

Abstract
Signal transduction by the ryanodine receptor (RyR) is essential in many excitable cells including all striated contractile cells and some types of neurons. While its transmembrane domain is a classic tetrameric, six‐transmembrane cation channel, the cytoplasmic domain is uniquely large and complex, hosting a multiplicity of specialized domains. The overall outline and substructure readily recognizable by electron microscopy make RyR a geometrically well‐behaved specimen. Hence, for the last two decades, the 3D structural study of the RyR has tracked closely the technological advances in electron microscopy, cryo‐electron microscopy (cryoEM), and computerized 3D reconstruction. This review summarizes the progress in the structural determination of RyR by cryoEM and, bearing in mind the leap in resolution provided by the recent implementation of direct electron detection, analyzes the first near‐atomic structures of RyR. These reveal a complex orchestration of domains controlling the channel's function, and help to understand how this could break down as a consequence of disease‐causing mutations.
Keywords: cryo electron microscopy, 3D reconstruction, ryanodine receptor, allosterism, calcium, excitation–contraction coupling
Statement
The ryanodine receptor, a large intracellular calcium channel, is essential for muscle contraction, neuron signaling and other signal transduction pathways. Cryo electron microscopy has played an unrivaled role in its 3D structural determination and now has given way to the first near‐atomic structures. This article summarizes this wealth of structural information and frames the earlier structural knowledge in the context of the new findings.
Introduction
With a gradient of Ca2+ ions across the endoplasmic reticulum membrane of several orders of magnitude, the large conductance RyR channel is a main gatekeeper of this intracellular Ca2+ store. The three mammalian isoforms are: RyR1, expressed in skeletal muscle, smooth muscle, brain, pancreas, and B cells; RyR2, expressed in heart, smooth muscle, brain, and T cells; and RyR3, expressed in brain and other tissues.1, 2, 3, 4 In skeletal muscle, RyR1 activation is under control of the action potential via voltage‐sensitive plasma membrane Ca2+ channels (Cav1.1 and auxiliary subunits, which form the dihydropyridine receptor or DHPR).5, 6, 7, 8 Ca2+ release through the RyR1 triggers contraction of the acto‐myosin fibers in the process known as excitation‐contraction coupling (EC coupling).9, 10 The interaction between RyR1 and DHPR takes place at the triad junctions, specialized regions of the muscle fiber of close apposition between endoplasmic/sarcoplasmic reticulum (ER/SR) and transverse (T) tubules (tubular invaginations of the plasma membrane).11, 12, 13, 14 In heart, RyR2 is activated by Cav1.2 without direct interaction.,15 via a calcium‐induced calcium release mechanism.16, 17
RyR mutations lead to deregulation of calcium homeostasis and can have severe consequences. Mutations in RyR1 result in myopathy (central core disease and multi‐minicore disease) and in malignant hyperthermia, a life threatening condition induced by inhalation of volatile anesthetics and by depolarizing muscle relaxants.18, 19, 20, 21, 22, 23 Mutations in RyR2 cause arrhythmia (polymorphic ventricular tachycardia and arrhythmogenic right ventricular cardiomyopathy) and heart failure.22, 24, 25, 26
Besides its primary effector, DHPR, RyR action is fine‐tuned by many other factors such as the concentration of soluble small molecules (Ca2+, Mg2+, ATP, oxidants) and diverse post‐translational modifications. In addition, multiple cytoplasmic and luminal ligands can bind to the receptor and further modulate it. The complex regulation of RyR1 has been reviewed extensively.1, 27, 28, 29 In this context RyR's 3D structure provides a platform encompassing the various binding sites and sensors, and importantly, a means to code the sensed information into a specific channel behavior using a highly sophisticated network of allosteric pathways.
RyR as a showcase protein for cryoEM
RyR, with a square shape, forms an electron dense structure on the surface of the sarcoplasmic reticulum and was first discerned by electron microscopy (TEM) in thin sections of skeletal muscle30 and later identified as the intracellular calcium release channel.10 Solubilized RyRs became a very good test specimen for the new “single particle” 3D reconstruction techniques whereby thousands of TEM images of the protein in different views are computationally combined to form a 3D reconstruction.31 Single particle techniques reach their maximum potential when used in combination with cryo‐electron microscopy (cryoEM),32, 33 which by imaging the specimen without any staining and under frozen‐hydrated conditions, gives access to the structure of the protein fully hydrated and in physiological buffer.34, 35 The first 3D reconstruction from negatively stained RyR using the “random conical” algorithm obtained in 198936 was soon improved by cryo EM;37 RyR1 was also the showcase for the “angular reconstitution” algorithm.38 This was followed by 3D difference mapping to localize several important domains: divergent regions between RyR isoforms, sequence regions, and biological ligands that modulate RyR's channel properties. These first‐generation 3D reconstructions, obtained with 3,500–10,000 particles, had resolutions of 30–35 Å; some examples will be described later.
The implementation of more brilliant and coherent field emission gun electron sources39 combined with contrast transfer function correction40 allowed recovering information in the higher frequency range. These developments, combined with sample preparation strategies to get a strong representation of RyR views in all orientations and the processing of more than 30,000 particles allowed to reach 10 Å resolution, which yielded the first visualization of α‐helices in the transmembrane domain.41
Use of electron energy filtering, which reduces the contribution from noisy inelastic scattering,42 led to a set of two 3D reconstructions of RyR1 at 8.5 Å and 6.1 Å resolution that revealed the entire alpha helical component of RyR1.43 More recently the direct electron detector (DED) technology, which enables higher resolution, has been implemented.44 The higher sensitivity of DEDs requires electron dose fractionation into successive exposures, which additionally allows correcting computationally for beam‐induced movement, thus increasing the attainable resolution further.32, 45 DED technology, often combined with automated data collection (yielding datasets of 100,000–1,000,000 particles) and faster computing, have catalyzed the race of cryoEM towards atomic resolution, and led to the first near‐atomic 3D structures of RyR1: one 3D map at 4.8 Å resolution, which enabled building a model of the C‐α backbone,46 and one at 3.8‐Å resolution revealing bulky side chains, which enabled building a near‐atomic model for the more rigid 70% of the structure.47 This review summarizes the structural features of RyR1 that are now resolved in the context of earlier findings. Knowing the different pieces, their architectural organization, and their motion is a significant step towards understanding the complex allosterism of this large ion channel.
The cytoplasmic domain of RyR1
The cytoplasmic domain has a flat square prism shape of 275 × 275 × 120 Å3. The handle and clamp domains form the sides and the corners of the square, respectively. Each of the four identical subunits contributes a triangular slice (Fig. 1) formed by multiple domains; these are described below following the sequence order. Their 3D locations are shown in Figures 1 and 2, and their sequence, alternative names and abbreviations in Table 1. All the high‐resolution features are based on the 4.8 and 3.8 Å resolution maps of RyR146, 47 except for the models derived from crystal structures.
Figure 1.
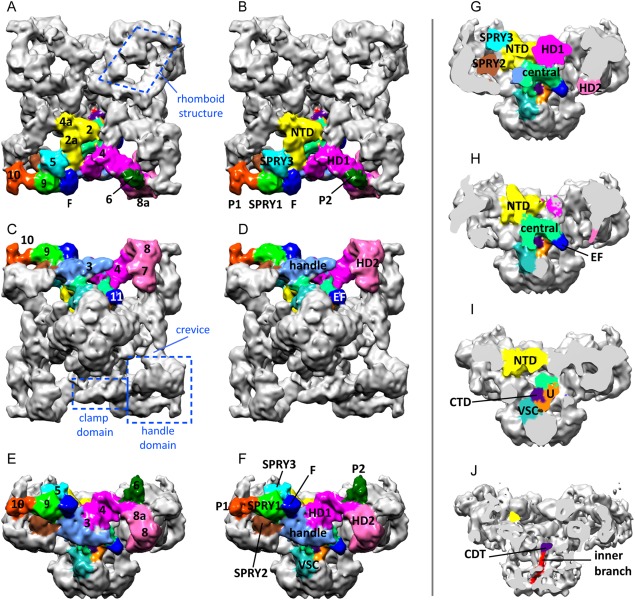
Multi‐domain structure of the cytoplasmic domain of RyR1. Cytoplasmic domain in three orthogonal views. The domains in one subunit are color‐coded. (A, B) Cytoplasmic view. (C, D) Endo/sarcoplasmic reticulum luminal view. (E, F) Side view. In (A‐F) the panels on the left show the traditional numerical designation and the panels on the right show the new names of the domains. (G–J) Successive slices towards the fourfold axis showing the internal domains. Based on the cryoEM map EMDB‐1606.
Figure 2.
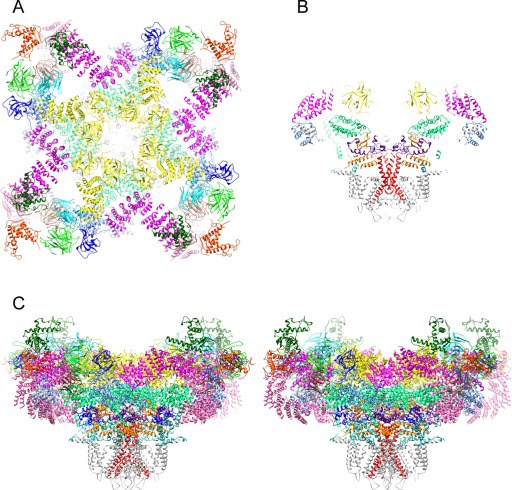
Near‐atomic structure of RyR1. (A) Cytoplasmic view; the transmembrane domain is omitted for clarity. (B) Central slice of the side view. (C) Stereo view. Subunits are color‐coded as in Figure 1 and S6 is colored in red. Based on the atomic model 3J8H.pdb.
Table 1.
Correspondence Between Domains and Their Sequence,a and Alternative Domain Denominations
| Domain name | Alternative names, abbreviations | Domain number | Sequence |
|---|---|---|---|
| N‐terminal domain | NTD; 4 NTS form the cytoplasmic vestibule | 2‐2a‐4a | 1–631 |
| SPRY1 | Part of clamp domain | 9 | 632–826, 1466–1491, 1615–1634 |
| P1 | Part of clamp domain, Ry12 | 10 | 851–1055 |
| SPRY2 | 827–845, 1071–1241 | ||
| SPRY3 | 5 | 1242–1465, 1492–1614 | |
| Handle | 3 | 1651–2145 | |
| HD1 | α−solenoid, includes crevice | 3, 4 | 2146–2712 |
| P2 | Phosphorylation domain, Ry34 | 6 | 2734–2940 |
| HD2 | Part of clamp domain, α−solenoid | 7‐8‐8a | 3016–3572 |
| Central domain | Part of outer branches/columns, CD | 3668–4251 | |
| CD‐EF hands | 11 | 4071–4132 | |
| CD–U motif | 4133–4251 | ||
| S1b | 4559–4579 | ||
| First luminal loop | S1‐S2 loop | 4580–4638 | |
| S2 | 4639–4662 | ||
| S2‐S3 loop | Part of outer branches/columns, VSC | 4663–4786 | |
| S3 | 4787–4805 | ||
| S4 | 4806–4819 | ||
| S4‐S5 linker | 4820–4833 | ||
| S5 | 4834–4858 | ||
| Second luminal loop | Luminal loop | 4859–4878 | |
| Pore helix | 4879–4893 | ||
| Selectivity filter | 4894–4900 | ||
| S6 ‐ transmembrane | Inner helix | 4904–4940 | |
| S6 ‐ hinge | 4934 | ||
| S6 ‐cytoplasmic gate | 4937 | ||
| S6 ‐ cytoplasmic | Inner branch | 4941–4956 | |
| C‐terminal domain | CTD; zinc‐finger; 4 CTDs form the cytoplasmic constriction | 4957–5037 |
Rabbit RyR1 sequence; sequence boundaries in the transmembrane helices according to a 3.8 Å cryoEM map.47
Light gray shading corresponds to the membrane‐embedded regions of the transmembrane domain; dark gray shading corresponds to luminal regions, no shading corresponds to cytoplasmic regions.
N‐terminal domain (domains 4a‐2‐2a)
The structure of the N‐terminal domain was first determined by X‐ray crystallography48 and has 3 subdomains: A and B formed by a beta trefoil structure each, and C with an all alpha‐helical structure. The four N‐terminal domains were docked in the cryoEM model of the full protein around the fourfold axis, forming a cytoplasmic vestibule.48 The cryoEM near atomic structures add more α‐helices at the C‐terminal end [Figs. 1(A,B,G–I), 2].
An earlier localization of the N terminus in the clamp domain based on GST and GFP insertions49 is now obsolete; mislocalization was due to a combination of low resolution and use of long linkers.50 Additional attempts to define the N terminal domain of the RyR1 involved an homology model based on an oxido‐reductase structure51 and a second model of this domain based on IP3R partial crystallographic structures,52 unfortunately in both cases the subsequent dockings of the model into the clamp region are also obsolete; they were based on low sequence identity (below 20%) which is known to give unreliable results.53
SPRY domains
These are protein–protein interaction modules consisting of a β‐sandwich comprised of two β‐sheets.54 RyR1 has three SPRY domains per subunit, SPRY1‐3.55
SPRY1 domain (domain 9)
SPRY1 protrudes on the perimeter of the cytoplasmic assembly and forms part of the binding site for FKBP12 [Figs. 1(A–F), 2(A,C)]. A subsequent crystallographic determination refined the core of the structure for regions not sufficiently resolved in the cryoEM map.56
SPRY2 domain
The SPRY2 domain was crystallized and docked into the cryoEM density map of RyR1,57 using the approximate location determined earlier by cryoEM and antibody labeling;58 this position has been confirmed in the 3.8 Å resolution structure. The peptide 1076–1112 of RyR1, which belongs to the SPRY2 domain, was found to interact specifically with the II–III loop of the Cav1.1 subunit of the DHPR, whereas the equivalent peptide of RyR2 did not, suggesting a role of this sequence in skeletal‐type EC coupling.59, 60 This was followed by experiments in vitro showing interactions between the SPRY2 core domain (1085–1208) and the II‐III loop,61 although more recent studies using the full SPRY2 domain (1070–1246) could not replicate this interaction.57 SPRY2 is situated behind SPRY1 and underneath SPRY3 [see Figs. 1(G), 2(A,C)]. Can the DHPR II‐III loop reach the SPRY2 domain, which is not on the outer surface of RyR1? 3D reconstruction and antibody labeling show that the 125‐residue II–III loop is a sizeable domain of the DHPR62 that should be able to reach RyR1's SPRY2 either gaining access through a surface groove or simply interacting from the side. In support of these two possibilities we found that three different antibodies against SPRY2 could bind to fully folded RyR1 both from the T‐tubule‐facing surface and from the side.58
SPRY3 domain (domain 5)
This domain sits on the RyR1 surface facing the T tubule, between SPRY1 and the N terminal domain [Figs. 1(A,B,E–G), 2(A,C)]. Although so far it has been less studied biochemically, given its location, participation of the SPRY3 domain in the RyR1‐DHPR interaction seems probable.
All three SPRY domains were swapped in the 7 Å43 and 4.8 Å46 resolution models, probably owing to their structural similarity. Interestingly, in the higher resolution 3D structure the sequence threads back and forth between the three SPRY domains and contributes additional short sequences of 20–25 residues at each pass, close‐knitting the three domains together47 (see Table 1). It is likely that this interlacing limits the flexibility in this region and has a role in RyR's long‐range allosteric pathways.
The DR2 region of divergence between RyR1 and RyR2 (residues 1298–1431 of RyR1) within the SPRY3 sequence has not been mapped in the near‐atomic maps but a GFP insertion visualized by cryoEM at low resolution places residue 1366 in domain 6 of RyR2 opposite of the region of contact with domain 5,63 thus presumably DR2 is in a region between domain 5 (SPRY3) and domain 6 (P2).
P1 domain (domain 10)
This domain corresponds to the “RyR repeat 1‐2” or the first tandem repeat.64 Its sequence emanates from the SPRY cluster and forms the small L‐shaped appendage at the end of domain 9, in the outer corner of the clamp domain [Figs. 1(A–F), 2(A,C)]. A crystal structure for this domain shows two additional β‐sheets.56
Handle domain (domain 3)
Domain 3 looks like a flat slab forming the side of the square [Figs. 1(A–F), 2]. It is formed by 16 α‐helices and encompasses the binding sites for FKBP12, apoCaM, and Ca2+‐CaM. The DR3 region of divergence between RyR1 and RyR2 (residues 1872–1923 of RyR1) is within the sequence of the handle domain but this specific stretch of the sequence is missing from the near‐atomic structures; this suggests that this segment with an unusual content of negatively charged residues is disordered and flexible. A 34‐Å resolution cryoEM study of RyR2 places a GFP inserted after residue 1874 in a region of domain 9 facing toward domain 3 and the FKBP12.6 binding site.65
Helical domains 1 and 2 (crevice or domain 4 and domains 8a‐8‐7)
These form an extended right‐handed α‐solenoid that includes the crevice and a 200 Å‐long self‐standing ribbon looping out, which connects the handle domain to the SPRY2 domain of the neighboring subunit [Figs. 1(A–G), 2]. Its inherent flexibility limits the resolution achievable for this region and only the Cα backbone has been assigned with the exception of a 60‐residue sequence stretching from the distal tip of the α‐solenoid back to the central domain where the sequence continues, which is missing entirely. The architecture and flexibility of the helical domains (HD) likely has a central role in the long‐range allosteric communication and confers elasticity and recoil to the overall structure. The outer loop of the α‐solenoid contacts P1 in the neighboring subunit. HD2, P1, and SPRY1 form the corners of the RyR1 known as clamp domains.
P2 (domain 6)
This region corresponding to “RyR repeat 3–4”or second tandem repeat64 was solved at atomic resolution by crystallography.66 Initially it was docked to domain 10 but this location is now obsolete: in view of the sequence path in the higher resolution cryoEM structures, the RyR repeat 3–4 was reassigned to domain 6.56 It contains the phosphorylation site Ser2843. The analogous region in RyR2 contains the phosphorylation sites 2808 and 2814. The sequence of P2 lies within HD1 and HD2, which agrees with an earlier localization at low resolution in RyR2 by GFP insertion,67 and forms the density in domain 6 that bridges towards domain 5 [Figs. 1(A,B,E,F), 2(A,C)]. This is the more variable region of RyR1's structure among different reconstructions and can only be seen when lowering the threshold, suggesting a region of high flexibility. Indeed, the loop of P2 containing the phosphorylation site was not resolved even in the crystal structure, which reflects the highly dynamic nature of this region. In addition, RyR1 from natural sources is expected to have heterogeneity in the level of phosphorylation that introduces further variability; however other factors must contribute to the observed flexibility, since dephosphorylation of RyR1 did not result in significant improvement of the appearance of this domain.46
Central domain (CD)
This is the only cytoplasmic domain that interacts with the transmembrane domain. The first section comprised of ∼19 α‐helices (residues 3668–4070) forms a horseshoe and is mostly internal, situated behind the handle domains and under the NTDs [Figs. 1(G,H), 2]; one alpha helix forms part of the outer branches or columns that extend towards the transmembrane domain. A second section forms two EF hands, visible as “domain 11” protruding from the stem of the cytoplasmic domain as it nears the membrane41 [Figs. 1(C–H), 2]. EF hands are presumed to confer Ca2+‐sensing ability to RyR1 by virtue of a large conformational change upon Ca2+ binding right in the core of the protein; other Ca2+ sensing domains outside the Central Domain further contribute to the complex regulation of RyR1 by Ca2+.68, 69 A third section of the central domain forms the U‐motif, a U‐shaped structure closer to the fourfold axis [Figs. 1(I), 2(B,C)].
Most of the DR1 region of divergence between RyR1 and RyR2 (residues 4354–4631 in RyR1) is in a ∼300 residue stretch that remains unmapped between the U‐motif and the first transmembrane segment (S1). A GFP inserted after residue 4365 of RyR2 mapped near the crevice (domain 4, HD1).70 While this could be plausible given the length of DR1, further work at higher resolution will be needed to establish the DR1 location with certainty. The last 100 residues of DR1 form the S1 and S2 transmembrane α‐helices.
After the sequence enters the membrane at residue ∼4559 there are two large stretches of the sequence that emerge back into the cytoplasm: the VSC domain and the last 100 residues of the sequence that include the inner branch and the CTD; these are described below. The membrane‐embedded regions will be described in a dedicated section.
S2–S3 loop (VSC domain)
VSC stands for “cytoplasmic subdomain in the voltage‐sensor like domain.”47 This 125‐residue sequence between the S2 and S3 transmembrane α‐helices contributes to the outer branches; it appears to make contact with the EF hands of the neighboring subunit [see Figs. 1(F–I), 2(B,C)].
Inner branches or cytoplasmic portion of S6
These are 25‐Å‐long extensions of the S6 helices running very close to the fourfold axis and forming four long fenestrations between them41, 71 [Figs. 1(J), 2(B,C)]. After the ion gate, these fenestrations likely constitute the next passageway for the Ca2+ ions entering the cytoplasmic space.
C terminal domain (CTD)
The CTDs are bulky domains at the C‐terminal tips of the inner branches [Fig. 1(J)]. The four CTDs contact each other in the closed state forming the “cytoplasmic constriction.”71 Each CTD encompasses a stabilizing zinc‐finger motif at its core and is surrounded, as if it was grasped, by a U‐motif from the same subunit [Figs. 1(I), 2(B,C)].
Binding sites of RyR1 ligands and quaternary interactions
As mentioned earlier, the main effector of RyR is the DHPR, which acts via direct interaction on RyR1, and via an indirect, calcium‐induced mechanism on RyR2. RyR is further regulated by cytoplasmic ligands such as FKBP and CaM and by SR proteins such as triadin and junctin. RyR1 and RyR2 share these and other effectors but it is often the case that the two isoforms do not respond exactly in the same way to the same effector. In addition, several tissues have their specific ligand isoforms. RyRs also self‐associate, forming planar 2D arrays in the SR that fine‐tune the Ca2+ release. A list of biological ligands that have been mapped to the 3D structure of RyR1 is listed below; their positions are illustrated in Figure 3.
Figure 3.
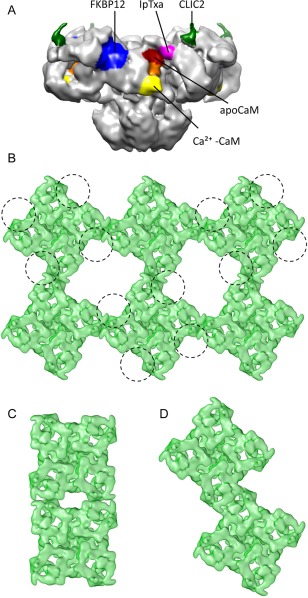
Quaternary interactions of RyR. (A) Side view showing the known binding sites for different RyR1 ligands: FKPB12 (blue), apoCaM (red), Ca2+‐CaM (yellow), approximate region of apo‐ and Ca2+‐CaM overlap (orange), CLIC2 (green) and IpTxa (magenta). (B) Cytoplasmic view showing an array of RyR1s and a tentative footprint of the DHPR tetrads (each DHPR represented with a circumference) interacting with every other RyR1. (C) Side‐by‐side interaction of RyR2. (D) Oblique interaction of RyR2.
FKBP12 is a 12 kDa protein that belongs to the immunophillin family; one FKBP12 binds per RyR1 subunit with nanomolar affinity.72 Removal of FKBP12 results in RyR1 activation and although there is general belief that it also results in subconductance levels of ¼, ½, and ¾ of RyR1's full conductance,73, 74 other studies suggest that conductance remains unaffected.75 Docking of the FKBP12 crystal structure in a cryoEM 3D difference map showed that FKBP12 binds at the interface between domains 9, 5, and 3 (i.e., the interface between the SPRY1, SPRY3 and the handle domains, respectively)76 [binding site shown in Fig. 3(A), and bound FKBP12 shown in Fig. 1(A–F), 2(A,C)]. This was identified as one of the “hinge” regions of RyR1 and suggests that presumably, one of the actions of FKBP12 is to stabilize this hinge.76 FKBP12.6, the cardiac isoform, binds to RyR2 at a similar region, as also determined by cryoEM and 3D difference mapping.77
Calmodulin (CaM) is a 17 kDa Ca2+‐sensing protein with four EF hands, two at the C‐terminus with high affinity for Ca2+ and two at the N‐terminus with lower affinity for Ca2+, separated by a long alpha helix. CaM's shape and specificity change dramatically upon exposure to Ca2+: at very low Ca2+ concentration, apoCaM is hydrophilic, whereas exposure to Ca2+ makes CaM become hydrophobic. In the case of RyR1 this results in a dual effect of CaM: apoCaM activates the channel, whereas Ca2+‐CaM inhibits it,78 thus counteracting slightly the effect that Ca2+ alone has on RyR1 (inhibition at submicromolar Ca2+, maximal activation at ∼50 μM Ca2+). Correspondingly, cryoEM of RyR1 with CaM under high and low Ca2+ conditions revealed two different binding sites.79, 80 Interestingly, these show partial overlap [see Fig. 3(A)] and given that the two modules of CaM have different affinity for Ca2+, it was proposed that CaM acts a mobile Ca2+‐sensing subunit of RyR1, moving with a two‐step mechanism.79 Recent FRET studies support a different position for the N‐terminal lobe of CaM bound to RyR1 under high and low Ca2+ conditions.81 Protection of two proteolytic sites by CaM led to the identification of the CaM binding site82 and subsequent crystallization of CaM with its RyR1 target peptide, residues 3614‐3643. Overall, the crystal structure reveals a standard configuration with CaM wrapped around the target peptide, though with larger separation between the two CaM lobes.83 Although the target sequence is not yet mapped in the near‐atomic structures, the closest sequence is in the handle domain at a position compatible both with apo‐ and Ca2+‐CaM's mapped locations. In the case of RyR2, both apoCaM and Ca2+‐CaM bind to a single position corresponding to the Ca2+‐CaM (inhibitory) site in RyR1,84 which is in agreement with the inhibitory effect of CaM on RyR2 both at high and low Ca2+ concentrations.
CLIC2 belongs to the chloride intracellular channel (CLIC) family and is expressed in skeletal muscle, cardiac muscle and brain. So far, its only known physiological function is an inhibitory action on RyR1 and RyR2.85, 86 In fact, a mutation in CLIC2 has been associated with heart failure, seizures and intellectual disability.87 In addition to the membrane‐embedded form, CLIC2 can also exist in a cytoplasmic, soluble form.88 The atomic structure of the soluble form of this 28 kDa protein revealed a gluthatione S‐transferase fold.88, 89 CryoEM and 3D difference mapping show that CLIC2 binds to RyR1 between domain 5 and domain 6, in the region now defined as P2 or phosphorylation domain85 [see Fig. 3(A)]. Consistent with its role as an effector of RyR, cryoEM also shows that binding of CLIC2 results in a change in conformation of RyR1.
Imperatoxin A is a ∼4 kDa scorpion toxin that binds with nanomolar affinity to RyR1, inducing subconductance states with long mean‐open times.90 Given its sequence and functional homology with a region of the II–III loop of the DHPR, it was proposed that IpTxa might emulate the native DHPR/RyR interaction.91 CryoEM and 3D difference mapping located IpTxa to the crevice region of the helical domain92 [Fig. 3(A)].
DHPR is the main effector of RyR1. In skeletal muscle, excitation–contraction coupling takes places at regions of close apposition between the terminal cisternae of the SR that host two parallel rows of RyR1s, and the T tubules that have distinctive groups of four DHPRs or tetrads. Every other RyR1 interacts with one DHPR tetrad following a quite reproducible geometry93, 94 [Fig. 3(B)]. Numerous functional studies95, 96, 97 suggest the involvement of almost all DHPR's cytoplasmic domains: Cav1.1's II–III loop (which is the most critical), the III‐VI loop and the C‐terminal domain, and the beta subunit, anchored to Cav1.1.98, 99, 100, 101 Electron microscopy and image processing show that the II–III loop, the C‐terminal domain, and the beta subunit are well‐folded domains that face RyR1.62, 102, 103 While the DHPR/RyR interaction is at the core EC coupling, further direct structural study of this interaction has proven elusive so far.
RyRs self‐associate through lateral interactions (inter‐RyR interactions). In skeletal muscle, they form checkerboard arrays through side‐by‐side interactions with an offset30, 104, 105 [Fig. 3(B)]. The interaction was thought to be through domain 6,104 but considering the higher resolution structures, the interacting position may involve more predominantly the helical domains and P1. In heart, RyR2s assemble into clusters of variable number,106, 107 whereby the mode of interaction and the number of RyR2s per cluster is predicted to influence SR Ca2+ release and kinetics.108, 109, 110 We found two modes of interaction that differ from the RyR1 interactions: side‐by‐side without offset [Fig. 3(C)], and oblique, whereby two RyR2s interact along their sides with an offset, their sides form an angle of ∼12°, and the P1 and SPRY1 domains, and perhaps FKBP12.6, are intertwined111 [Fig. 3(D)]. Both the helical domains and the SPRY domains mediating these RyR homologous interactions are known protein‐protein interaction modules.112, 113
The transmembrane domain
When RyR was cloned, the hydropathy profile combined with alpha helical prediction assigned up to 10 transmembrane domains per subunit;64 the most commonly accepted prediction114 is indicated in Figure 4(A). This assignment was remarkably accurate and also predicted an alpha helix that does not cross the membrane, the pore helix. The homology between the selectivity filter of the K+ and RyR channels also lead to predict the selectivity filter of RyR.116 A cryoEM 3D reconstruction of RyR1 in the closed state (10.3 Å resolution for the entire protein and 9.0 Å for the transmembrane domain) yielded the first view of four inner helices forming a helical bundle, in an architecture akin to the K+ channel family, and four inner branches41 [see Fig. 1(J)]. This was followed by the first visualization of the S1–S4, S5, pore helix and S4–S5 linker, and the proposition that the architecture of RyR1 corresponds to that of the super family of 6‐TM voltage‐gated cation channels71 [Figs. 6(B), 7]. This information together with the use of secondary structure prediction programs led to an improved sequence assignment [Fig. 4(B)] and a model of the entire transmembrane domain.115 The cryoEM structures at 4.8 Å and 3.8 Å resolution confirm the 6‐TM architecture; although there is a frame shift of 5 residues spanning the stretch between S3 and S5 between these two sequence assignments [Fig. 4(C,D)]. The model corresponding to the 3.8 Å map, which should provide higher accuracy, is shown in Figure 5. The cytoplasmic ion gate, i.e. narrowest point of the S6 helical bundle, corresponds to Ile 4937. Despite the shared architecture with that of the voltage‐gated ion channels, being intracellular RyR1 is not gated by voltage, and in fact S4 has no positively charged residues in the 3.8 Å resolution model [Fig. 4(D)].
Figure 4.
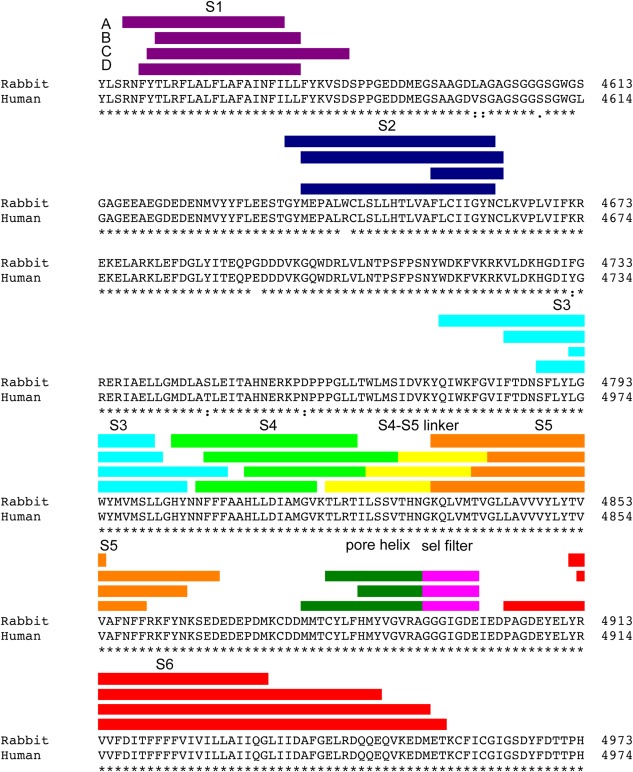
Transmembrane domain sequence assignment. Predictions of the boundaries of the transmembrane segments of RyR1 based on (A) hydropathy plots,114 (B) refined secondary structure prediction,115 (C) modeling based on a 4.8 Å cryoEM map,46 and (D) modeling based on a 3.8 Å resolution cryoEM map.47
Figure 6.
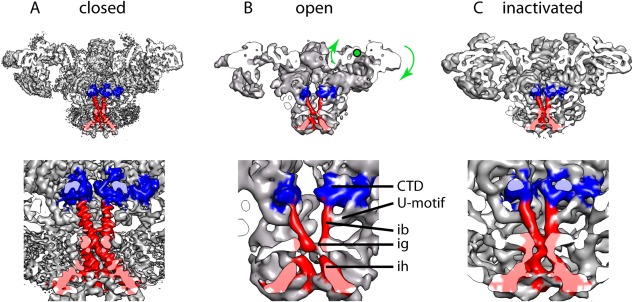
The closed, open and inactivated conformations of RyR1. (A) RyR1 closed at 3.8 Å resolution (cryoEM map EMDB‐2807); map filtered to 7 Å for easier comparison. The extra mass surrounding the transmembrane domain corresponds to the detergent micelle. (B) RyR1 in open conformation at 10‐Å resolution (cryoEM map EMDB‐1607). Green arrows indicate the swivel movement of the rhomboid structure in going form the closed to the open conformation, green dot shows the approximate center of rotation. (C) RyR1 inactivated at 8.5‐Å resolution (cryoEM map EMDB‐2752). The side view is cut along the fourfold axis, the panels below show a magnified image of the regions surrounding the ion gate (ig). The C‐terminal domain is highlighted in blue and S6, formed by the inner helices (ih) and inner branches (ib), is highlighted in red in all four subunits. The CTD and the region of the U‐motif contacting the inner branches are also highlighted.
Figure 5.
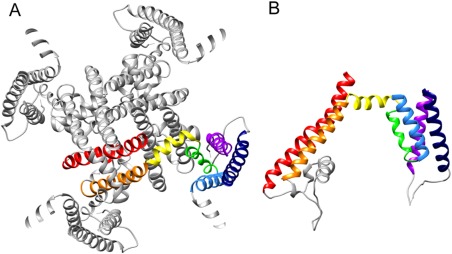
Structure of the transmembrane domain of RyR1. (A) Transmembrane domain seen from the cytoplasm; α‐helices are color‐coded as indicated in Figure 4. (B) Side view of the transmembrane domain for one subunit; cytoplasmic side is above and SR luminal side is below. The atomic model corresponds to 3J8H.pdb.
Until the near‐atomic structure of RyR1 was known, there was a controversy between the first report of closed RyR1 at 10.3‐Å resolution and a second report at 9.6‐Å resolution. Despite being prepared in same closed state conditions, in the second report the inner helices had an open conformation and the inner branches were absent. This controversy was fueled by the different criteria used to determine the resolution (the first study used the 0.143 cutoff of the FSC resolution curve and the second study used the more conservative 0.5 cutoff). Because ∼9 Å resolution are needed to resolve alpha helices,117 ascertaining the “real” resolution was crucial to determine which of the two maps was correct. With more careful analysis, the resolution of the open‐like structure was later amended to 12 Å by the authors;118 one likely explanation is that in some cases data over‐refinement and noise bias can shift the FSC resolution curve yielding better apparent resolution values,119, 120 and may result in incorrect structural interpretation even when using the 0.5 cutoff. When there is no over‐refinement the 0.143 cutoff criterion is appropriate as judged by the observation of features that require the stated resolution; this criterion has been used recently to report the resolution of near‐atomic maps of RyR1.46, 47 Likewise the clear identification of the inner helices and inner branches (which together constitute S6) in the Samso et al. map41 indicates that the reported resolution of 10.3 Å and corresponding interpretation was correct.
A major deviation from canonical 6‐TM channels is that the S6 helices are very long, ∼63 Å, and extend into the cytoplasmic domain forming the inner branches. This is a feature found more rarely in K+ channels, such as it is the case for KcsA.121 In the closed state the transition of S6 from inner helix to inner branch is accompanied by a bend in its mid‐section to accommodate the change in angle from ∼45° tilt to being more perpendicular to the membrane on the cytoplasmic side.
On the luminal side, the first luminal loop (between S1 and S2) and the second luminal loop (between S5 and the pore helix) protrude into the lumen; both have multiple negative charges. The luminal loops organize the quaternary interactions with triadin and junctin, which in turn anchor calsequestrin in the SR lumen.122, 123, 124, 125
Transmission of allosteric changes from the cytoplasmic domains to the ion gate and channel opening
The coordination of more than 40 domains makes RyR the quintessential allosteric machine. The input from the DHPR is probably sensed at the SPRY complex, the helical domains, P2, and perhaps the N‐terminal domains. Inputs from CaM, FKBP and neighboring RyRs are received at SPRY1, SPRY3, handle and helical domains. In a very simplistic account, these signals are transmitted to the central domain and then to the transmembrane domain through two pathways: the U‐motif, which by grasping the CTDs can control directly the inner branches (S6 helices) and the ion gate; and the outer branches/columns that are also in direct contact with the transmembrane domain. Ca2+ concentration sensing at the EF hands, in the stem of the cytoplasmic domain, would be closer in hierarchy towards the transmembrane domain.
The first detailed structure available of RyR1 in an open conformation, with 10.2‐Å resolution, was prepared with 50 μM Ca2+ and 10 μM PCB and using 0.5% CHAPS as the detergent; this yields a Po close to 0.96, and was functionally validated using BLM single channel recording.71 The transitions from the closed to the open state of RyR1 revealed a generalized conformational change. Most domains move away from the fourfold axis: the cytoplasmic vestibule formed by the NTDs relaxes, the 10 Å diameter cytoplasmic constriction formed by the four CTDs becomes unfastened, the inner branches separate, the four S4–S5 linkers move outwards, and the inner helices separate [Figs. 6(A,B), 7(A–F)]. The pore helices were visible and pointing towards the fourfold axis in the open state but they were not resolved in the closed state. By displaying the 3D reconstructions at lower threshold, the ion gate can be seen blocking the pore in the closed state, whereas the narrower point in the open state is the selectivity filter [Fig. 7(G,H)].
Figure 7.
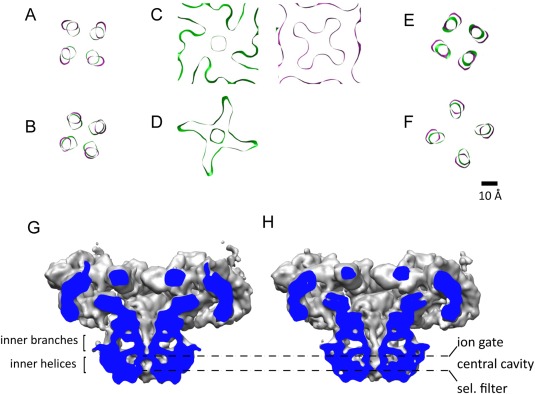
A change in trajectory of S6 opens the channel passage. (A–F) Cross‐sections of S6 in the closed (green, cryoEM map EMDB‐1606) and open (magenta, cryoEM map EMDB‐1607) conformations, in going from the cytoplasm to the SR lumen, perpendicular to the fourfold axis. (A–B) Inner branches (C, D) Ion gate; the flexibility of the inner helices in the open state results in loss of continuity in section (D). (E, F) Inner helices. (G, H) Cross‐section of RyR1 in the closed and open conformations displayed at a lower threshold to reveal the different profiles of the channel passage.
A more intricate description of the conformational change of S6 and the ion gate upon opening is as follows. The ion gate is where the two helical bundles (i.e., the inner branches and the inner helices) meet, and relaxation of the two helical bundles opens the ion gate. On one hand, the inner branches move 6‐Å apart (considering the centers of two helices facing diagonally) [Fig. 7(A–B)]. On the other hand, the inner helices “straighten up” by tilting 5° toward the vertical, and bend outwards when approaching the ion gate [Fig. 7(E,F)]. Just before the ion gate, the density of the inner helices in the open state disappears [Figs. 6(B), 7(D)] implying a flexible region of S6, proposed to be Gly 4934, which accommodates the conformational change. The ion gate, clearly visible in both conformations [Fig. 7(C)], increases its diameter in 4 Å in going from the closed to the open state.71 Noticeably, the cross‐section of the open ion gate shows four openings radiating from the center [Fig. 7(C)]. This appears to result from the juxtaposition of S6 next to the S4–S5 linker, which forms an almost square frame surrounding the inner helices. The pore profile for the open conformation is shown in Figure 7(H).
The large movements of the cytoplasmic domain are more obvious from the side view. A group of domains forming the “rhomboid” structure appears to move as a rigid block. The rhomboid structure is formed by: (a) the NTD, SPRY complex, and P1 domains from one subunit, and (b) the HD2 and P2 domains from the neighboring subunit [Fig. 1(A)]. Upon opening, the four rhomboids swivel, as if they were “prying open” the RyR.71 Thus, while the inner tip of the rhomboid (the NTD) moves away from the transmembrane domain and away from the fourfold axis, the outer tip of the rhomboid moves towards them. The rotation of this rhomboid structure with respect to the fourfold axis is 5° and the net movement is 5 Å away from the fourfold axis (measured at P2) and 8 Å toward the SR (measured at P1),71 see curved arrows in Figure 6(B). Several central core disease/malignant hyperthermia mutations map to the interfaces between the NTDs from each subunit, which hold the rhomboid structures together in the closed state, suggesting that mutations may lower the stability of the cytoplasmic vestibule, and offering an explanation for the “leaky” phenotype of these RyR1 mutants.126 The handle domains undergo little movement, defining two hinges. These form the binding site of effectors: FKBP12 binds to the “left” hinge, and CaM and IpTxa bind to the “right” hinge, suggesting that controlling the hinge regions may be an efficient way to modulate the function of the channel.
During revision of this article, a report of RyR1 in exactly the same open‐state conditions using a DED yielded a 3D reconstruction at 5.7‐Å resolution in the open conformation, and several related conformers.127 The 3D reconstruction of the open state, defined as the conformer that has an open pore, reproduces the conformational changes aforementioned, with very good superimposition of all the domains at the common limiting resolution of 10.2 Å. In the cytoplasmic domain, the “breathing” motion of the helical domains and the NTDs referenced in the article corresponds to the movement of the rhomboid structures described above. The entire trajectory of S6 is visible in the new reconstruction as a continuous helix and, when compared to the closed state, reveals an unchanged conformation for the luminal half of S6, followed by a 15° bend after which there is a dilation of S6 resulting in an increase of pore diameter by 5 Å, as well as a separation of the CTD domains (these two structures together with the VSC are termed “O‐ring in the new study”). By comparison to the closed 3.8‐Å resolution structure of RyR1, the position of the bend is also proposed to be Gly 4934. Complex conformational changes in the central domain are proposed to couple the movements in the cytoplasmic and transmembrane domains. There is no mention to the EF hands in this article.
Also during revision of this article, another report appeared of a 3D reconstruction of RyR1 obtained using a DED and yielding 4.9‐Å resolution (4.2 Å for the core region). This was in a “blocked‐open” state obtained by incubation of the channels in a solution containing 100 μM Ca2+ followed by the addition of 10 μM ruthenium red;128 Amphipol A8‐35 was used instead of detergent. The cytoplasmic domain of RyR1 also adopts the open‐like conformation found in the previous open reconstructions, and structural rearrangements in the central domain appear to be initiated by a conformational change in the EF hands (defined as residues 4071–4132). The study also suggests Gly 4934 as the S6 hinge. In contrast with the previous two studies, the major conformational change of S6 takes place at the luminal half, while the movement of the remainder of S6 is of lesser magnitude, and the S4–S5 linker remains in a closed‐like conformation. In this 3D reconstruction, the selectivity filter becomes very wide, becoming 16.7 Å at the narrowest point. To account for ion selectivity, an electrical field provided by the negative residues at the luminal end of the selectivity filter (Asp 4899 and Glu 4900) is suggested. Nonetheless, given that ruthenium red appears to be lodged into the pore, it is possible that it could artificially widen the selectivity filter, thus conclusions regarding this region should be interpreted carefully in this case. Another novel structural feature of this model is a second short pore helix downstream of the selectivity filter.
A 3D structure of RyR1 at 8.5‐Å resolution obtained in 10 mM Ca2+ was presented as an open state.43 However considering the bell‐shaped curve of RyR1's Po with respect to Ca2+ concentration, whereby RyR1 is maximally activated at ∼50 μM Ca2+ and fully inhibited beyond 1 mM Ca2+,129, 130 the structure must represent a fully inhibited, inactivated state. The structure shows a conformational change that is “half way” of that found from the closed to the open state, with an increase in ion gate diameter of 2 Å and a rocking movement of the clamp domains 5 Å toward the SR membrane [Fig. 6(C)]. The significance of this conformation is unclear since the Ca2+ concentration is above the physiological range experienced by RyR1.
Future prospects
CryoEM combined with single‐particle image reconstruction has been the main driving force pushing the structural discovery of RyR. The first near‐atomic model for 70% of the protein arising from this big effort will enable advancing functional studies such as more precise insertion of FRET probes for dynamic studies, design of cleverer mutations to test function, and having a frameset for data interpretation. On the other hand cryoEM will continue to provide more high resolution data, which will be integrated with the information from X‐ray structures, and provide larger coverage of the structure of RyR under different physiological settings. Comparing these multiple 3D reconstructions will start shedding light on RyR's complex allosterism. In addition, next‐generation cryo electron tomography should provide a picture of RyR in the context of the membrane and its relationship with the DHPR. In summary, it is anticipated that cryoEM will continue to bring great excitement to the RyR field during the next few years.
References
- 1. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL (2010) Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2:a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewarchik CM, Orabi AI, Jin S, Wang D, Muili KA, Shah AU, Eisses JF, Malik A, Bottino R, Jayaraman T, Husain SZ (2014) The ryanodine receptor is expressed in human pancreatic acinar cells and contributes to acinar cell injury. Am J Physiol Gastrointest Liver Physiol 307:G574–5G581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ogawa Y, Kurebayashi N, Murayama T (2000) Putative roles of type 3 ryanodine receptor isoforms (RyR3). Trends Cardiovasc Med 10:65–70. [DOI] [PubMed] [Google Scholar]
- 4. Hosoi E, Nishizaki C, Gallagher KL, Wyre HW, Matsuo Y, Sei Y (2001) Expression of the ryanodine receptor isoforms in immune cells. J Immunol 167:4887–4894. [DOI] [PubMed] [Google Scholar]
- 5. Beam KG, Knudson CM, Powell JA (1986) A lethal mutation in mice eliminates the slow calcium current in skeletal muscle cells. Nature 320:168–170. [DOI] [PubMed] [Google Scholar]
- 6. Schneider MF (1981) Membrane charge movement and depolarization‐contraction coupling. Annu Rev Physiol 43:507–517. [DOI] [PubMed] [Google Scholar]
- 7. Tanabe T, Beam KG, Powell JA, Numa S (1988) Restoration of excitation‐contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature 336:134–139. [DOI] [PubMed] [Google Scholar]
- 8. Rios E, Brum G (1987) Involvement of dihydropyridine receptors in excitation–contraction coupling in skeletal muscle. Nature 325:717–720. [DOI] [PubMed] [Google Scholar]
- 9. Fill M, Ma JJ, Knudson CM, Imagawa T, Campbell KP, Coronado R (1989) Role of the ryanodine receptor of skeletal muscle in excitation–contraction coupling. Ann N Y Acad Sci 560:155–162. [DOI] [PubMed] [Google Scholar]
- 10. Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G (1988) Purification and reconstitution of the calcium release channel from skeletal muscle. Nature 331:315–319. [DOI] [PubMed] [Google Scholar]
- 11. Franzini‐Armstrong C, Nunzi G (1983) Junctional feet and particles in the triads of a fast‐twitch muscle fibre. J Muscle Res Cell Motil 4:233–252. [DOI] [PubMed] [Google Scholar]
- 12. Flucher BE (1992) Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol 154:245–260. [DOI] [PubMed] [Google Scholar]
- 13. Meissner G, Lu X (1995) Dihydropyridine receptor‐ryanodine receptor interactions in skeletal muscle excitation‐contraction coupling. Biosci Rep 15:399–408. [DOI] [PubMed] [Google Scholar]
- 14. Block BA, Imagawa T, Campbell KP, Franzini‐Armstrong C (1988) Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol 107:2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun XH, Protasi F, Takahashi M, Takeshima H, Ferguson DG, Franzini‐Armstrong C (1995) Molecular architecture of membranes involved in excitation‐contraction coupling of cardiac muscle. J Cell Biol 129:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fabiato A (1983) Calcium‐induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol 245:C1–14. [DOI] [PubMed] [Google Scholar]
- 17. Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205. [DOI] [PubMed] [Google Scholar]
- 18. Yang T, Ta TA, Pessah IN, Allen PD (2003) Functional defects in six ryanodine receptor isoform‐1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation‐contraction coupling. J Biol Chem 278:25722–25730. [DOI] [PubMed] [Google Scholar]
- 19. Quane KA, Healy JM, Keating KE, Manning BM, Couch FJ, Palmucci LM, Doriguzzi C, Fagerlund TH, Berg K, Ording H, Bendixen D, Mortier W, Linz U, Muller CR, McCarthy TV. (1993) Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet 5:51–55. [DOI] [PubMed] [Google Scholar]
- 20. Dirksen RT, Avila G (2004) Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys J 87:3193–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 27:977–989. [DOI] [PubMed] [Google Scholar]
- 22. Benkusky NA, Farrell EF, Valdivia HH (2004) Ryanodine receptor channelopathies. Biochem Biophys Res Commun 322:1280–1285. [DOI] [PubMed] [Google Scholar]
- 23. Yang T, Riehl J, Esteve E, Matthaei KI, Goth S, Allen PD, Pessah IN, Lopez JR (2006) Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock‐in mouse. Anesthesiology 105:1164–1175. [DOI] [PubMed] [Google Scholar]
- 24. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino VV, Danieli GA (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103:196–200. [DOI] [PubMed] [Google Scholar]
- 25. Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K (2001) Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103:485–490. [DOI] [PubMed] [Google Scholar]
- 26. George CH, Jundi H, Thomas NL, Fry DL, Lai FA (2007) Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol 42:34–50. [DOI] [PubMed] [Google Scholar]
- 27. Song DW, Lee JG, Youn HS, Eom SH, Kim do H (2011) Ryanodine receptor assembly: a novel systems biology approach to 3D mapping. Prog Biophys Mol Biol 105:145–161. [DOI] [PubMed] [Google Scholar]
- 28. Hernandez‐Ochoa EO, Pratt SJ, Lovering RM, Schneider MF (2015) Critical role of intracellular RyR1 calcium release channels in skeletal muscle function and disease. Front Physiol 6:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Capes EM, Loaiza R, Valdivia HH (2011) Ryanodine receptors. Skelet Muscle 1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferguson DG, Schwartz HW, Franzini‐Armstrong C (1984) Subunit structure of junctional feet in triads of skeletal muscle: a freeze‐drying, rotary‐shadowing study. J Cell Biol 99:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frank J (2009) Single‐particle reconstruction of biological macromolecules in electron microscopy–30 years. Q Rev Biophys 42:139–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng Y, Grigorieff N, Penczek PA, Walz T (2015) A primer to single‐particle cryo‐electron microscopy. Cell 161:438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Skiniotis G, Southworth DR (2016) Single‐particle cryo‐electron microscopy of macromolecular complexes. Microscopy (Oxf) 65:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P (1988) Cryo‐electron microscopy of vitrified specimens. Q Rev Biophys 21:129–228. [DOI] [PubMed] [Google Scholar]
- 35. Cabra V, Samso M (2014) Do's and don'ts of cryo‐electron microscopy: a primer on sample preparation and high quality data collection for macromolecular 3D reconstruction. J Vis Exp 95:e52311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wagenknecht T, Grassucci R, Frank J, Saito A, Inui M, Fleischer S (1989) Three‐dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature 338:167–170. [DOI] [PubMed] [Google Scholar]
- 37. Radermacher M, Wagenknecht T, Grassucci R, Frank J, Inui M, Chadwick C, Fleischer S (1992) Cryo‐EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys J 61:936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serysheva II, Orlova EV, Chiu W, Sherman MB, Hamilton SL, van Heel M (1995) Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat Struct Biol 2:18–24. [DOI] [PubMed] [Google Scholar]
- 39. Orlova EV, Saibil HR (2004) Structure determination of macromolecular assemblies by single‐particle analysis of cryo‐electron micrographs. Curr Opin Struct Biol 14:584–590. [DOI] [PubMed] [Google Scholar]
- 40. Zhu J, Penczek PA, Schroder R, Frank J (1997) Three‐dimensional reconstruction with contrast transfer function correction from energy‐filtered cryoelectron micrographs: procedure and application to the 70S Escherichia coli ribosome. J Struct Biol 118:197–219. [DOI] [PubMed] [Google Scholar]
- 41. Samso M, Wagenknecht T, Allen PD (2005) Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo‐EM. Nat Struct Mol Biol 12:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Angert I, Majorovits E, Schroder RR (2000) Zero‐loss image formation and modified contrast transfer theory in EFTEM. Ultramicroscopy 81:203–222. [DOI] [PubMed] [Google Scholar]
- 43. Efremov RG, Leitner A, Aebersold R, Raunser S (2015) Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517:39–43. [DOI] [PubMed] [Google Scholar]
- 44. Faruqi AR, McMullan G (2011) Electronic detectors for electron microscopy. Q Rev Biophys 44:357–390. [DOI] [PubMed] [Google Scholar]
- 45. Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y (2013) Electron counting and beam‐induced motion correction enable near‐atomic‐resolution single‐particle cryo‐EM. Nat Methods 10:584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR (2015) Structure of a mammalian ryanodine receptor. Nature 517:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T, Peng W, Yin CC, Li X, Scheres SH, Shi Y, Yan N (2015) Structure of the rabbit ryanodine receptor RyR1 at near‐atomic resolution. Nature 517:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tung CC, Lobo PA, Kimlicka L, Van Petegem F (2010) The amino‐terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 468:585–588. [DOI] [PubMed] [Google Scholar]
- 49. Liu Z, Zhang J, Sharma MR, Li P, Chen SR, Wagenknecht T (2001) Three‐dimensional reconstruction of the recombinant type 3 ryanodine receptor and localization of its amino terminus. Proc Natl Acad Sci USA 98:6104–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhong X, Liu Y, Zhu L, Meng X, Wang R, Van Petegem F, Wagenknecht T, Chen SR, Liu Z (2013) Conformational dynamics inside amino‐terminal disease hotspot of ryanodine receptor. Structure 21:2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker ML, Serysheva II, Sencer S, Wu Y, Ludtke SJ, Jiang W, Hamilton SL, Chiu W (2002) The skeletal muscle Ca2+ release channel has an oxidoreductase‐like domain. Proc Natl Acad Sci USA 99:12155–12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Serysheva II, Ludtke SJ, Baker ML, Cong Y, Topf M, Eramian D, Sali A, Hamilton SL, Chiu W (2008) Subnanometer‐resolution electron cryomicroscopy‐based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc Natl Acad Sci USA 105:9610–9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Topf M, Sali A (2005) Combining electron microscopy and comparative protein structure modeling. Curr Opin Struct Biol. 15:578–585. [DOI] [PubMed] [Google Scholar]
- 54. D'Cruz AA, Babon JJ, Norton RS, Nicola NA, Nicholson SE (2013) Structure and function of the SPRY/B30.2 domain proteins involved in innate immunity. Protein Sci 22:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ponting C, Schultz J, Bork P (1997) SPRY domains in ryanodine receptors (Ca(2+)‐release channels). Trends Biochem Sci 22:193–194. [DOI] [PubMed] [Google Scholar]
- 56. Yuchi Z, Yuen SM, Lau K, Underhill AQ, Cornea RL, Fessenden JD, Van Petegem F (2015) Crystal structures of ryanodine receptor SPRY1 and tandem‐repeat domains reveal a critical FKBP12 binding determinant. Nat Commun 6:7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lau K, Van Petegem F (2014) Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat Commun 5:5397. [DOI] [PubMed] [Google Scholar]
- 58. Peralvarez‐Marin A, Tae H, Board PG, Casarotto MG, Dulhunty AF, Samso M (2011) 3D Mapping of the SPRY2 domain of ryanodine receptor 1 by single‐particle cryo‐EM. PLoS One 6:e25813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leong P, MacLennan DH (1998) A 37‐amino acid sequence in the skeletal muscle ryanodine receptor interacts with the cytoplasmic loop between domains II and III in the skeletal muscle dihydropyridine receptor. J Biol Chem 273:7791–7794. [DOI] [PubMed] [Google Scholar]
- 60. Leong P, MacLennan DH (1998) Complex interactions between skeletal muscle ryanodine receptor and dihydropyridine receptor proteins. Biochem Cell Biol 76:681–694. [DOI] [PubMed] [Google Scholar]
- 61. Cui Y, Tae HS, Norris NC, Karunasekara Y, Pouliquin P, Board PG, Dulhuny AF, Casarotto MG (2009) A dihydropyridine receptor alpha1S loop region critical for skeletal muscle contraction is intrinsically unstructured and binds to a SPRY domain of the type 1 ryanodine receptor. Int J Biochem Cell Biol 41:677–686. [DOI] [PubMed] [Google Scholar]
- 62. Szpyt J, Lorenzon N, Perez CF, Norris E, Allen PD, Beam KG, Samso M (2012) Three‐dimensional localization of the alpha and beta subunits and of the II–III loop in the skeletal muscle L‐type Ca2+ channel. J Biol Chem 287:43853–43861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu Z, Zhang J, Wang R, Wayne Chen SR, Wagenknecht T (2004) Location of divergent region 2 on the three‐dimensional structure of cardiac muscle ryanodine receptor/calcium release channel. J Mol Biol 338:533–545. [DOI] [PubMed] [Google Scholar]
- 64. Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, Meissner G, MacLennan DH (1990) Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem 265:2244–2256. [PubMed] [Google Scholar]
- 65. Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, Wagenknecht T, Chen SR (2003) Three‐dimensional localization of divergent region 3 of the ryanodine receptor to the clamp‐shaped structures adjacent to the FKBP binding sites. J Biol Chem 278:14211–14218. [DOI] [PubMed] [Google Scholar]
- 66. Yuchi Z, Lau K, Van Petegem F (2012) Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure 20:1201–1211. [DOI] [PubMed] [Google Scholar]
- 67. Meng X, Xiao B, Cai S, Huang X, Li F, Bolstad J, Trujillo R, Airey J, Chen SR, Wagenknecht T, Liu Z (2007) Three‐dimensional localization of serine 2808, a phosphorylation site in cardiac ryanodine receptor. J Biol Chem 282:25929–25939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Y, Xue S, Zou J, Lopez JR, Yang JJ, Perez CF (2014) Myoplasmic resting Ca2+ regulation by ryanodine receptors is under the control of a novel Ca2+‐binding region of the receptor. Biochem J 460:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fessenden JD, Feng W, Pessah IN, Allen PD (2004) Mutational analysis of putative calcium binding motifs within the skeletal ryanodine receptor isoform, RyR1. J Biol Chem 279:53028–53035. [DOI] [PubMed] [Google Scholar]
- 70. Liu Z, Zhang J, Li P, Chen SR, Wagenknecht T (2002) Three‐dimensional reconstruction of the recombinant type 2 ryanodine receptor and localization of its divergent region 1. J Biol Chem 277:46712–46719. [DOI] [PubMed] [Google Scholar]
- 71. Samso M, Feng W, Pessah IN, Allen PD (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol 7:980–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument‐Bromage H, Tempst P, Marks AR (1992) FK506 binding protein associated with the calcium release channel (ryanodine receptor). J Biol Chem 267:9474–9477. [PubMed] [Google Scholar]
- 73. Ondrias K, Marx SO, Gaburjakova M, Marks AR (1998) FKBP12 modulates gating of the ryanodine receptor/calcium release channel. Ann N Y Acad Sci 853:149–156. [DOI] [PubMed] [Google Scholar]
- 74. Ahern GP, Junankar PR, Dulhunty AF (1997) Subconductance states in single‐channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys J 72:146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mei Y, Xu L, Kramer HF, Tomberlin GH, Townsend C, Meissner G (2013) Stabilization of the skeletal muscle ryanodine receptor ion channel‐FKBP12 complex by the 1,4‐benzothiazepine derivative S107. PLoS One 8:e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Samso M, Shen X, Allen PD (2006) Structural characterization of the RyR1‐FKBP12 interaction. J Mol Biol 356:917–927. [DOI] [PubMed] [Google Scholar]
- 77. Sharma MR, Jeyakumar LH, Fleischer S, Wagenknecht T (2006) Three‐dimensional visualization of FKBP12.6 binding to an open conformation of cardiac ryanodine receptor. Biophys J 90:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tripathy A, Xu L, Mann G, Meissner G (1995) Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys J 69:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Samso M, Wagenknecht T (2002) Apocalmodulin and Ca2+‐calmodulin bind to neighboring locations on the ryanodine receptor. J Biol Chem 277:1349–1353. [DOI] [PubMed] [Google Scholar]
- 80. Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S (1997) Locations of calmodulin and FK506‐binding protein on the three‐dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem 272:32463–32471. [DOI] [PubMed] [Google Scholar]
- 81. Cornea RL, Nitu F, Gruber S, Kohler K, Satzer M, Thomas DD, Fruen BR (2009) FRET‐based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc Natl Acad Sci USA 106:6128–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moore CP, Rodney G, Zhang JZ, Santacruz‐Toloza L, Strasburg G, Hamilton SL (1999) Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry 38:8532–8537. [DOI] [PubMed] [Google Scholar]
- 83. Maximciuc AA, Putkey JA, Shamoo Y, Mackenzie KR (2006) Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure 14:1547–1556. [DOI] [PubMed] [Google Scholar]
- 84. Huang X, Fruen B, Farrington DT, Wagenknecht T, Liu Z (2012) Calmodulin‐binding locations on the skeletal and cardiac ryanodine receptors. J Biol Chem 287:30328–30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Meng X, Wang G, Viero C, Wang Q, Mi W, Su XD, Wagenknecht T, Williams AJ, Liu Z, Yin CC (2009) CLIC2‐RyR1 interaction and structural characterization by cryo‐electron microscopy. J Mol Biol 387:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dulhunty AF, Pouliquin P, Coggan M, Gage PW, Board PG (2005) A recently identified member of the glutathione transferase structural family modifies cardiac RyR2 substate activity, coupled gating and activation by Ca2+ and ATP. Biochem J 390:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Takano K, Liu D, Tarpey P, Gallant E, Lam A, Witham S, Alexov E, Chaubey A, Stevenson RE, Schwartz CE, Board PG, Dulhunty AF (2012) An X‐linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum Mol Genet 21:4497–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cromer BA, Gorman MA, Hansen G, Adams JJ, Coggan M, Littler DR, Brown LJ, Mazzanti M, Breit SN, Curmi PM, Dulhunty AF, Board PG, Parker MW (2007) Structure of the Janus protein human CLIC2. J Mol Biol 374:719–731. [DOI] [PubMed] [Google Scholar]
- 89. Mi W, Liang YH, Li L, Su XD (2008) The crystal structure of human chloride intracellular channel protein 2: a disulfide bond with functional implications. Proteins 71:509–513. [DOI] [PubMed] [Google Scholar]
- 90. Tripathy A, Resch W, Xu L, Valdivia HH, Meissner G (1998) Imperatoxin A induces subconductance states in Ca2+ release channels (ryanodine receptors) of cardiac and skeletal muscle. J Gen Physiol 111:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gurrola GB, Arevalo C, Sreekumar R, Lokuta AJ, Walker JW, Valdivia HH (1999) Activation of ryanodine receptors by imperatoxin A and a peptide segment of the II‐III loop of the dihydropyridine receptor. J Biol Chem 274:7879–7886. [DOI] [PubMed] [Google Scholar]
- 92. Samso M, Trujillo R, Gurrola GB, Valdivia HH, Wagenknecht T (1999) Three‐dimensional location of the imperatoxin A binding site on the ryanodine receptor. J Cell Biol 146:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Paolini C, Protasi F, Franzini‐Armstrong C (2004) The relative position of RyR feet and DHPR tetrads in skeletal muscle. J Mol Biol 342:145–153. [DOI] [PubMed] [Google Scholar]
- 94. Protasi F, Paolini C, Nakai J, Beam KG, Franzini‐Armstrong C, Allen PD (2002) Multiple regions of RyR1 mediate functional and structural interactions with alpha(1S)‐dihydropyridine receptors in skeletal muscle. Biophys J 83:3230–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Papadopoulos S, Leuranguer V, Bannister RA, Beam KG (2004) Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein‐yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J Biol Chem 279:44046–44056. [DOI] [PubMed] [Google Scholar]
- 96. Polster A, Ohrtman JD, Beam KG, Papadopoulos S (2012) Fluorescence resonance energy transfer (FRET) indicates that association with the type I ryanodine receptor (RyR1) causes reorientation of multiple cytoplasmic domains of the dihydropyridine receptor (DHPR) alpha(1S) subunit. J Biol Chem 287:41560–41568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Carbonneau L, Bhattacharya D, Sheridan DC, Coronado R (2005) Multiple loops of the dihydropyridine receptor pore subunit are required for full‐scale excitation‐contraction coupling in skeletal muscle. Biophys J 89:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rebbeck RT, Karunasekara Y, Board PG, Beard NA, Casarotto MG, Dulhunty AF (2014) Skeletal muscle excitation‐contraction coupling: who are the dancing partners? Int J Biochem Cell Biol 48:28–38. [DOI] [PubMed] [Google Scholar]
- 99. Opatowsky Y, Chen CC, Campbell KP, Hirsch JA (2004) Structural analysis of the voltage‐dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42:387–399. [DOI] [PubMed] [Google Scholar]
- 100. Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J (2004) Structural basis of the alpha1‐beta subunit interaction of voltage‐gated Ca2+ channels. Nature 429:675–680. [DOI] [PubMed] [Google Scholar]
- 101. Van Petegem F, Clark KA, Chatelain FC, Minor DL, Jr. (2004) Structure of a complex between a voltage‐gated calcium channel beta‐subunit and an alpha‐subunit domain. Nature 429:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N (2003) Visualization of the domain structure of an L‐type Ca2+ channel using electron cryo‐microscopy. J Mol Biol 332:171–182. [DOI] [PubMed] [Google Scholar]
- 103. Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M, Yan N (2015) Structure of the voltage‐gated calcium channel Cav1.1 complex. Science 350:aad2395. [DOI] [PubMed] [Google Scholar]
- 104. Yin CC, Blayney LM, Lai FA (2005) Physical coupling between ryanodine receptor‐calcium release channels. J Mol Biol 349:538–546. [DOI] [PubMed] [Google Scholar]
- 105. Yin CC, Lai FA (2000) Intrinsic lattice formation by the ryanodine receptor calcium‐release channel. Nat Cell Biol 2:669–671. [DOI] [PubMed] [Google Scholar]
- 106. Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C (2009) Optical single‐channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci USA 106:22275–22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hou Y, Jayasinghe I, Crossman DJ, Baddeley D, Soeller C (2015) Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes. J Mol Cell Cardiol 80:45–55. [DOI] [PubMed] [Google Scholar]
- 108. Walker MA, Williams GS, Kohl T, Lehnart SE, Jafri MS, Greenstein JL, Lederer WJ, Winslow RL (2014) Superresolution modeling of calcium release in the heart. Biophys J 107:3018–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cannell MB, Kong CH, Imtiaz MS, Laver DR (2013) Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J 104:2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS (2002) Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium‐induced calcium release. Biophys J 83:59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cabra V, Murayama T, Samso M (2016) Ultrastructural analysis of self‐associated RyR2s. Biophys J 110:2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tewari R, Bailes E, Bunting KA, Coates JC (2010) Armadillo‐repeat protein functions: questions for little creatures. Trends Cell Biol 20:470–481. [DOI] [PubMed] [Google Scholar]
- 113. Kuang Z, Yao S, Xu Y, Lewis RS, Low A, Masters SL, Willson TA, Kolesnik TB, Nicholson SE, Garrett TJ, Norton RS (2009) SPRY domain‐containing SOCS box protein 2: crystal structure and residues critical for protein binding. J Mol Biol 386:662–674. [DOI] [PubMed] [Google Scholar]
- 114. Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH (2002) Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc Natl Acad Sci USA 99:16725–16730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ramachandran S, Chakraborty A, Xu L, Mei Y, Samso M, Dokholyan NV, Meissner G (2013) Structural determinants of skeletal muscle ryanodine receptor gating. J Biol Chem 288:6154–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gao L, Balshaw D, Xu L, Tripathy A, Xin C, Meissner G (2000) Evidence for a role of the lumenal M3‐M4 loop in skeletal muscle Ca(2+) release channel (ryanodine receptor) activity and conductance. Biophys J 79:828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Fujiyoshi Y (1998) The structural study of membrane proteins by electron crystallography. Adv Biophys 35:25–80. [DOI] [PubMed] [Google Scholar]
- 118. Ludtke SJ, Serysheva, II (2013) Single‐particle cryo‐EM of calcium release channels: structural validation. Curr Opin Struct Biol 23:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Stewart A, Grigorieff N (2004) Noise bias in the refinement of structures derived from single particles. Ultramicroscopy 102:67–84. [DOI] [PubMed] [Google Scholar]
- 120. Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R (2013) High‐resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cortes DM, Cuello LG, Perozo E (2001) Molecular architecture of full‐length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen Physiol 117:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR (1997) Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 272:23389–23397. [DOI] [PubMed] [Google Scholar]
- 123. Wei L, Gallant EM, Dulhunty AF, Beard NA (2009) Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int J Biochem Cell Biol 41:2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Boncompagni S, Thomas M, Lopez JR, Allen PD, Yuan Q, Kranias EG, Franzini‐Armstrong C, Perez CF (2012) Triadin/Junctin double null mouse reveals a differential role for Triadin and Junctin in anchoring CASQ to the jSR and regulating Ca(2+) homeostasis. PLoS One 7:e39962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lee JM, Rho SH, Shin DW, Cho C, Park WJ, Eom SH, Ma J, Kim DH (2004) Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J Biol Chem 279:6994–7000. [DOI] [PubMed] [Google Scholar]
- 126. Kimlicka L, Lau K, Tung CC, Van Petegem F (2013) Disease mutations in the ryanodine receptor N‐terminal region couple to a mobile intersubunit interface. Nat Commun 4:1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bai XC, Yan Z, Wu J, Li Z, Yan N (2016) The central domain of RyR1 is the transducer for long‐range allosteric gating of channel opening. Cell Res 26:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wei R, Wang X, Zhang Y, Mukherjee S, Zhang L, Chen Q, Huang X, Jing S, Liu C, Li S, Wang G, Xu Y, Zhu S, Williams AJ, Sun F, Yin CC (2016) Structural insights into Ca(2+)‐activated long‐range allosteric channel gating of RyR1. Cell Res 26:977–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gomez AC, Yamaguchi N (2014) Two regions of the ryanodine receptor calcium channel are involved in Ca(2+)‐dependent inactivation. Biochemistry 53:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Meissner G (2002) Regulation of mammalian ryanodine receptors. Front Biosci 7:d2072–20d2080. [DOI] [PubMed] [Google Scholar]