

Abstract
With the ability to resolve structures of macromolecules at atomic resolution, X‐ray crystallography has been the most powerful tool in modern structural biology. At the same time, recent technical improvements have triggered a resolution revolution in the single particle cryo‐EM method. While the two methods are different in many respects, from sample preparation to structure determination, they both have the power to solve macromolecular structures at atomic resolution. It is important to understand the unique advantages and caveats of the two methods in solving structures and to appreciate the complementary nature of the two methods in structural biology. In this review we provide some examples, and discuss how X‐ray crystallography and cryo‐EM can be combined in deciphering structures of macromolecules for our full understanding of their biological mechanisms.
Keywords: cryo‐EM, X‐ray crystallography, structural biology, structure determination methods, protein structure determination, X‐ray and electron scattering
Abbreviations
- cryo‐EM
cryo‐electron microscopy
- microED
micro‐electron diffraction
- NCS
non‐crystallographic symmetry
- RdRP
RNA‐dependent RNA polymerase
Introduction
Since its birth in 1950s, structural biology has revolutionized our understanding of life by revealing the shape and structure of biological molecules in great detail.1 Toward the aim to determine the spatial relationship of the hundreds of thousands of atoms, and the change of their relative locations within a biological macromolecule, multiple methodologies with very different physical principles were implemented and exploited in the past half century. These include X‐ray crystallography, NMR spectroscopy, cryo‐electron microscopy (cryo‐EM), X‐ray solution scattering, neutron diffraction, and various spectroscopic techniques. Among these, X‐ray crystallography has played a dominating role in solving molecule structures at atomic resolution. In recent years, cryo‐EM has experienced dramatic technical advancement and is starting to become instrumental in high‐resolution structure determination of macromolecular complexes, especially supra‐assemblies.2 The past few years have seen a tendency of contrasting X‐ray crystallography and cryo‐EM as being mutually exclusive techniques. In this review, we would like to emphasize the complementary nature of the two methods in structural biology.
Principles of X‐Ray Crystallography and Cryo‐EM
X‐ray crystallography's foundation principle lies in Bragg's Law of X‐ray diffraction by crystals, i.e. by well‐ordered packing of homogenous molecules in three‐dimension (3D). Illuminated by a beam of X‐ray light, the crystal can diffract the light at various angles, some of which have stronger intensity than others [Fig. 1(A)]. This kind of intensity variation at different angles can be recorded on media as a “diffraction pattern”. The diffraction pattern, normally appearing as a series of sharp spots, reflects the structural arrangement of atoms within the crystal and therefore can be used to deduce the original structure of the crystal using Bragg's Law. In order to solve the structure, besides the measured intensities of the diffraction spots, additional information called phases of the spots is required. The phase information is normally obtained by other experimental or computational means such as SIR/MIR, SAD/MAD, SIRAS/MIRAS, or molecular replacement.3 The intensity and phase information of multiple diffraction patterns of the crystal can then be reconstructed and Fourier transformed in a computer to generate a virtual structure for interpretation. The quality of the structure heavily depends on the sharpness of the diffraction spots, which in turn is determined by the degree of order of the crystal. Therefore, modern X‐ray crystallography's essential step is to obtain highly‐ordered three‐dimensional crystals. In order to achieve this, a large amount of highly purified macromolecules may be necessary when screening a large number of crystallization conditions. Also engineering of the molecules, e.g. stability promotion, side‐chain modification, proteolysis, may be important to improve the crystal quality.4 Until now, most of the atomic resolution macromolecular structures have been solved by X‐ray crystallography.
Figure 1.
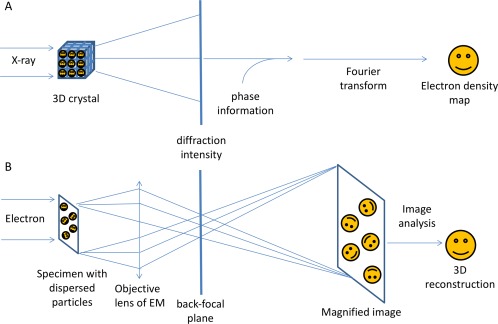
Technical difference between X‐ray crystallography and single particle cryo‐EM. A: illustrates the physics and mathematical principles of X‐ray crystallography to solve a structure. Periodic arrays of molecules in a regular three‐dimensional lattice resulted in a “diffraction pattern” when illuminated by X‐rays. Because of the lack of focus lens for X‐rays, only intensities can be recorded, which resulted in the phase problem in X‐ray crystallography. Part (B) illustrates the physics and mathematical principles of EM in solving a structure, because of an electron's strong interaction with each atom's Coulomb potential, individual molecules within a specimen can be imaged directly. The virtue density of a biological molecule can be reconstructed from a set of 2D projections of the molecule with common structure, imaged at various orientations by the electron microscope. In the back‐focal plane of the objective lens, the diffraction pattern of a biological specimen can also be obtained.
Cryo‐EM uses high energy electrons as source to illuminate very thin specimens in a transmission electron microscope and takes advantage of the negatively charged electron's trajectory bending by magnetic field to focus the electron beam passing through the specimen. Cryo‐EM therefore uses a magnetic objective lens to produce both the diffraction pattern of a specimen at the back‐focal plane and the magnified image in the image plane [Fig. 1(B)]. Because of an electron's strong interaction with each atom's Coulomb potential, individual molecules within a specimen can be imaged and the magnified image already includes the full structural information of the molecule. Therefore, cryo‐EM can be used to examine non‐crystalline structure. As a matter of fact, the most recent trend of cryo‐EM in structural biology is to solve near‐atomic resolution structures from isolated macromolecules, i.e. single particles, directly from their images. From hundreds of thousands of single particle cryo‐EM images of one type of macromolecule, we can perform statistical analysis to classify, align, and average the images to finally reconstruct the common features among all the single particle images into a 3D virtual structure (normally called 3D cryo‐EM map) in a computer. How fine the structure can be defined (resolution of the structure) depends on various factors such as the microscopy performance (coherence of illumination, detector quantum efficiency), the similarity among the individual images (homogeneity), the distribution of molecule orientations in the specimen, the number of images, the robustness of the statistical analysis, etc. Because cryo‐EM reconstructs the 3D structure directly from a lot of 2D projections, depending on the limits imposed by instrumental conditions, it can provide structural information at different resolution levels from ∼3 Å to ∼3 nm. In contrast to X‐ray crystallography, which needs large amount of materials to optimize the crystallization condition, cryo‐EM needs much smaller macromolecule samples to work with and normally does not need rigorous molecular engineering. More importantly, cryo‐EM specimen is made by fast freezing biological samples in liquid nitrogen temperature directly from the solution, therefore maintaining the macromolecules in their soluble states in comparison with a state in the crystal packing constraint. This lends cryo‐EM the advantage to reveal structures in more close‐to‐native state than X‐ray crystallography but also the difficulty of dealing with intrinsic structural heterogeneity due to the thermal fluctuation.
Nowadays, crystallographic and cryo‐EM data may complement each other for structural determination in several ways, among which two major ways are most widely used. In one way, a low‐resolution cryo‐EM map of an entire molecule provides the overall shape of the molecule, whose sub‐components or their homologues may be solved by X‐ray crystallography or other methods. The crystallographic atomic models of the components can be docked into the cryo‐EM map for interpretation of the entire molecule. In the other way, a macromolecule can be crystallized into well‐ordered crystals which can diffract to high resolution. The cryo‐EM reconstruction of the macromolecule at a reasonable resolution may serve as an initial model to solve the phasing problem of the crystals for high resolution structural determination. The two schemes are discussed in the following sections.
Docking of X‐Ray Crystallographic Structures Within Cryo‐EM Maps
In the past decade, the most widely accepted combinatorial approach of X‐ray crystallography and cryo‐EM technique has been the practice of docking X‐ray crystallographic atomic models into a cryo‐EM map at low resolution.5 This practice started in 1990s when crystal structures were attempted to be docked into low‐resolution cryo‐EM maps of icosahedral viruses or cytoskeletal helical filaments.6 In the past two decades, several dozen docking software packages have been developed but only a few are still being actively developed today. The docking methods can be classified mainly into two categories: rigid‐body docking and flexible docking. Rigid‐body docking is used to find the optimal orientation and position of sub‐component atomic crystallographic structures in the cryo‐EM map. The most popular docking software packages include Situs,7 EMfit,8 and UCSF Chimera docking utility.9 In cases where some minor conformational differences exist between the crystal structure of an individual component and its counterpart in the cryo‐EM map, flexible docking algorithms, such as normal mode analysis and molecular dynamic simulation, are used by introducing conformational changes to the X‐ray structure to fit into the cryo‐EM map while maintaining the stereo‐chemistry of the atomic models. Some flexible docking software packages include Situs, Flex‐EM,10 MDFF,11 iMODFIT12 and more recently Rosetta.13
The docking can be quite useful in defining the protein location and protein–protein interface within a complex and new interaction modes that are not revealed by X‐ray crystallography. If the cryo‐EM density is of good enough quality, the docking can be quite precise even at lower resolution. In a recent example, the crystal structure of Ryanodine receptor 1's SPRY2 domain (∼50 kDa) was docked as a rigid body into a 10 Å resolution cryo‐EM map of the entire complex (∼1.5 MDa) (Fig. 2).14 The best scored result by the Colores program of Situs turned out to define the domain's location and orientation quite precisely to agree with the same domain in the high resolution structure of the complex in a 3.8 Å structure solved more recently by single particle cryo‐EM.15 The RMSD between the Cα atoms of the docked SPRY2 model and the high resolution atomic model of the complex was only 2.1 Angstrom. This underscores both the quality of the low‐resolution cryo‐EM map and the robustness of the docking algorithm.
Figure 2.
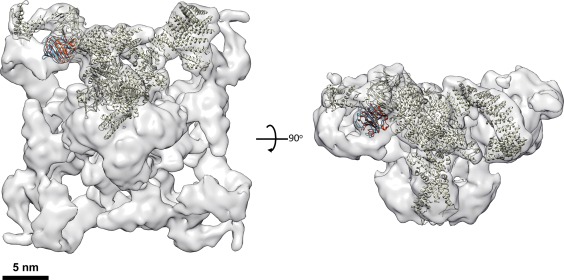
Docking of atomic models into the 3D cryo‐EM map of Ryanodine receptor 1 complex at 10 Angstrom resolution. The 3D EM map of the Ryanodine receptor 1 (EMD‐5041) is shown in semi‐transparent rendering in two orthogonal views. The atomic model of a ryanodine receptor monomer (grey) is docked in the map. The atomic model of SPRY2 domain docked in the map with rigid docking algorithm is shown in red color. Its equivalent domain in the atomic model of the full‐length protein is shown in blue color. The atomic models of the two SPRY2 domain are in very similar orientation and location.
In our study, elucidating the architecture of yeast RNA exosome complex, docking of atomic models into low‐resolution EM 3D reconstruction maps was critical for us to uncover new mechanistic insights of the complex's function. We solved a 3D reconstruction of the yeast exosome complex comprising ten subunits using single particle EM at about 18 Angstrom resolution, in which we were able to dock homologous atomic models of the nine‐subunit human core complex and bacterial RNase III protein determined by X‐ray crystallography.16 This allowed us to reveal the mode of interaction between the Rrp44 subunit and the core complex. In a later study, we docked the crystal structure of RNA‐bound yeast exosome 10‐mer17 into a 3D EM reconstruction of the complex in conformations induced by different lengths of RNA substrates with high fidelity.18 Alternatively, we revealed a different conformation of the exosome bound by RNAs with short 3′‐ss tails and built atomic models by docking available crystal structures of the core complex and Rrp44 separately in the 3D EM reconstruction. The two docking results allowed us to reveal distinct pathways of RNA substrate recruitment and processing within the complex. Interestingly, the most recent near‐atomic‐resolution structure of the apo‐exosome complex (PDBID: 5G06)19 verified the interfaces between the subunits calculated from the previous docking models in the low‐resolution cryo‐EM map of the apo‐exosome complex.
Solving the Phases of X‐Ray Crystallographic Diffraction Data Using Cryo‐EM Maps
Because cryo‐EM uses images to analyze the structure, the cryo‐EM reconstruction includes phase information of the structural factors in Fourier space. In contrast, X‐ray crystallographic diffraction data provide precise amplitude information but lack phases. As cryo‐EM reconstruction approaches higher resolution, sufficient resolution overlap between the cryo‐EM and crystallographic data of the same or homologous macromolecule may lead to successful crystal structure determination. In this approach, the low‐resolution cryo‐EM reconstruction is used as the initial phasing model.20 In phasing the high‐resolution X‐ray data with low‐resolution EM reconstruction by molecular replacement, once the search EM map has been positioned, theoretical phases can be calculated up to the EM reconstruction resolution. This has been used in determining the structures of some large complexes such as viruses, ribosomes, and others.21, 22 A few years ago, in order to generate enough overlap between the low resolution of cryo‐EM map and the crystallographic diffraction data, a greater camera length or a smaller beam blocker had to be used to collect low frequency signals.20
The current progress of single particle cryo‐EM reconstruction allows reconstruction of relatively small molecules at sub‐nanometer resolution.23 This brings the opportunity to solve the phasing problem of regular crystallographic diffraction data from molecules with much smaller size than before. We have applied this practice to a 280 kDa complex that is composed of the ecto,‐LRR domain of the Toll‐like receptor 13 (TLR13) and its RNA substrate.24 TLR13 recognizes a stretch of conserved nucleotides from bacterial 23S ribosomal RNA with stringent specificity to trigger immune response. The TLR13 ecto‐LRR domain in complex with a 13‐nucleotide RNA oligomer (ssRNA13) could be crystallized and the crystals diffracted X‐rays to 2.3 Å. Unfortunately, the phasing problem could not be solved by molecular replacement with homologue structures, heavy atom derivative preparations, or even selenium substitution. We therefore performed single particle cryo‐EM reconstruction and obtained the structure at 4.8 Å resolution of TLR13 bound with a 25‐nucleotide RNA oligomer (ssRNA25) [Fig. 3(A)]. Using this medium‐resolution reconstruction, we successfully solved the phases of the TLR13‐ssRNA13 X‐ray diffraction data to 2.3 Å resolution [Fig. 3(B)].24
Figure 3.
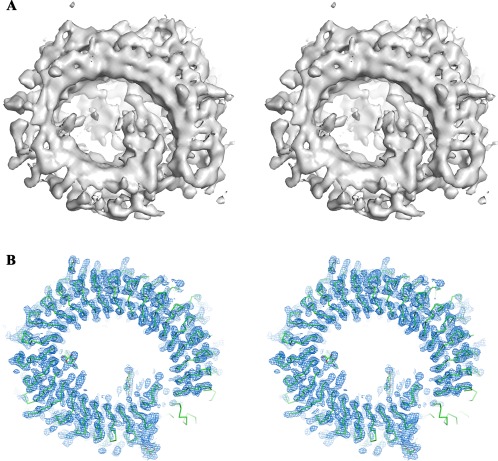
Cryo‐EM 3D reconstruction helps phasing in X‐ray crystallography. A: is the single particle cryo‐EM 3D reconstruction of the TLR13‐ssRNA at 4.8 Å resolution. Part (B) is the electron density map (blue mesh) of the TLR13‐ssRNA after phase extension from the initial cryo‐EM map to X‐ray crystallographic data at 2.3 Angstrom resolution. The main chain of the atomic model of TLR13 protein in the density is shown in green sticks. A few glycosylated sites with the sugar molecules are shown in stick model. Both (A) and (B) are shown in stereo pairs.
Phase extension to higher resolution provided by the X‐ray diffraction data was usually achieved by iterated density‐modification procedures, especially non‐crystallographic symmetry (NCS) map averaging. This strategy has been successfully applied to the study of macromolecular particles with a high‐degree of internal symmetry, e.g. virus crystallography, which results in a good‐quality electron density map for interpretation even at a resolution of around 3 Å because of the power of the NCS averaging. However, in the case of TLR13, there are only two copies of TLR13 molecules in the asymmetric unit of the crystal, which degrades the power of the usual phase extension method. To solve this problem, we have developed an improved method for phasing crystal structures with no or low non‐crystallographic symmetry using cryo‐EM data, which iterates the phase improvements through the combination of a reciprocal‐space bias‐removal method and a real‐space fragment extraction method.25 Additional benefits would be available with the cryo‐EM phasing X‐ray method if the Bijvoet differences were measured for some weak anomalous scattering atoms, which could help determine the correct absolute hand of the cryo‐EM map.26
The fast‐developing micro‐electron diffraction (microED) technique has shown the power to collect high quality diffraction data from micro crystals using an electron beam to solve protein or polypeptide structures at high resolution.27 The phasing problem of microED is currently solved mainly by molecular replacement from available homologous structures,28 or directly from intensities by so‐called direct methods if the data are measured to atomic resolution. It is predictable in the future to use single particle cryo‐EM reconstructions as initial models to solve the phasing problem of the microED data.
Cryo‐EM and X‐Ray Crystallographic Data Complement Each Other in Deciphering Structures
The most recent technical breakthroughs in cryo‐EM have resulted in a major leap in solving structure of macromolecules at high resolution. Because cryo‐EM does not need crystallization of the target molecules, many molecules, especially the super‐complexes that are either very hard to produce in large quantity or almost impossible to crystallize, are now possible to be determined at reasonably high resolution. A major hurdle for single particle reconstruction at present is the presence of conformational heterogeneity in the sample because of the flexible nature of the molecule. Therefore, most of the single particle reconstructions suffer from uneven distribution of resolutions within the 3D EM maps, with the molecular core region normally having the highest resolution whereas the molecule periphery being worse resolved. Algorithms dealing with heterogeneity by stringent classification and local masking have been developed but are still not powerful enough to solve all the flexible domains in atomic detail.29 In contrast, crystallization of molecules or their partial components, such as a subunit or a domain, locks the molecules in the highly ordered crystal lattice and thus enforces them to stay in a rigid conformation. For instance, the single particle cryo‐EM reconstruction of influenza RNA‐dependent RNA polymerase (RdRP)30 revealed the near atomic model of the PB1 subunit and C‐terminal domain of PA subunit but could not solve the structure of the N‐terminal domain of the PA subunit due to its flexibility in the tetrameric single particle complex [Fig. 4(A)]. The same N‐terminal domain of the PA subunit, however, is well characterized in the crystal structure of a monomeric complex of the influenza RdRP.31 It is clear that the lattice packing in the crystal caused the N‐terminal domain of PA subunit in one RdRP to interact with the PB1 domain of an adjacent RdRP, thus stabilizing the complex in a rigid conformation [Fig. 4(B)]. Therefore, crystallography has the advantage to freeze macromolecules in ordered states. This works even better for well‐defined domains that are normally easy to be crystallized and solved by crystallography possibly with the initial phases provided by the corresponding part of cryo‐EM map.
Figure 4.
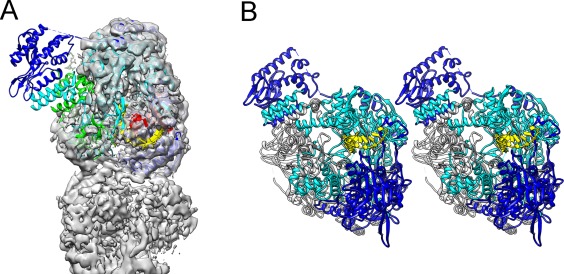
Domain structure locked in rigid state by crystal packing. Part (A) is a partial map from the single particle cryo‐EM 3D reconstruction of the influenza RdRP tetramer at 4.3 Å resolution in semi‐transparent rendering, with the crystal structure of an RdRP monomer docked in the map. The N‐terminal domain of PA subunit is not resolved in the 3D map so the atomic model protruding out from the map. Part (B) shows two adjacent RdRP monomers packed in the crystal lattice for X‐ray crystallography. The N‐terminal domain of PA subunit is in close contact with the PB subunit of the adjacent monomer. In both (A) and (B), PA subunit is in blue color, PB subunit is in cyan color, and the RNA is in yellow color.
Because macromolecules may be prepared under different biochemical conditions for cryo‐EM analysis or crystallization, the corresponding structures solved thus can represent different biological states, providing a more complete structural picture of the molecules. The crystal structure of a macromolecule can serve as a good starting model to fit into heterogeneous conformations solved by cryo‐EM. Cryo‐EM reconstructions can in turn provide important guidance to engineer the macromolecule for crystallization at high order. Currently, X‐ray crystallography is the major tool in elucidating the detail mechanisms at chemistry level such as the enzymatic catalytic reaction or ion transportation. Summarizing the advantages of both cryo‐EM and X‐ray crystallography methods, a future structural biology study of a macromolecular complex may be a scenario combining the two technologies along the following lines. First, the structure of the whole complex as an entity is solved using single particle cryo‐EM at near atomic resolution with the overall architecture as well as subunit and domain boundaries defined. Some peripheral portion or flexible part of the complex may not be defined well but have a rough shape and location in the molecule. The cryo‐EM map and architecture can guide purification and crystallization of sub‐complexes or domains that can be solved at atomic resolution by crystallography. The atomic models can then be fit back to the cryo‐EM map to envision more details of the entire complex.
As cryo‐EM is approaching higher and higher resolution, will X‐ray crystallography finally be replaced by cryo‐EM to reveal the fine structure of biological molecules? Our discussions in the previous sections already argue against this statement. More importantly, we should keep in mind that X‐rays and electron beams interact with substances in different ways. X‐ray photons interact with the electrons around an atom while a high energy electron beam deviates by the Coulomb potential around an atom. The mathematical function describing a structure solved by X‐ray crystallography reflects the electron cloud distribution within the molecule and is called “electron density”. In contrast, the structural mathematical function solved by cryo‐EM reflects the Coulomb potential distribution within the molecule and is called “Coulomb potential density”. The two types of density represent different physical properties of a molecule. At a resolution below 2 Å, the electron density and Coulomb potential density of a molecule have very similar shape and features. This might be the reason that “electron density” was misused in some literatures describing a 3D cryo‐EM reconstruction. When the resolution is higher than 1 Å, the electron density of a molecule will appear different from its Coulomb potential. In that range, the X‐ray and electron will give distinct outcomes to a molecule's structure reflecting different physical properties. This is underlined by the most recent work of ultra‐high resolution X‐ray crystallography of an iron–sulfur protein, in which the electron orbitals can be clearly identified by X‐ray.32 We speculate that structural biology will move in at least two directions. In one direction, the structures can be solved with high precision in larger scale due to the fast development of cryo‐EM.33 In the other direction, the structures can be solved at ultra‐high resolution by both cryo‐EM and X‐ray diffraction, therefore revealing more in‐depth chemical and physical mechanisms behind the principle of life.34
References
- 1. Shi Y (2014) A glimpse of structural biology through X‐ray crystallography. Cell 159:995–1014. [DOI] [PubMed] [Google Scholar]
- 2. Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 3. Taylor GL (2010) Introduction to phasing. Acta Cryst D66:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newman J (2006) A review of techniques for maximizing diffraction from a protein crystal in stilla. Acta Cryst D62:27–31. [DOI] [PubMed] [Google Scholar]
- 5. Rossmann MG (2000) Fitting atomic models into electron‐microscopy maps. Acta Cryst D56:1341–1349. [DOI] [PubMed] [Google Scholar]
- 6. Baker TS, Johnson JE (1996) Low resolution meets high: towards a resolution continuum from cells to atoms. Curr Opin Struct Biol 6:585–594. [DOI] [PubMed] [Google Scholar]
- 7. Wriggers W, Milligan RA, Mccammon JA (1999) Situs: A package for docking crystal structures into low‐resolution maps from electron microscopy. J Struct Biol 125:185–195. [DOI] [PubMed] [Google Scholar]
- 8. Rossmann MG, Bernal R, Pletnev SV (2001) Combining electron microscopic with X‐ray crystallographic structures. J Struct Biol 136:190–200. [DOI] [PubMed] [Google Scholar]
- 9. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 10. Pandurangan AP, Topf M (2012) Finding rigid bodies in protein structures: application to flexible fitting into cryoEM maps. J Struct Biol 177:520–531. [DOI] [PubMed] [Google Scholar]
- 11. Simchaev L, McCarty NA, Ford B, Senderowitz H (2015) Molecular dynamics flexible fitting (MDFF) simulations identify models of close‐state CFTR. Pediat Pulmon 50:201–201. [DOI] [PubMed] [Google Scholar]
- 12. Lopez‐Blanco JR, Chacon P (2013) iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J Struct Biol 184:261–270. [DOI] [PubMed] [Google Scholar]
- 13. Wang RY, Kudryashev M, Li X, Egelman EH, Basler M, Cheng Y, Baker D, Dimaio F (2015) De novo protein structure determination from near‐atomic‐resolution cryo‐EM maps. Nat Meth 12. [PAGE #S]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lau K, Van Petegem F (2014) Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat Commun 5:5397. [DOI] [PubMed] [Google Scholar]
- 15. Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T, Peng W, Yin CC, Li X, Scheres SH, Shi Y, Yan N (2015) Structure of the rabbit ryanodine receptor RyR1 at near‐atomic resolution. Nature 517:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang HW, Wang J, Ding F, Callahan K, Bratkowski MA, Butler JS, Nogales E, Ke A (2007) Architecture of the yeast Rrp44 exosome complex suggests routes of RNA recruitment for 3' end processing. Proc Natl Acad Sci USA 104:16844–16849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Makino DL, Baumgartner M, Conti E (2013) Crystal structure of an RNA‐bound 11‐subunit eukaryotic exosome complex. Nature 495:70–75. [DOI] [PubMed] [Google Scholar]
- 18. Liu JJ, Bratkowski MA, Liu X, Niu CY, Ke A, Wang HW (2014) Visualization of distinct substrate‐recruitment pathways in the yeast exosome by EM. Nat Struct Mol Biol 21:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu JJ, Niu CY, Wu Y, Tan D, Wang Y, Ye MD, Liu Y, Zhao W, Zhou K, Liu QS, Dai J, Yang X, Dong MQ, Huang N, Wang HW (2016) CryoEM structure of yeast cytoplasmic exosome complex. Cell Res 26:822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jenni S, Ban N (2009) Imperfect pseudo‐merohedral twinning in crystals of fungal fatty acid synthase. Acta Cryst D65:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dodson EJ (2001) Using electron‐microscopy images as a model for molecular replacement. Acta Cryst D57:1405–1409. [DOI] [PubMed] [Google Scholar]
- 22. Stuart DI, Abrescia NG (2013) From lows to highs: using low‐resolution models to phase X‐ray data. Acta Cryst D69:2257–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, Subramaniam S (2016) Breaking cryo‐EM resolution barriers to facilitate drug discovery. Cell 165:1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song W, Wang J, Han Z, Zhang Y, Zhang H, Wang W, Chang J, Xia B, Fan S, Zhang D, Wang J, Wang H‐W, Chai J (2015) Structural basis for specific recognition of single‐stranded RNA by toll‐like receptor 13. Nat Struct Mol Biol 22:782–787. [DOI] [PubMed] [Google Scholar]
- 25. Wang J, Wang W, Song W, Han Z, Zhang H, Chai J, Wang H, Wang J (2015) An improved method for phasing crystal structures with low non‐crystallographic symmetry using cryo‐electron microscopy data. Prot Cell 6:919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J, Chai J, Wang H (2016) Structure of the mouse toll‐like receptor 13 ectodomain in complex with a conserved sequence from bacterial 23S ribosomal RNA. Febs J 283:1631–1635. [DOI] [PubMed] [Google Scholar]
- 27. Nannenga BL, Gonen T (2014) Protein structure determination by MicroED. Curr Opin Struct Biol 27:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi D, Nannenga BL, Iadanza MG, Gonen T (2013) Three‐dimensional electron crystallography of protein microcrystals. Elife 2:e01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scheres SHW. Processing of structurally heterogeneous Cryo‐EM data in RELION. Methods Enzymol. Doi: http://dx.doi.org/10.1016/bs.mie.2016.04.012 [DOI] [PubMed] [Google Scholar]
- 30. Chang S, Sun D, Liang H, Wang J, Li J, Guo L, Wang X, Guan C, Boruah BM, Yuan L, Feng F, Yang M, Wang L, Wang Y, Wojdyla J, Li L, Wang J, Wang M, Cheng G, Wang HW, Liu Y (2015) Cryo‐EM structure of influenza virus RNA polymerase complex at 4.3 A resolution. Mol Cell 57:925–935. [DOI] [PubMed] [Google Scholar]
- 31. Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crepin T, Hart D, Lunardi T, Nanao M, Ruigrok RW, Cusack S (2014) Structural insight into cap‐snatching and RNA synthesis by influenza polymerase. Nature 516:361–366. [DOI] [PubMed] [Google Scholar]
- 32. Hirano Y, Takeda K, Miki K (2016) Charge‐density analysis of an iron–sulfur protein at an ultra‐high resolution of 0.48 Å. Nature 534:281–284. [DOI] [PubMed] [Google Scholar]
- 33. Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B (2009) Appion: An integrated, database‐driven pipeline to facilitate EM image processing. J Struct Biol 166:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coppens P, Hermansson K (1997) X‐ray charge densities and chemical bonding. International Union of Crystallography. Oxford University Press: New York. [Google Scholar]