and spleen. As shown in Figure 4A,B, q‐PCR analysis revealed that propranolol does not significantly alter mRNA levels of cytokines in the analysed tissues. Next, the cytokine and chemokine protein profiling in the sera and tumour samples derived from control and propranolol‐treated melanoma‐bearing mice was assessed by a proprietary spotted antibody array capable of assessing 40 different cytokines and chemokines. Thirteen serum proteins and 11 tumour proteins were identified. Compared with controls, propranolol at the antitumour dose of 20 mg·kg−1 caused only small variations in the protein expression (less than a twofold change in chemoluminescent signal), which agree well with q‐PCR results (Figure 4C,D). Collectively, these results suggest that anti‐inflammatory mechanisms do not appear to have a primary role in the anti‐melanoma activity of propranolol.
Figure 4.
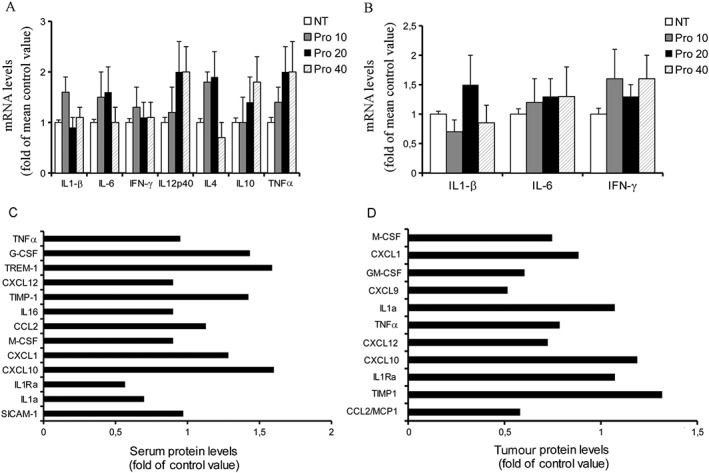
Propranolol does not affect the expression of key inflammatory cytokines. (A) mRNA levels of cytokines in B16F10 tumours. Propranolol treatment did not significantly change mRNA expression of IL‐1β, IL‐4, IL‐6, IL‐10, IL‐12, IFN‐γ and TNF‐α in tumour samples (n = 10 per group). (B) mRNA levels of IL‐1β, IL‐6 and IFN‐γ in the spleen of melanoma‐bearing mice. Again, propranolol did not significantly change mRNA expression of cytokines (n = 10 per group). Cytokine and chemokine protein profiling in the sera (C) and tumour tissues (D). The samples derived from control and propranolol‐treated melanoma‐bearing mice were pooled (5 mice per group) and analysed by a dot blot immunoassay. Compared to control, variations of protein expression in the group treated with propranolol (20 mg·kg−1·day−1) were small with all proteins showing a fold change of less than a factor of two.
Propranolol exhibits a biphasic effect on SVR
Propranolol has a short half‐life and its effects occur and usually disappear within 5–6 h after administration. Thus, we evaluated propranolol effects on SVR in healthy C57BL/6 mice before and 20 min after its administration. After carotid artery cannulation, two different selected doses of propranolol were tested: 20 mg·kg−1, which is the dose corresponding to its optimal antitumour activity, and 40 mg·kg−1, a dose devoid of antitumour activity. Intraperitoneal administration of propranolol at 20 mg·kg−1 did not affect mean arterial pressure (MAP, from 72 ± 2 to 71 ± 2 mmHg) but significantly decreased HR (from 417 ± 16 to 356 ± 12 bpm) (Figure 5A,C). In contrast, administration of 40 mg·kg−1 significantly decreased both MAP (from 73 ± 3 to 60 ± 2 mmHg) and HR (from 415 ± 21 to 340 ± 19 bpm) (Figure 5B,D). Administration of 20 or 40 mg·kg−1 had minimal effects on fractional shortening (FS, from 29.0 ± 1.1 to 26.5 ± 1.2% and from 29.2 ± 1.1 to 26.1 ± 1.4%, respectively) (Figure 5E–G). In contrast, both doses significantly decreased CO by about 20% (Figure 5H). As compared with the respective basal values, SVR was significantly increased by 20 mg·kg−1 propranolol, whereas it returned to basal values after treatment with 40 mg·kg−1 (Figure 5I). Together, these results indicate that there is a direct relationship between systemic vascular tone and anti‐melanoma activity of propranolol and that the fall in CO is crucial for the BP‐lowering activity of propranolol.
Figure 5.
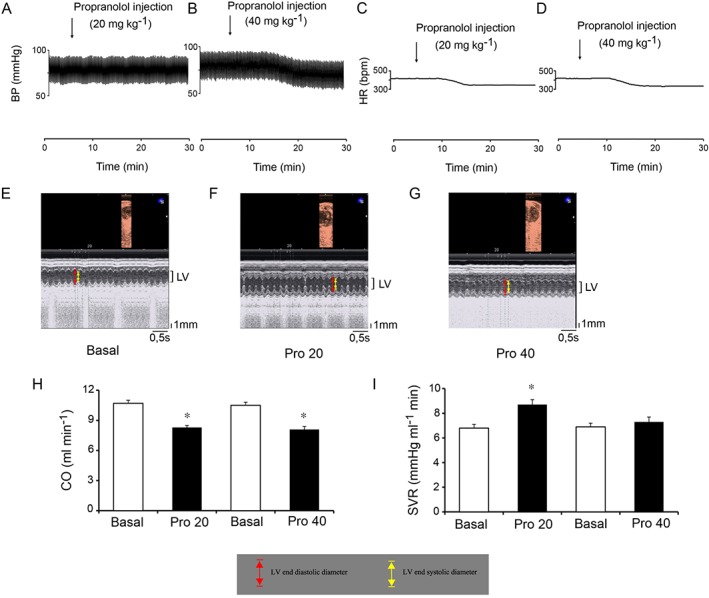
Low‐dose propranolol causes systemic peripheral vasoconstriction. (A–D) Representative tracings of propranolol effects on arterial BP and HR. Intraperitoneal administration of 20 mg·kg−1 of propranolol had no effect on BP whereas 40 mg·kg−1 decreased it (A,B). Both doses of propranolol reduced HR (C,D). (E–G) Representative echocardiographic M‐mode images of the LV. Administration of 20 or 40 mg·kg−1 of propranolol had minimal effects on fractional shortening (see Results section). (H) Propranolol effects on CO. Both doses of propranolol significantly reduced CO by about 20%. CO was calculated as the product of SV and HR; n = 5 per group. * P < 0.05, significantly different from the respective basal values. (I) Propranolol effects on SVR. SVR was increased by 20 mg·kg−1 of propranolol, whereas it returned to basal values after administration of 40 mg·kg−1. SVR was calculated as mean arterial pressure (MAP) divided by CO. n = 5 per group; * P < 0.05, significantly different from the respective basal values).
Discussion
In recent years, inhibition of β‐AR signalling has emerged as a potential strategy to suppress tumour growth, and propranolol, the prototype of β‐blockers, has been found to have potent anticancer activity in preclinical models of melanoma (Glasner et al., 2010; Barbieri et al., 2012; Wrobel and Le Gal, 2015). However, no study has examined whether there is a linear relationship between propranolol dose and inhibition of melanoma growth. This relationship is a fundamental and essential concept in pharmacology, and its knowledge is critical for the safe and effective use of drugs in patients. In this study, we show that propranolol inhibits tumour growth in B16F10 melanoma bearing‐mice not in a linear manner, but in a biphasic relationship with a U‐shaped configuration. Optimal inhibition of tumour growth was obtained by i.p. injection of 20 mg−1·kg−1·day−1, beyond which further dose increases resulted in less activity or even in complete loss of the antitumour activity when the β‐blocker was administered at 40 mg−1·kg−1·day−1.
A second, equally important result of this study is that there is a direct relationship between the anticancer activity of propranolol and its effects on SVR. Indeed, we found that propranolol has a biphasic effect on vascular resistance with low and high doses producing vasoconstriction and vasodilation, respectively, which also implies that vasoconstrictor doses of propranolol have anti‐melanoma activity whereas vasodilating doses do not.
Vasoactive agents have been used to modify tumour flow with the goal of improving cancer detection and treatment. However, given that the intratumour vasculature is structurally and functionally abnormal with vessels lacking anatomical elements required for normal vasoactive responses, a direct and predictable regulation of tumour blood flow would be extremely difficult, if not impossible. As such, modulating tumour perfusion with drugs that can indirectly regulate tumour blood flow through vasoconstriction or vasodilation of normal vascular beds can represent an attractive alternative strategy. Several studies indicate that the tumour behaves as a passive resistance, that is tumour flow is inversely related to that in the host periphery. Administration of hydralazine or NO donors causes peripheral vasodilation, which drives a redistribution of blood away from the tumour vasculature through a steal effect and reduces tumour perfusion (Zlotecki et al., 1995; Sonveaux et al., 2005; Isenberg et al., 2008). Conversely, peripheral vasoconstriction induced by adrenaline or angiotensin II increases tumour flow through an inverse steal effect (Zlotecki et al., 1995, Isenberg et al., 2008, Wright et al., 2011). Given that the abnormal tumour environment is an important contributor to tumour progression and resistance to treatment, improvements in intratumour perfusion and hypoxia may promote a more normalized tumour environment thus inhibiting tumour progression. Here, we show that vasoconstrictor doses of propranolol inhibit melanoma growth whereas vasodilating doses are ineffective and stimulate tumour arteriogenesis, an indicator of severe tissue hypoxia (Boroujerdi et al., 2012). A possible mechanism for antitumour activity is that the β‐blocker constricts microvessels within the tumour thus reducing the blood supply available to the tumour and slowing down tumour growth. As hypoxia and inadequate blood supply may promote an aggressive phenotype and contribute to anticancer therapy resistance (Harris, 2002; Carmeliet and Jain, 2011), another possible mechanism is that propranolol causes constriction of normal vessels in parallel tissues, which ‘pushes’ blood into the tumour, increases tumour blood flow and inhibits tumour progression by improving tumour oxygenation. Considering the vasoactive properties of normal vascular beds, the latter hypothesis seems more likely and is consistent with the previous observation that intravenous administration of 10 mg·kg−1 of propranolol in a syngeneic mouse sarcoma model causes a significant increase of about 100% in tumour perfusion (Bomber et al., 1986). However, whether antitumour activity of propranolol is primarily mediated by peripheral vasoconstriction and whether propranolol causes increased tumour perfusion and reduced tumour hypoxia in melanoma need to be demonstrated in future studies.
The mechanism responsible for biphasic effect of propranolol on vascular resistance also remains unclear. A possible explanation is that propranolol may affect multiple signalling pathways, and that vasoconstrictor activity requires doses which only inhibit β‐adrenoceptor signalling. Given that peripheral vascular resistance increases as a result of reflex sympathetic activation due to the fall in CO, it is possible that high doses of propranolol cause a non‐specific central sympatholytic effect that opposes peripheral vasoconstriction producing the return of vascular resistance to basal values and the decrease in arterial BP. This would be in accordance with the observation that the hypotensive effect of propranolol is paralleled by the reduction in central sympathetic nervous activity (Lewis and Haeusler, 1975).
In the present study, we observed a paradoxical effect of propranolol on tumour growth and vascular resistance, that is low doses of propranolol cause poor tumour growth and vasoconstriction, whereas high doses cause the opposite effect. The observation that the treatment with β‐blockers can cause paradoxical effects is not surprising. In fact, it is well known that the β‐blocker administration in subjects with heart failure produces symptomatic worsening at the onset of therapy, but haemodynamics and mortality improve with chronic use. Furthermore, in an antigen‐driven murine model of asthma, some β‐blockers increased the airway constrictor response to cholinergic stimulation when administered acutely but significantly attenuated the airway constrictor response with chronic use (Nguyen et al., 2009). In addition, β‐blockers are not the first drugs to exhibit biphasic effects. Strong regulators of angiogenesis have recently been reported to display a U‐shaped dose response curve. These include, among others, thrombospondin, angiostatin, endostatin and rapamycin (Reynolds, 2010).
Recent studies show that propranolol has an antiangiogenic activity. Indeed, short‐term administration of propranolol inhibits the formation and growth of pathological ocular neovascularization (Ristori et al., 2011; Lavine et al., 2013) suggesting that propranolol treatment may be useful in those conditions where VEGF‐induced neovascularization threatens the integrity of vision. This effect however remains controversial. In fact another study reported that propranolol fails to prevent retinopathy development in a mouse model of oxygen‐induced retinopathy (Chen et al., 2012). Here, we show that propranolol administration in immunocompetent mice has no effect on B16F10 tumour angiogenesis but increases tumour arteriolar remodelling at the highest dose. This result is in accordance with the observation that genetic deletion of β1/2‐adrenoceptors increases tumour vascularization in the B16F10 melanoma model (Sereni et al., 2015). Conversely, a recent study performed in immunodeficient mice showed that propranolol administered in drinking water decreases intratumour vessel density in a preclinical model of human melanoma (Wrobel and Le Gal, 2015). One possible explanation of this discrepancy lies in the different method of evaluating vessel density. Wrobel and Le Gal (2015) assessed vessel density by measuring vascular area as a percentage of total area. This approach does not distinguish between vasoconstriction and reduced neovascularization. Thus, if propranolol induces vasoconstriction, the number of intratumour vessels will not change, but the vascular area expressed as a percentage of total area will decrease as constricted vessels become narrower. Another possible explanation is that overall angiogenic activity within a tumour is influenced by immune cells and that the effects of propranolol on angiogenesis are dependent on the degree of immune system impairment.
In conclusion, we have shown that propranolol treatment affects melanoma growth in a U‐shaped biphasic manner and that there is a direct relationship between the anti‐melanoma activity of propranolol and its effects on peripheral vascular resistance. The present results also provide guidance for the clinical application of this drug in melanoma treatment.
Author contributions
S.M., M.B., A.R., M.S., D.M., L.G., T.S., C.B. and L.C. performed the research and data acquisition. R.R., L.G. and G.M. analysed the data and designed the artwork. L.G. and G.M. designed the research study, interpreted the data and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by the Ministero della Salute (RF 2011, grant no. 02351158 to G.M.).
Maccari, S. , Buoncervello, M. , Rampin, A. , Spada, M. , Macchia, D. , Giordani, L. , Stati, T. , Bearzi, C. , Catalano, L. , Rizzi, R. , Gabriele, L. , and Marano, G. (2017) Biphasic effects of propranolol on tumour growth in B16F10 melanoma‐bearing mice. British Journal of Pharmacology, 174: 139–149. doi: 10.1111/bph.13662.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A, Palma G, Rosati A, Giudice A, Falco A, Petrillo A et al. (2012). Role of endothelial nitric oxide synthase (eNOS) in chronic stress‐promoted tumour growth. J Cell Mol Med 16: 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham MM, Saltzman B, Vo NJ, Perkins JA (2012). Propranolol reduces infantile hemangioma volume and vessel density. Otolaryngol Head Neck Surg 147: 338–344. [DOI] [PubMed] [Google Scholar]
- Boroujerdi A, Welser‐Alves JV, Tigges U, Milner R (2012). Chronic cerebral hypoxia promotes arteriogenic remodeling events that can be identified by reduced endoglin (CD105) expression and a switch in β1 integrins. J Cereb Blood Flow Metab 32: 1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomber P, McCready R, Hammersley P (1986). Propranolol hydrochloride enhancement of tumor perfusion and uptake of gallium‐67 in a mouse sarcoma. J Nucl Med 27: 243–245. [PubMed] [Google Scholar]
- Buoncervello M, Borghi P, Romagnoli G, Spadaro F, Belardelli F, Toschi E et al. (2012). Apicidin and docetaxel combination treatment drives CTCFL expression and HMGB1 release acting as potential antitumor immune response inducers in metastatic breast cancer cells. Neoplasia 14: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvani M, Pelon F, Comito G, Taddei ML, Moretti S, Innocenti S et al. (2015). Norepinephrine promotes tumor microenvironment reactivity through β3‐adrenoreceptors during melanoma progression. Oncotarget 6: 4615–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK (2011). Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10: 417–427. [DOI] [PubMed] [Google Scholar]
- Chen J, Joyal JS, Hatton CJ, Juan AM, Pei DT, Hurst CG et al. (2012). Propranolol inhibition of β‐adrenergic receptor does not suppress pathologic neovascularization in oxygen‐induced retinopathy. Invest Ophthalmol Vis Sci 53: 2968–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Monte M, Casini G, Filippi L, Nicchia GP, Svelto M, Bagnoli P (2013). Functional involvement of β3‐adrenergic receptors in melanoma growth and vascularization. J Mol Med 91: 1407–1419. [DOI] [PubMed] [Google Scholar]
- Flacco N, Segura V, Perez‐Aso M, Estrada S, Seller JF, Jiménez‐Altayó F et al. (2013). Different β‐adrenoceptor subtypes coupling to cAMP or NO/cGMP pathways: implications in the relaxant response of rat conductance and resistance vessels. Br J Pharmacol 169: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasner A, Avraham R, Rosenne E, Benish M, Zmora O, Shemer S et al. (2010). Improving survival rates in two models of spontaneous postoperative metastasis in mice by combined administration of a β‐adrenergic antagonist and a cyclooxygenase‐2 inhibitor. J Immunol 184: 2449–2457. [DOI] [PubMed] [Google Scholar]
- Harris AL (2002). Hypoxia‐a key regulatory factor in tumour growth. Nat Rev Cancer 2: 38–47. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Saiki I (2002). Psychosocial stress augments tumor development through β‐adrenergic activation in mice. Jpn J Cancer Res 93: 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G et al. (2005). Ischemic neoangiogenesis enhanced by β2‐adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res 97: 1182–1189. [DOI] [PubMed] [Google Scholar]
- Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC et al. (2008). Thrombospondin 1 and vasoactive agents indirectly alter tumor blood flow. Neoplasia 10: 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J et al. (2011). Pivotal role of cardiomyocyte TGF‐β signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121: 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ (2013). Localized immunotherapy via liposome‐anchored Anti‐CD137 + IL‐2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res 73: 1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy S, Lachambre MP, Lord‐Dufour S, Béliveau R (2010). Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol 53: 200–208. [DOI] [PubMed] [Google Scholar]
- Lavine JA, Sang Y, Wang S, Ip MS, Sheibani N (2013). Attenuation of choroidal neovascularization by β2‐adrenoreceptor antagonism. JAMA Ophthalmol 131: 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PJ, Haeusler G (1975). Reduction in sympathetic nervous activity as a mechanism for hypotensive effect of propranolol. Nature 256: 440. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti S, Massi D, Farini V, Baroni G, Parri M, Innocenti S et al. (2013). β‐adrenoceptors are upregulated in human melanoma and their activation releases pro‐tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab Invest 93: 279–290. [DOI] [PubMed] [Google Scholar]
- Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ et al. (2009). β2‐adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci U S A 106: 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizio M, Musumeci M, Stati T, Fasanaro P, Palazzesi S, Catalano L et al. (2007). Propranolol causes a paradoxical enhancement of cardiomyocyte foetal gene response to hypertrophic stimuli. Br J Pharmacol 152: 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AR (2010). Potential relevance of bell‐shaped and U‐shaped dose‐responses for the therapeutic targeting of angiogenesis in cancer. Dose Response 8: 253–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristori C, Filippi L, Dal Monte M, Martini D, Cammalleri M, Fortunato P et al. (2011). Role of the adrenergic system in a mouse model of oxygen‐induced retinopathy: antiangiogenic effects of beta‐adrenoreceptor blockade. Invest Ophthalmol Vis Sci 52: 155–170. [DOI] [PubMed] [Google Scholar]
- Sereni F, Dal Monte M, Filippi L, Bagnoli P (2015). Role of host β1‐ and β2‐adrenergic receptors in a murine model of B16 melanoma: functional involvement of β3‐adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 388: 1317–1331. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Kaz AM, Snyder SA, Richardson RA, Cárdenas‐Navia LI, Braun RD et al. (2005). Oxygen regulation of tumor perfusion by S‐nitrosohemoglobin reveals a pressor activity of nitric oxide. Circ Res 96: 1119–1126. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C et al. (2006). Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 12: 939–944. [DOI] [PubMed] [Google Scholar]
- Tournoux F, Petersen B, Thibault H, Zou L, Raher MJ, Kurtz B et al. (2011). Validation of noninvasive measurements of cardiac output in mice using echocardiography. J Am Soc Echocardiogr 24: 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandersmissen I, Craps S, Depypere M, Coppiello G, van Gastel N, Maes F et al. (2015). Endothelial Msx1 transduces hemodynamic changes into an arteriogenic remodeling response. J Cell Biol 210: 1239–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ et al. (2010). Guidelines for the welfare and use of animals in cancer research. Br J Cancer 102: 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel LJ, Le Gal FA (2015). Inhibition of human melanoma growth by a non‐cardioselective β‐blocker. J Invest Dermatol 135: 525–531. [DOI] [PubMed] [Google Scholar]
- Xu Q, Jennings NL, Sim K, Chang L, Gao XM, Kiriazis H et al. (2015). Pathological hypertrophy reverses β2‐adrenergic receptor‐induced angiogenesis in mouse heart. Physiol Rep 3: e12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KC, Ravoori MK, Dixon KA, Han L, Singh SP, Liu P et al. (2011). Perfusion CT assessment of tissue hemodynamics following hepatic arterial infusion of increasing doses of angiotensin II in a rabbit liver tumor model. Radiology 260: 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotecki RA, Baxter LT, Boucher Y, Jain RK (1995). Pharmacologic modification of tumor blood flow and interstitial fluid pressure in a human tumor xenograft: network analysis and mechanistic interpretation. Microvasc Res 50: 429–443. [DOI] [PubMed] [Google Scholar]