

Abstract
Background
heart failure has become increasingly prevalent along with the aging population and the increased survival of acute ischemic heart events. Impairments of mitochondrial function in the heart are intricately linked to the development of heart failure but there is no therapy for mitochondrial dysfunction in the clinic.
Methods and Results
we report that NAD+ redox imbalance (increased NADH/NAD+) and protein hyperacetylation, previously observed in genetic models of defective mitochondrial function, are also present in human failing hearts as well as in mouse hearts with pathological hypertrophy. Elevation of NAD+ levels by stimulating the NAD+ salvage pathway suppressed mitochondrial protein hyperacetylation and cardiac hypertrophy, and improved cardiac function in responses to stresses. Acetylome analysis identified a subpopulation of mitochondrial proteins that was sensitive to changes in the NADH/NAD+ ratio. Hyperacetylation of mitochondrial malate-aspartate shuttle proteins impaired the transport and oxidation of cytosolic NADH in the mitochondria, resulting in altered cytosolic redox state and energy deficiency. Furthermore, acetylation of oligomycin-sensitive conferring protein at lysine-70 in ATP synthase complex promoted its interaction with cyclophilin D, and sensitized the opening of mitochondrial permeability transition pore. Both could be alleviated by normalizing the NAD+ redox balance either genetically or pharmacologically.
Conclusions
we show that mitochondrial protein hyperacetylation due to NAD+ redox imbalance contributes to the pathological remodeling of the heart via two distinct mechanisms. Our preclinical data demonstrate a clear benefit of normalizing NADH/NAD+ imbalance in the failing hearts. These findings have a high translational potential as the pharmacological strategy of increasing NAD+ precursors are feasible in human.
Keywords: heart failure, hypertrophy, metabolism, apoptosis
Cardiovascular disease is a leading cause of death worldwide1. As the life expectancy increases and the mortality of acute ischemic events decreases, the incidence of heart failure is mounting at a pace of 900,000 per year2. However, medical therapy for heart failure has been stalled for nearly two decades. Novel concepts and strategies in the treatment of heart failure are urgently needed.
The heart is a high energy-consuming organ. Mitochondrion is the powerhouse of the cell and mitochondrial dysfunction is a well-recognized maladaptive mechanism during the development of heart failure3, 4. Targeting mitochondria for heart failure therapy has long been sought; however, previous work focusing on improving mitochondrial energy production and reducing reactive oxygen species yielded few successful clinical applications5. In recent years, protein lysine acetylation emerged as an important mechanism linking mitochondrial metabolism to cellular pathologies6–8. The level of protein acetylation reflects the balance of acetylation and deacetylation. While the former is dependent on the abundance of acetyl-CoA and/or the activity of acetyl-transferase, the later is determined by the deacetylase activity, and primarily Sirtuins in the mitochondria. The Sirtuin deacetylases consume NAD+ as a co-substrate9; mitochondrial function is critical for setting the NADH/NAD+ balance thus the NAD+ available for Sirtuin activity.
Using a mouse model with primary mitochondrial dysfunction (cardiac-specific deletion of a Complex I protein, Ndufs4; cKO), we recently found that elevation in NADH/NAD+ ratio induces mitochondrial protein hyperacetylation and renders the hearts highly susceptible to stresses10. In this study we defined the molecular intermediaries linking specific NAD+-sensitive hyperacetylation targets to the development of heart failure and demonstrated the relevance of these mechanisms in human heart failure. Furthermore, we showed that restoring the NADH/NAD+ ratio by genetic and pharmacological approaches is an effective and potentially translatable strategy for the treatment of heart failure in clinical practice.
Methods
Animal care, surgical procedures and echocardiography
All procedures involving animal use were performed with the approval of IACUC of the University of Washington. Detailed procedures for animal care and crossing, surgeries and echocardiography were described in supplementary methods.
Ex vivo measurements of cardiac function and energetics
Langendorff perfused mouse hearts were isolated as previously described11, 12. Detailed methods were described in supplementary methods.
Mitochondrial isolation, proteome, and acetylome analyses
Mitochondria were isolated as described13. Detailed methods of proteome and acetylome analyses can be found in supplementary materials.
Molecular docking calculation
Crystal structure data from PDB (2BIU and 2WSS, chain S) for CypD and OSCP proteins were subjected to rigid body molecular docking using online platform PATCHDOCK http://bioinfo3d.cs.tau.ac.il/PatchDock/14. Detailed methods were described in supplementary materials
Mitochondrial calcium uptake assay and biochemical assays
Detailed methods of mitochondrial calcium uptake assay in isolated mitochondria or permeabilized cells were described in supplementary methods. All biochemical assays used in this study were described in supplementary methods.
Antibodies, Western blot and immunoprecipitation
Detailed methods of antibodies, Western blot and immunoprecipitation were described in supplementary materials.
Statistical analysis
Comparisons among the multiple groups were performed by 1-way ANOVA, followed by Newman-Keuls multiple comparison test. For comparisons only involving two groups, unpaired 2-tailed t-tests were used. For repeated measurements of multiple groups, 2-way repeated measure ANOVA was performed. All analyses were performed using GraphPad Prism 6.0. All data are expressed as mean ± SEM and a p<0.05 was considered significant.
Results
Protein hyperacetylation in the failing heart was reversed by expanding the NAD+ pool
We previously showed that increased NADH/NAD+ ratio and inhibition of NAD+-dependent protein deacetylation caused by mitochondrial Complex I deficiency (cKO) increased cardiac susceptibility to stress10. We here sought to test whether protein hyperacetylation occurs during the development of heart failure. In cardiac tissues of heart failure patients with ischemic or dilated cardiomyopathy (Table S1), we observed higher acetylation levels comparing to non-failing human hearts (Figure 1A). In addition, pressure overload generated by transverse aortic constriction (TAC) elevated cardiac NADH/NAD+ ratio and mitochondrial protein hyperacetylation (Figure 1B–C, Figure S1A–B), further supporting a positive correlation of NAD+-sensitive protein acetylation and heart failure development. Next, we tested whether restoring NADH/NAD+ balance by either genetic or pharmacological approach can normalize protein acetylation. We elevated NAD+ synthesis via the NAD+ salvage pathway by supplementing NAD+ precursor NMN or overexpressing the rate-limiting enzyme, nicotinamide phosphoribosyltransferase, in the mouse hearts (cNAMPT, Figure 1D; Figure S1C)15. Both measures increased NAD+ level10, 15 without affecting cardiac function (fractional shortening, FS) in unstressed mice (Figure S1D–H). NMN administration normalized the NADH/NAD+ ratio and importantly, reversed mitochondrial protein hyperacetylation in control mice after TAC (Figure 1B–C Figure S1A–B). Similarly, NMN administration also attenuated the NAD+ redox imbalance and protein hyperacetylation induced by primary mitochondrial dysfunction in cKO hearts (Figure 1E–F, Figure S1I–J). The data from multiple models collectively indicate that the NAD+-dependent protein hyperacetylation is present in failing hearts and/or hearts with mitochondrial dysfunction, which can be abrogated by elevation of NAD+ levels and restoration of the NADH/NAD+ balance.
Figure 1. Protein hyperacetylation in the failing hearts was normalized by restoring the NADH/NAD+ ratio.
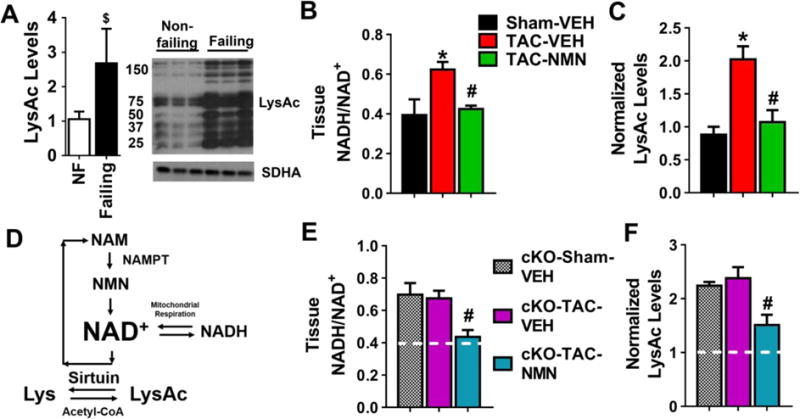
(A) Acetylation of human failing hearts were estimated by Western blotting. N=8–10. The ratio of heart failure patients with ischemic vs dilated cardiomyopathy is 1:1. (B) Tissue NADH/NAD+ ratio and (C) mitochondrial protein acetylation levels by Western Blotting of control mice four weeks after sham or TAC surgeries with and without NMN treatment were measured. N=3–4. (D) Sirtuin deacetylase reaction requires NAD+. Mitochondrial NAD+ level is determined by the NADH/NAD+ ratio and the rate of NAD+ synthesis via salvage pathway. Nicotinamide Phosphoribosyltransferase (NAMPT), the rate limiting enzyme of this pathway, catalyzes the conversion of nicotinamide (NAM) into nicotinamide mononucleotide (NMN). Over-expression of NAMPT and supplementation of NMN were used to elevate NAD+ levels in this study. (E) Tissue NADH/NAD+ ratio and (F) mitochondrial protein acetylation in cardiac tissues of the cKO mice were measured. N=3–4. White lines represent levels in control mice with sham-operation*: P<0.05 compared to Sham-VEH; #: P<0.05 compared to TAC-VEH. $: P<0.05 compared to non-failing. All data are expressed as means ±SEM.
Normalization of protein acetylation blunted the development of heart failure during chronic stresses
Next, we sought to determine if the reversal of protein hyperacetylation by NMN would improve cardiac function and reduce pathological hypertrophy induced by pressure overload. TAC caused significant cardiac hypertrophy, left ventricular (LV) dilation and decline in fractional shortening (FS) in control mice (Figure 2A–D, Figure S2A). NMN administration improved FS (Figure 2A), LV dilation and hypertrophy (Figure 2B–D) in TAC-stressed mice. In addition, the accelerated course of heart failure in cKO mice after TAC (Figure S2B)10 was also abrogated by NMN administration. NMN treatment preserved contractile function (Figure 2E), reduced cardiac hypertrophy, LV dilation, and lung edema (Figure 2F–H, S2C–D). The data strongly supported the efficacy of NMN administration in delaying the development of heart failure in mice with mitochondrial dysfunction, either primary or acquired.
Figure 2. Nicotinamide mononucleotide (NMN) protected hearts from pathological hypertrophy and contractile dysfunction induced by chronic pressure overload.
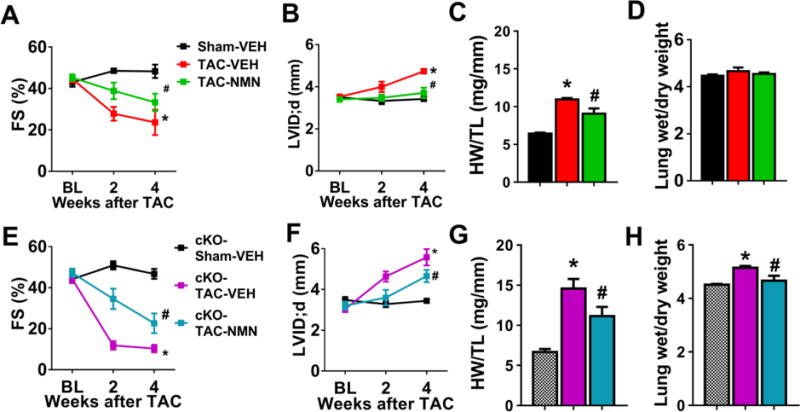
(A) Fractional shortening (FS), (B) LV dilation (left ventricular internal dimension at diastole; LVID;d), (C) Cardiac hypertrophy (heart weight/tibia length, HW/TL, at 4-week end-point) and (D) Lung edema (wet/dry lung weight at 4-week end-point) of control mice after sham or TAC surgeries with or without NMN treatment were assessed. N=5–8. (E) FS, (F) LV dilation, (G) cardiac hypertrophy and (H) Lung edema of cKO mice after indicated treatments were assessed. N=5–6. *: P<0.05 compared to corresponding Sham; #: P<0.05 compared to corresponding TAC-VEH. All data are expressed as means ±SEM.
We further tested whether elevation of NAD+ levels specifically in heart by increasing NAMPT activity would also be effective to prevent cardiac dysfunction and hypertrophy induced by another stressor with distinct mechanism, isoproterenol (ISO) stimulation. Transgenic mice over-expressing NAMPT only in the hearts (cNAMPT)15 were crossed with control and cKO mice to obtain mice with cNAMPT expression. We stressed the mice by chronic β-adrenergic stimulation with ISO, which has been shown to cause cardiac myocyte death and pathological hypertrophy16. ISO (30 mg/kg/day) delivered by osmotic mini-pump for two weeks (Figure S3A) induced significant cardiac dysfunction and hypertrophy in both control (Figure 3A–D, S3B) and cKO mice (Figure 3E–H, S3C) while cKO mice presented worse phenotypes. Elevation of cardiac NAD+ levels by cNAMPT protected both mice from ISO-induced cardiac dysfunction, LV dilation and hypertrophy (Figure 3, S3). The data further supported that targeting the NAD+ salvage pathway to elevate cellular NAD+ levels represents a viable intervention strategy to improve cardiac function in response to chronic stresses. Moreover, the cNAMPT mice provide a useful tool to dissect the mechanistic roles of NADH/NAD+-sensitive protein acetylation in heart failure propensity.
Figure 3. Cardiac NAMPT expression reversed ISO-induced cardiac dysfunction and hypertrophy.
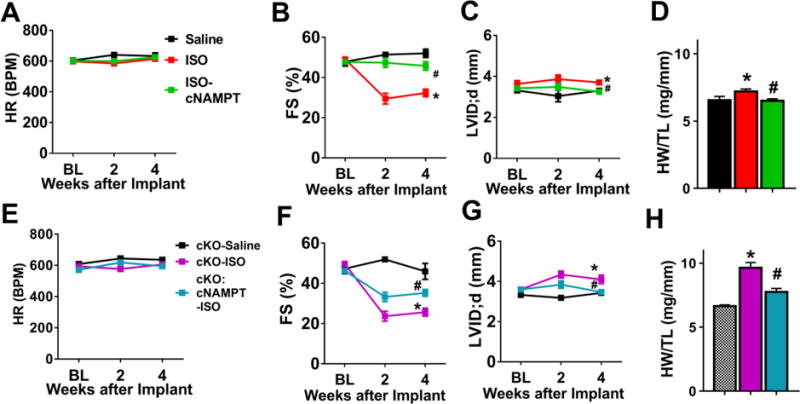
(A) HR, (B) FS, and (C) LVID;d and (D) cardiac hypertrophy (HW/TL) of control or control+cNAMPT mice challenged with saline or isoproterenol (ISO, 30 mg/kg/day) for two weeks were measured. N=5–9. (E) HR, (F) FS, (G) LVID;d, ratio of (H) HW/TL of cKO or cKO+cNAMPT mice challenged with saline or ISO were recorded. N=5–8. *: P<0.05 compared to corresponding Saline; #: P<0.05 compared to corresponding ISO. Data are expressed as means ±SEM.
Acetylome analyses identified NADH/NAD+-sensitive changes in acetylation landscape
To test the hypothesis that protein acetylation that is sensitive to NADH/NAD+ ratio modulates cardiac sensitivity to stress, we performed acetylome analysis of cKO hearts with and without cNAMPT. We first compared the mitochondrial proteomes from control and cKO hearts to rule out the possibility that increases in acetylated proteins in cKO were due to increases in the total protein amount. We did not observe significant up-regulation of mitochondrial protein levels while multiple proteins in mitochondrial Complex-I were downregulated (Table S2–3; Figure S4A–B). This is consistent with the observation that deletion of Ndusf4 resulted in poor assembly of Complex-I and hence degradation of Complex-I proteins10. Despite the minimal changes in protein levels, the number of acetylated proteins (Figure 4A) and acetylation levels of peptides increased in cKO hearts (Figure 4B), consistent with increased acetylation shown by Western blot (Figure 4C, S4C). Hyperacetylation of some known protein targets were validated by Western blots (Figure S4D–E). Increased acetylation levels in cKO hearts were not attributable to alterations in the levels of acetyl-CoA, mitochondrial acetyltransferase GCN5L1, or sirtuin expressions (Figure S4F–G). As expected, cNAMPT overexpression normalized the NADH/NAD+ ratio and hyperacetylation in cKO hearts (Figure 4C, S4C,H). The number of acetylation proteins as well as the levels of acetylated peptides in cKO hearts were reduced by cNAMPT (Figure 4A–B, left-shift from cKO). Although mitochondrial proteins only constituted ~1/3 of the 616 acetylated proteins identified (Figure 4D), a disproportionately high fraction of hyperacetylated peptides were originated from mitochondrial proteins in cKO (Figure 4E), and they responded robustly to the normalization of NADH/NAD+ ratio by cNAMPT (Figure 4F, left-shift from cKO). These observations further support a critical role of NADH/NAD+ ratio in connecting mitochondrial function and acetylation landscape. Interestingly, acetylation levels of proteins from non-mitochondrial compartments were also elevated in cKO hearts (Figure 4E, right-shift from 1). Expression of cNAMPT moderately reduced the acetylation of non-mitochondrial proteins (Figure S4I). This observation raised an intriguing possibility that the NAD+ redox imbalance caused by mitochondrial dysfunction could affect other cellular compartments, and as such contributed to pathological remodeling of the heart by altering cytosolic redox state and whole cell hyperacetylation.
Figure 4. Acetylome analysis identified NADH/NAD+-sensitive changes in acetylation landscape.
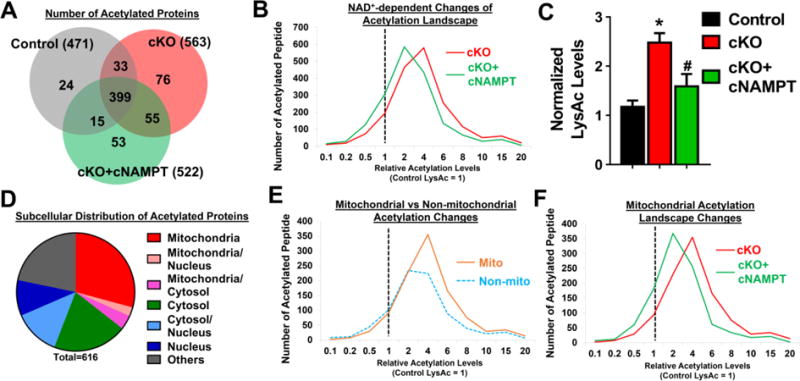
(A) Venn diagram showing the overlapping of acetylated proteins identified in control, cKO and cKO+cNAMPT hearts. Numbers in bracket represent the total number of acetylated proteins identified in each group. (B) Acetylated peptides of cKO and cKO+cNAMPT hearts were quantified by LC-MS and normalized to control values. Distribution of the acetylation changes was plotted. Dotted line at 1 was set to the mean of control peptide levels. Distribution shift to the right of 1 represented increased acetylation levels compared to control. (C) Acetylation levels from indicated hearts were assessed by Western blot. *: P<0.05 compared to control. #: P<0.05 compared to cKO. (D) Pie chart showing subcellular localization of acetylation proteins identified in cKO hearts. (E) Changes of acetylation in mitochondrial (mito) versus non-mitochondrial (non-mito) compartments. (F) Mitochondrial acetylation landscape changes of cKO and cKO+cNAMPT hearts were compared.
Acetylation of Malate Aspartate Shuttle Regulated Cytosolic NAD+ Redox Balance and Cardiac Energetics
Among the subpopulation of proteins whose acetylation responded robustly to NADH/NAD+ ratio, hyperacetylation of the mitochondrial isoforms of malate aspartate shuttle (MAS) proteins (Figure 5A, Table S4) is of interest. MAS is a key player in the communication of cytosolic and mitochondrial NAD+ redox states17, carrying electrons from cytosolic NADH into mitochondria for oxidative phosphorylation18. Decreased MAS activity was found in cKO and ISO-treated hypertrophic hearts, and in both cases could be restored by overexpressing cNAMPT (Figure 5B–C). Acetylation of a number of lysines in the MAS proteins was reduced towards normal level by overexpressing cNAMPT (Table S4). Moreover, incubation of mitochondrial proteins with acetyl-coA promoted acetylation of mitochondrial glutamate oxaloacetate transaminase (GOT2) and inhibited its activity (Figure S5A–C), further supporting the notion that protein hyperacetylation suppressed the shuttle activity. The lactate/pyruvate ratio, a marker for cytosolic NADH/NAD+ ratio, was elevated in ISO-treated and TAC-stressed hearts and was lowered by cNAMPT or NMN (Figure 5D–E). The data collectively suggest that hyperacetylation of MAS decreases the import of cytosolic NADH into mitochondria for oxidation hence altered cytosolic NADH/NAD+ ratio.
Figure 5. Inhibition of malate aspartate shuttle (MAS) by acetylation altered cytosolic redox state and cardiac energetics.
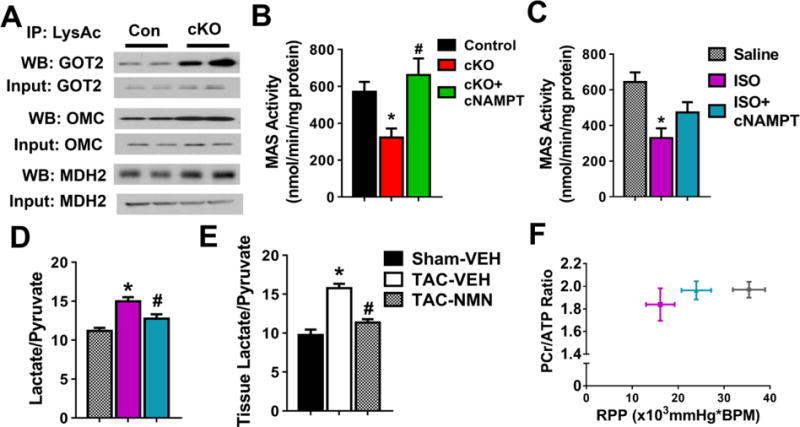
(A) Acetylation levels of mitochondrial isoforms of MAS proteins were assessed by immunoprecipitation/Western blot (IP-WB) analysis. N=4. MAS activity of mitochondria isolated from (B) control, cKO and cKO+cNAMPT and (C) ISO-treated hearts were measured. N=4. Tissue lactate/pyruvate ratio of (D) ISO-treated or (E) TAC-stressed hearts were measured. N=4. (F) The relationship between cardiac energetics, estimated by phosphocreatine to ATP ratio (PCr/ATP) and contractile function, estimated by rate pressure product (RPP), measured simultaneously in isolated perfused hearts by 31P NMR spectroscopy. N=4. *: P<0.05 compared to corresponding control/Saline/Sham-VEH; #: P<0.05 compared to corresponding cKO/ISO/TAC-VEH. All data are expressed as means ±SEM.
Upregulation of glycolysis is a hallmark of metabolic remodeling in pathological hypertrophy19, 20. Elevation of MAS flux under these conditions would facilitate aerobic glycolysis for ATP production19–23. It was shown previously that elevated MAS flux at early stage of pathological hypertrophy was attenuated as the heart transitions into energetic and contractile failure, even though the MAS protein levels were unaltered23. To test whether inhibition of MAS by acetylation is partially responsible for the impaired energetics in pathological hypertrophy, we performed 31P NMR spectroscopy of isolated perfused hearts to simultaneously measure myocardial high energy phosphate content and contractile function. We observed a downward-left shift in the relationship of myocardial energetic status assessed by phosphocreatine to ATP ratio (PCr/ATP) and contractile function assessed by the rate pressure product (RPP) in ISO-treated hearts (Figure 5F). The impairments were improved by partially restored MAS activity via overexpressing cNAMPT (Figure 5C,F, upward-right shift in plot). These results suggest that acetylation of MAS proteins is a key mechanism through which mitochondrial dysfunction impacts energetics via redox-sensitive regulations during pathological remodeling.
Acetylation of OSCP and CypD increased mPTP Sensitivity in the Failing Heart
We previously showed that increased cardiac susceptibility to stresses in cKO hearts is partly attributable to the hypersensitivity of calcium-triggered mitochondrial permeability transition pore (mPTP) opening10. Here we found that similar to cKO, mitochondria isolated from TAC hearts demonstrated a lower calcium retention capacity (CRC), indicating increased sensitivity of mPTP opening, which were normalized by elevation of NAD+ levels (Figure 6A, Figure S6A). Several hyperacetylated proteins identified by acetylome analysis participate in mPTP regulation (Table S5) such as cyclophilin D (CypD) and oligomycin-sensitive conferring protein (OSCP), and majority of the hyperacetylated sites on these proteins were responsive to NADH/NAD+ ratio (Table S5). Although the physical identity of the mPTP remains elusive, CypD is an undisputed regulator of the mPTP24, 25. Recent studies suggest that mitochondrial ATP synthase forms the mPTP26, and the opening of mPTP is regulated through the interaction of CypD with OSCP subunit of ATP synthase26–28. We observed increased acetylation of both CypD and OSCP in cKO mitochondria and in human failing hearts (Figure 6B–C and Figure S6B, Table S5). Furthermore, increased acetylation was associated with increased interaction of OSCP-CypD in cKO, TAC-stressed, or ISO-treated hearts (Figure 6D–E, Figure S6C). The increased interaction of OSCP-CypD in cKO was alleviated by cyclosporine A, an established mPTP inhibitor (CsA; Figure 6D), indicating the importance of such an interaction in regulating mPTP sensitivity27. The data strongly suggest that NAD+-sensitive acetylation modulates the OSCP-CypD interaction and thus the mPTP sensitivity in hearts with pathological hypertrophy.
Figure 6. Acetylation promoted the interaction between OSCP and CypD and increased the sensitivity of mPTP.
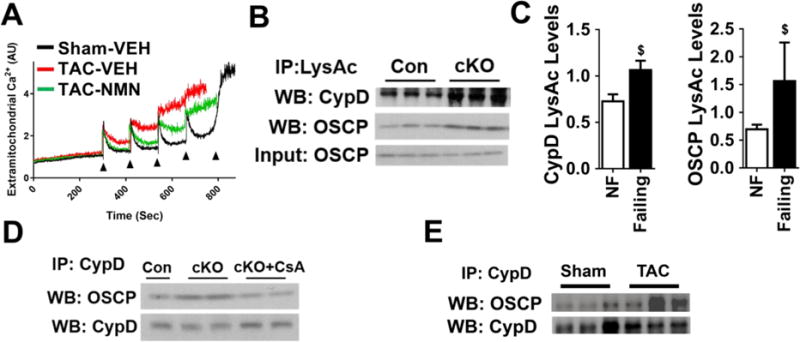
(A) Representative experiment of calcium-stimulated mPTP opening in mitochondria isolated from sham or TAC hearts with and without NMN treatment. N=3–4. Arrows represent each calcium pulse. (B) Acetylation levels of CypD and OSCP in control (Con) and cKO mitochondria determined by IP-WB. N=3. (C) Acetylation levels of CypD and OSCP from non-failing and failing human hearts were quantified. N=8–10. $: P<0.05 compared to NF. (D) Interaction of CypD with OSCP were determined by IP-WB in mitochondria isolated from cKO hearts. N=3–4. CsA: cyclosporine A. CsA was added to mitochondria at final concentration of 1 μM. (E) Interaction of CypD with OSCP in heart tissues after sham/TAC surgeries was assessed by IP-WB.
Acetylation of OSCP-K70 Promoted OSCP-CypD Interaction and the mPTP Opening
To identify specific acetylation sites on OSCP and CypD responsible for regulating the protein-protein interaction, crystal structures of CypD29 and OSCP of ATP synthase30 were subjected to molecular docking14. Docking results from the highest scoring solutions consistently identified an interaction interface of OSCP with variable CypD orientations (Figure S7A). The conserved interface coincided with an empty space next to OSCP in F1-ATP synthase, which serves as a potential interaction interface for CypD binding on the intact F1-ATP complex (Figure S7A–C). Acetylated lysine residues identified in the two proteins from the acetylome analysis were mapped to the docked models (Figure 7A, left panel magenta spheres, Table S5). Analysis of the top-ten scoring docking solutions revealed that OSCP-K70 was always present in the putative interaction interface. Acetylation of OSCP-K70 (OSCP-K70Ac) was normalized by cNAMPT expression in cKO (Table S5). In addition, OSCP-K70 is highly conserved among species surveyed (Figure S7D, red asterisk) while other lysines (16 out of 21 lysines) are not. Seven out of the top-ten docking solutions showed that several different lysine residues of CypD could be in close contact with OSCP-K70 (data not shown). In two of these solutions, OSCP-K70 was in close proximity (3.1 Å) of CypD-K66 which would result in charge repulsion, destabilizing the OSCP-CypD interaction (Figure 7A right panel, magenta sticks). Previous studies suggest that hyperacetylation of CypD-K166 modulate mPTP sensitivity31, 32. Although CypD-K166Ac was not detected in our acetylome analysis (Table S5), it was one of the lysines from CypD found within the interaction interface out of the highest scoring docking solutions. This suggests that CypD-K166 may cause repulsion with OSCP-K70 and regulate mPTP opening.
Figure 7. Acetylation of OSCP-K70 was a critical determinant of mPTP sensitivity.
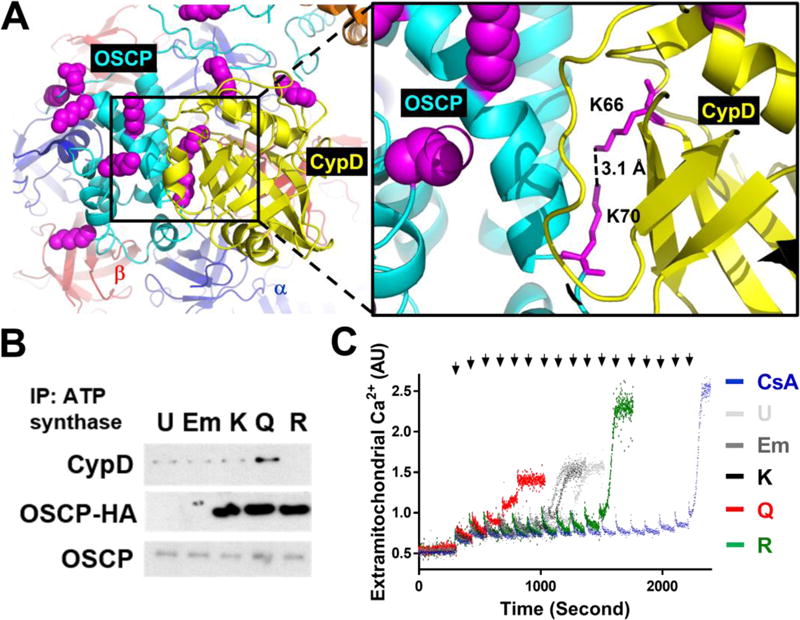
(A) Docking of C-V ATP synthase (PDB: 2WSS) and CypD (yellow, PDB: 2BIU) with Patchdock and predefined constraint (see methods). Left: overall view of OSCP and CypD interface with acetylated lysine residues highlighted as magenta spheres. Two lysines are located at the predicted interaction interface (black box). Right: magnified image of the putative interaction interface depicting the proposed regulation of interaction by acetylation on K70 of OSCP and K66 of CypD (magenta sticks). The distance between the two ε-nitrogen atoms of K70 of OSCP and K66 of CypD is measured by Pymol (dotted line). (B) Expression vectors indicated were transfected into HEK293 cells. Cell lysates were immunoprecipitated by ATP synthase antibodies and the presence of OSCP, HA-tagged OSCP and CypD was assessed by Western blot. U: untransfected, Em: empty pcDNA3.1 vector transfected, K: wild-type HA-tagged OSCP, Q: K70Q mutant of OSCP, R: K70R of OSCP. Representative blots from at least 3 independent experiments were shown in b. (C) Representative calcium-stimulated mPTP opening of permeabilized HEK293 cells after indicated transfection was shown. CsA (1 uM) was added after permeabilization of untransfected HEK293 cells. Arrows represent each 5 nmol calcium pulse.
Electrostatic interaction was suggested in regulating the OSCP-CypD interaction26. Together with the structural information (Figure 7A, S7A–C), we hypothesized that OSCP-K70Ac promotes the OSCP-CypD interaction and sensitizes mPTP opening by alleviating electrostatic repulsion. To determine the significance of OSCP-K70Ac, wild-type (K), K70Q (Q) and K70R (R) mutants of OSCP-K70 were expressed in HEK293 cells (Figure S7E). Expression of acetylation-mimetic K70Q mutant showed increased interaction with CypD and sensitized mPTP opening (lowered CRC), while expression of acetylation-insensitive K70R showed the opposite effects (Figure 7B–C, Figure S7F–G). The data collectively indicate a critical role of OSCP-K70Ac in determining mPTP sensitivity via its interaction with CypD.
Discussion
In this study we demonstrated that protein hyperacetylation induced by mitochondrial dysfunction is a positive regulator of pathological remodeling in mouse hearts with primary or acquired mitochondrial dysfunction as well as in human failing hearts. Our study here identified two distinct mechanisms that hyperacetylated protein targets, i.e. the MAS and the regulators of mPTP, mediate increased propensity to heart failure. Importantly, we demonstrated that normalization of NADH/NAD+ ratio in the heart by genetic or pharmacological approaches alleviated hyperacetylation, abrogated the two pathogenic mechanisms, and blunted the course of heart failure in mouse models, thus suggesting a novel therapy for heart failure.
Mitochondrial dysfunction has long been recognized as a maladaptive mechanism during the development of heart failure3, 4. However, there has been no effective therapy in the clinic5. Based on findings obtained from a genetic model of defective mitochondrial function, the present study identified and targeted pathogenic mechanisms caused by the imbalance of NADH production and oxidation in the mitochondria of hypertrophied and failing heart. We found that expanding the NAD+ pool could normalize NADH/NAD+ ratio, restore protein acetylation and mitigate the development of heart failure in multiple mouse models of pathological hypertrophy and failure. The strategy represents a novel therapeutic approach that targets the modification of protein functions consequent of mitochondrial dysfunction. Indeed, the NAD+ precursors such as NMN or nicotinamide riboside (NR) have yielded beneficial outcomes in animal models of diabetes33; obesity34 and aging35. By comparing mouse models and human failing hearts in this study, we provide strong evidence that a similar mechanism applies to human heart failure, hence indicate a highly translatable therapeutic strategy. Although NMN has poor oral bioavailability, oral administration of NR, a recently approved nutritional supplement, is effective in rising blood NAD+ levels in healthy volunteers (Airhart et. al., unpublished). Further study of the safety and tolerability of expanding NAD+ pool in patient population is thus highly warranted.
Previous studies have shown that the level of protein acetylation is closely related to the metabolic state of the cell, and it fluctuates with nutritional status36. Persistent hyperacetylation in the heart, such as in mice with deletion of Sirt3 or Ndusf4, resulted in increased sensitivity to stress while the unstressed heart is normal10, 31. We previously observed that increased NADH/NAD+ ratio in the Ndusf4 deficient heart inhibited Sirt3 resulting in protein hyperacetylation10. Restoring Sirt3 activity through normalization of NADH/NAD+ ratio in this study reverses mitochondrial protein hyperacetylation in cKO hearts as well as in hearts with pathological hypertrophy by pressure overload or isoproterenol stimulation. Acetylome analyses from studies of Sirt3-null mice37–39 and failing hearts40 have identified thousands of mitochondrial acetylation sites but only a handful of their functions have been biochemically characterized. The specific mechanisms connecting protein acetylation and increased sensitivity to stress remain to be poorly understood. Here we identified a subgroup of mitochondrial proteins that are highly sensitive to NADH/NAD+ ratio, among which hyperacetylation of malate aspartate shuttle and regulators of mPTP were shown to be causally linked to the development of heart failure. Furthermore, by expanding the NAD+ pool we were able to normalize but not overcorrect the NADH/NAD+ ratio, suggesting that such an approach is desirable from bioenergetics point of view.
It is conceivable that the disease mechanisms mediated by protein hyperacetylation involve other protein targets beyond the two groups reported here. Previous studies have identified other molecular targets of acetylation which are sensitive to NAD+ precursor supplementation in diabetes and mitochondrial diseases33–35, 41. Furthermore, we found that downregulation of malate aspartate shuttle (MAS) activity resulted in significant changes of cytosolic redox state. The MAS transfers the electron from NADH generated by glycolysis into mitochondria. Inhibition of MAS by acetylation serves as a feedback mechanism to protect mitochondrial compartment from further increases in NADH/NAD+ ratio but at the cost of altering cytosolic redox environment and sirtuin activities in the non-mitochondrial compartment. In addition, multiple redox dependent regulatory mechanisms, such as cysteine oxidation, glutathionylation, redox-mediated phosphorylation, have been shown to play important roles in the development of cardiac dysfunction42, 43. Of note, ATP citrate lyase, an enzyme that catalyzes the cytoplasmic conversion of mitochondrial citrate into the acetyl-coA in the cytosol, is activated at reduced state44, thus increasing substrate supply for protein acetylation. Consistently, a significant number of hyperacetylated proteins are found in the non-mitochondrial compartments of the Complex-I deficient hearts suggesting that mitochondrial dysfunction could ultimately affect whole cell acetylation. Although the scope of this study did not allow us to determine the specific contribution by each of these mechanisms in the progression of heart failure, it provides compelling evidence for targeting NADH/NAD+ ratio for therapy. Moreover, observations made here open a new avenue for investigating mitochondrial-cytosolic redox communications in chronic diseases involving mitochondrial dysfunction.
Cell death caused by the opening of mitochondrial permeability transition pore (mPTP) is an important mechanism in the development of heart failure45. We and others have shown previously that increased mitochondrial protein acetylation sensitizes the mPTP10, 31 but the specific molecular targets are unknown due to the lack of physical identity of the mPTP. By combining the computational and mutagenesis approaches, we here identified the acetylation of K70 on OSCP as a key determinant of mPTP sensitivity via its interaction of CypD. This finding also reconciles with prior reports suggesting that acetylation of CypD at K166 could increase the sensitivity of mPTP31, 32. Our computation models propose that acetylation of several lysine residues on CypD, including K66 that is at the closest proximity and K166 that is present at the putative interface of one top scoring model, could further reduce the repulsion between the two proteins. OSCP is located at the matrix side of the F1 sub-complex of the ATP synthase, making stable interaction with proteins of both F1 and the peripheral stalk (Figure S6B–C). Recent studies suggest that either the dimer of F1F0-ATP synthase26 or F0 sub-complex46, 47 forms the mPTP under specific conditions. Our results, although in line with this model, do not confirm the specific identity of the physical pore. They, nevertheless, provide a novel target to manipulate the mPTP sensitivity for therapy.
In summary, using a mouse model of mitochondrial Complex-I deficiency as the discovery tool we have unveiled novel mechanisms by which mitochondrial dysfunction modulates cellular stress response through NADH/NAD+-sensitive protein acetylation. The findings were validated in multiple mouse models of pathological hypertrophy, as well as in human failing hearts. Our preclinical data not only demonstrate a clear benefit of expanding NAD+ pool in heart failure therapy, the currently available NAD+ precursor compounds also make our findings immediately translatable.
Supplementary Material
Acknowledgments
We thank Dr. Michael Sack for GCN5L1 antibody and Dr. Junichi Sadoshima for providing cardiac-specific NAMPT mice.
Funding sources
This study is supported by grants from the National Institutes of Health (HL110349 and HL067970 to RT; R01GM086688 to JEB; 2T32DK007247 to NDR; T32AG000057 to YAC; S10RR029021 to 14T High Resolution Imaging Facility), the American Heart Association (postdoctoral fellowship 13POST16200007 to CFL), the Ellison/American Federation for Aging Research (Postdoctoral Fellowship to YAC) and the Glenn/AFAR Postdoctoral Fellowship for Translational Research on Aging (to YAC).
Footnotes
Disclosures
None.
References
- 1.Roth GA, Forouzanfar MH, Moran AE, Barber R, Nguyen G, Feigin VL, Naghavi M, Mensah GA, Murray CJ. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med. 2015;372:1333–41. doi: 10.1056/NEJMoa1406656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C and Stroke Statistics S Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 3.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2013;55:31–41. doi: 10.1016/j.yjmcc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang W, Karamanlidis G, Tian R. Novel targets for mitochondrial medicine. Sci Transl Med. 2016;8:326rv3. doi: 10.1126/scitranslmed.aac7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nature reviews Molecular cell biology. 2014;15:536–50. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 7.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee CF, Tian R. Mitochondrion as a Target for Heart Failure Therapy- Role of Protein Lysine Acetylation. Circ J. 2015;79:1863–70. doi: 10.1253/circj.CJ-15-0742. [DOI] [PubMed] [Google Scholar]
- 9.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 10.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–50. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–28. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolwicz SC, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-Specific Deletion of Acetyl CoA Carboxylase 2 (ACC2) Prevents Metabolic Remodeling During Pressure-Overload Hypertrophy. Circ Res. 2012 doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K. Increased uncoupling proteins and decreased efficiency in palmitate-perfused hyperthyroid rat heart. Am J Physiol Heart Circ Physiol. 2001;280:H977–83. doi: 10.1152/ajpheart.2001.280.3.H977. [DOI] [PubMed] [Google Scholar]
- 14.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–7. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–91. doi: 10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Errami M, Galindo CL, Tassa AT, Dimaio JM, Hill JA, Garner HR. Doxycycline attenuates isoproterenol- and transverse aortic banding-induced cardiac hypertrophy in mice. J Pharmacol Exp Ther. 2008;324:1196–203. doi: 10.1124/jpet.107.133975. [DOI] [PubMed] [Google Scholar]
- 17.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaNoue KF, Williamson JR. Interrelationships between malate-aspartate shuttle and citric acid cycle in rat heart mitochondria. Metabolism: clinical and experimental. 1971;20:119–40. doi: 10.1016/0026-0495(71)90087-4. [DOI] [PubMed] [Google Scholar]
- 19.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 20.Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004;44:662–7. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- 21.Wambolt RB, Lopaschuk GD, Brownsey RW, Allard MF. Dichloroacetate improves postischemic function of hypertrophied rat hearts. J Am Coll Cardiol. 2000;36:1378–85. doi: 10.1016/s0735-1097(00)00856-1. [DOI] [PubMed] [Google Scholar]
- 22.Rupert BE, Segar JL, Schutte BC, Scholz TD. Metabolic adaptation of the hypertrophied heart: role of the malate/aspartate and alpha-glycerophosphate shuttles. J Mol Cell Cardiol. 2000;32:2287–97. doi: 10.1006/jmcc.2000.1257. [DOI] [PubMed] [Google Scholar]
- 23.Lewandowski ED, O’Donnell JM, Scholz TD, Sorokina N, Buttrick PM. Recruitment of NADH shuttling in pressure-overloaded and hypertrophic rat hearts. Am J Physiol Cell Physiol. 2007;292:C1880–6. doi: 10.1152/ajpcell.00576.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 26.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–92. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–8. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chinopoulos C, Adam-Vizi V. Modulation of the mitochondrial permeability transition by cyclophilin D: moving closer to F(0)–F(1) ATP synthase? Mitochondrion. 2012;12:41–5. doi: 10.1016/j.mito.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 29.Schlatter D, Thoma R, Kung E, Stihle M, Muller F, Borroni E, Cesura A, Hennig M. Crystal engineering yields crystals of cyclophilin D diffracting to 1.7 A resolution. Acta Crystallogr D Biol Crystallogr. 2005;61:513–9. doi: 10.1107/S0907444905003070. [DOI] [PubMed] [Google Scholar]
- 30.Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci U S A. 2009;106:21597–601. doi: 10.1073/pnas.0910365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–23. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bochaton T, Crola-Da-Silva C, Pillot B, Villedieu C, Ferreras L, Alam MR, Thibault H, Strina M, Gharib A, Ovize M, Baetz D. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J Mol Cell Cardiol. 2015;84:61–9. doi: 10.1016/j.yjmcc.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 33.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD(+) Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012;15:838–47. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, Vaitheesvaran B, Hartil K, Robinson AJ, Hoopmann MR, Eng JK, Kurland IJ, Bruce JE. The fasted/fed mouse metabolic acetylome: N6-acetylation differences suggest acetylation coordinates organ-specific fuel switching. J Proteome Res. 2011;10:4134–49. doi: 10.1021/pr200313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–5. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–7. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 39.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, Verdin E. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2011;12:654–61. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vege RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC, Jr, Margulies KB, Coon JJ, Kelly DP. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016 doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042–9. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res. 2013;112:382–92. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, Steenbergen C. Signaling by S-nitrosylation in the heart. J Mol Cell Cardiol. 2014;73:18–25. doi: 10.1016/j.yjmcc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wells TN, Saxty BA. Redox control of catalysis in ATP-citrate lysate from rat liver. Eur J Biochem. 1992;204:249–55. doi: 10.1111/j.1432-1033.1992.tb16631.x. [DOI] [PubMed] [Google Scholar]
- 45.Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ Res. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–5. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–83. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.