

Abstract
Metachronous gastric cancer occurs frequently following endoscopic removal of an early gastric cancer. H. pylori eradication significantly reduces that risk. While, the pathogenesis of this phenomenon remains unclear, it is clear that the natural history of metachronous gastric cancer is altered following H. pylori eradication. Genetic instability of host cells induced by inflammation, H. pylori, host or environmental factors can result in the production of malignant cells. H. pylori eradication reduces and alters the inflammation, and can reverse epigenetic damage and abnormal expression of miRNA's. Fundamentally, H. pylori eradication stops the progression and may reverse some of the damage to the mucosa resulting in improved acid secretion and improving the gastric microbiome. Because the risk of developing metachronous cancer varies among patients, prospective research is needed to identify reliable biomarkers to predict development of metachronous cancer as well as to define surveillance methods, intervals, and duration. Some candidate examples of prognostic or predictive biomarkers for the prediction of subsequent risk include the presence or absence, titers, and changes in anti-H. pylori IgG and or anti-CagA antibodies, serum pepsinogens, gastrin, and miRNAs.
Introduction
Helicobacter pylori (H. pylori) infection is a class I human carcinogen and a necessary but insufficient causative factor in gastric cancer. The pathogenesis of the gastric cancer is multifactorial with cancer being an end product of the decades-long interaction between the organisms and the host. The major drivers of H. pylori-related gastric carcinogenesis are chronic inflammation along with H. pylori-mediated genetic and epigenetic changes resulting in genetic instability in host cells1,2). Environmental factors are important as changes in diet and food preservation can enhance or reduce the severity of H. pylori-induced gastric mucosal damage and result in a change in the primary outcome of the infection3, 4). The local environment in the stomach is also important in that H. pylori inflammation and mucosal damage can result in hypochlorhydria/achlorhydria which allows intragastric growth of non-H. pylori bacteria that promote biotransformation of ingested or secreted compounds (eg, nitrates) to produce carcinogens5-7).
H. pylori infection promotes field cancerization. Field cancerization was described by Slaughter et al. in the early 1950's to explain the presence of multicentric cancers occurring in oral stratified squamous epithelium8). The concept is based on the fact that carcinogens act widely within a mucosal field thus resulting in multi-centric disease. In 1961 at the 4th National Cancer Conference Proceedings, Leslie Foulds summary of field cancerization provides an excellent introduction to gastric cancer. He wrote, “The carcinogen establishes a state of incipient neoplasia coextensive with the area of exposure to a carcinogen. At this stage, corresponding with initiation, there is not necessarily any multiplication of cells or other specific clinical or histologic abnormality, yet the tissue is permanently altered and has acquired new capacities for progression to overt neoplasia. After considerable delay, with or without further extrinsic stimulation, visible lesions emerge focally or multifocally within the region. Malignant tumors emerge still later; some develop by progression in the lesions established in the preceding stage, but some emerge at places where no precursor lesion has been seen; they may emerge consecutively for long periods after those lesions have regressed. The early lesions are subject to diverse fates; some regress, some persist indolently, some grow progressively as benign tumors and some, usually a small minority, undergo progression to malignant neoplasia. They are “precancerous” only in the sense of having a certain statistical probability of progression to carcinoma. Carcinoma in situ is interpreted as an imperfect carcinoma whence invasive carcinoma may develop by a progression that confers the property of invasiveness which was not present in the earlier lesion.9) as reported by Bockus10) page 748. This succinct description provides an excellent overview of progressive nature of H. pylori-induced gastric mucosal damage resulting in cancer and provides a basis for understanding why metachronous tumors often appear after removal of an initial early gastric cancer.
H. pylori causes progressive gastric mucosal damage
The association of atrophic gastritis and gastric cancer has been a focus of research since the late 19th century11). Initially H. pylori infection and mucosal damage is most severe in the antrum and later progresses proximally into the gastric corpus as an advancing band of inflammation that results in an ever enlarging “lawn” of metaplastic pyloric type epithelium. This epithelium was originally termed pseudopyloric metaplasia by Stoerk to describe the mucosa that repopulated the stomach following diphtheria-induced acute gastritis (as related by Faber). When stained with hematoxylin and eosin pseudopyloric mucosa is easily confused with antral mucosa and unless the pathologist is informed that the biopsy was definitely from corpus mucosa, it is not likely to be recognized as a metaplastic epithelium. Until recently, the primary method of identifying pseudopyloric metaplasia in gastric biopsy specimens was to stain for the presence of pepsinogen I secreting cells which are normally restricted to the corpus13). Pseudopyloric metaplasia is now easily recognized because it stains positive for spasmolytic polypeptide and after decades of being ignored it is now a focus of research as spasmolytic polypeptide expressing epithelium14-16). This metaplastic epithelium is thought to arise by transdifferentiation of chief cells16, 17) and it has been suggested that both intestinal metaplasia and gastric cancer may arise from spasmolytic polypeptide expressing epithelium16-18). However, many no longer consider intestinal metaplasia a definite precursor of gastric cancer but rather as an easily recognized manifestation of atrophic gastritis19, 20) {Hattori, 1986 17952 /id.} It remains unclear whether the transdifferentiation is reversible; this will be discussed briefly below.
Pathogenesis of gastric mucosal damage and genetic instability
As noted above, the pathogenesis of gastric cancer depends on the presence of mutated gastric cells (eg, genetic instability) coupled with their acquisition of the ability to evade immune destruction, suppress the immune response and persist. Genetic instability of the gastric mucosa arises from three principle mechanisms: as a consequence of H. pylori-induced acute and chronic inflammation, because of direct bacterial host interactions, and because of interactions with exogenous factors to produce carcinogens locally in the stomach1). Gastric cancer is an inflammation-associated cancer that occurs as a result of the infection which causes chronic, often life-long, gastric mucosal inflammation histologically characterized as acute-on-chronic inflammation (Figure 1)1, 2).
Figure 1. Inflammation-host-bacterial interactions leading to gastric cancer (Adapted from reference1), with permission).
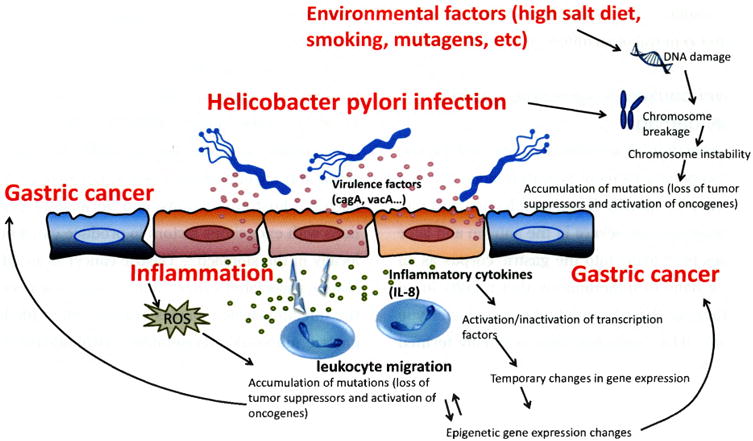
As noted above, H. pylori is a necessary but insufficient cause of gastric cancer which is typical of infectious causes of cancer such as human papilloma virus and cervical cancer or hepatitis C infection and liver cancer in that a malignancy only occurs in a subset of patients and the risk is often enhanced by the presence of environmental factors that enhance carcinogenesis. Bacterial, host, environmental interactions are responsible for the marked geographic differences in gastric cancer incidence as well as for the rapid changes in gastric cancer incidence that have been noted in association with changes in diet and food preservation in the West and more recently in Asia3, 4). A high incidence of gastric cancer in a population is associated with environment factors conducive to the early development of atrophic gastritis and hypo/achlorhydria. Risk is further enhanced by the presence of a more virulent strain of H. pylori21). A number of putative virulence factors such as the cag pathogenicity island (eg, CagA positive strain) have been identified as associated with an increased risk of a clinical outcome such as peptic ulcer or gastric cancer. While none of the virulence factors has disease specificity, they typically are associated with an increased host inflammatory response21). Importantly, no avirulent strain has been described; all H. pylori cause gastric inflammation and can result in gastric cancer. The difference in cancer risk between the most and the least virulent is probably less than 3-fold and leading to the recommendation that all H. pylori infections be eradicated21).
H. pylori infection also stimulates immune lymphocytes in the gastric mucosa and immune cells constituting mucosa-associated lymphoid tissue (MALT) which migrate to and infiltrate the sites of H. pylori infection in the stomach22) where chronically produced inflammatory cytokines may contribute to tumor promotion and progression23). In an inflammatory state, cell turnover is high rate and the micro-environment is often highly oxidative and nitrosative which results in increasing the opportunities for DNA damage and somatic mutation2, 24).
H. pylori-induced gastric cancer
Genetic instability, whether induced by inflammation, H. pylori, host or environmental factors can result in the production of malignant cells. The host is normally protected by innate and adaptive immune responses from the attack of pathogens. In the inflammatory microenvironment produced by the infection, there is a balance between antitumor immunity and tumor-originated pro-inflammatory activity25). These cells interact with the immune system which attempt to destroy them as “non-self”26). If potential malignant cells survive, they may remain as a small focus held in check by the immune system and require additional genetic (immuno-editing) changes to escape the immune system and become visible as an intramucosal neoplasia. Accumulation of additional mutations can result in acquisition of invasiveness which allows an early gastric cancer to metastasize and become the cause of the host's demise.
H. pylori itself can directly cause genetic instability (Figure 2). For example, H. pylori infection can induce methylation of multiple CpG islands including methylation of E-cadherin which is a tumor suppressor gene27). This hypermethylation is potentially reversible following H. pylori eradication28-30). H. pylori also stimulates activation-induced cytidine deaminase (AID) which induces nucleotide alterations involved in DNA mutations31)/ Aberrant AID expression is widely detectable not only in cancer lesions but also in H. pylori chronic gastritis32, 33). H. pylori infection has also been shown to lead to double-stranded DNA breaks in host DNA as well as to disordered regulation of microRNA production, all of which may increase genetic instability and be related to the increased risk of gastric cancer1, 2). Importantly all these events are present in all H. pylori infected individuals emphasizing the potentially critical role of host and environmental interactions in possibly affecting repair mechanisms resulting in differences in the rate of accumulation of genetic damage.
Figure 2.
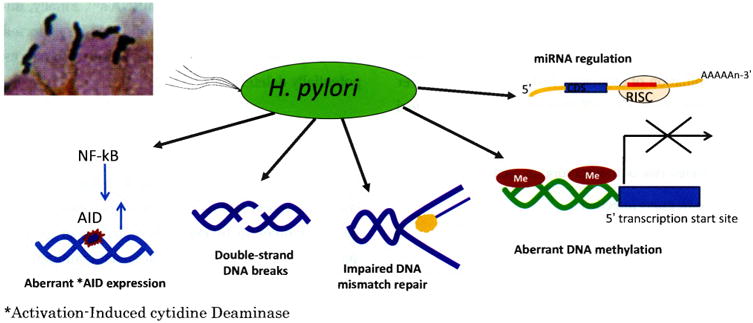
H. pylori host-cell interactions causing genetic instability (Adapted from reference 1), with permission)
Effect of H. pylori eradication on gastric cancer risk
H. pylori eradication stops the progression and may reverse some of the damage to the mucosa. The risk of developing gastric cancer is related to the development and extent of atrophic gastritis such that cure of the infection while non-atrophic mucosa stops the process and cancer risk remains minimal to absent. Thus, the benefit obtained by eradication is proportional to the baseline cancer risk at the time H. pylori eradication occurs. The natural history of H. pylori gastritis is progression of damage which results in an exponential increase in risk. H. pylori eradication is expected to stop the progression and to stabilize or reduce the risk. In addition, if parietal cells remain, but are suppressed by inflammatory mediators (eg, IL-1 β), improved acid secretion occurs would further reduce or eliminate any untoward effects associated with overgrowth with non-H. pylori bacteria34). These expectations can be seen in recent large studies. For example, a large-scale cohort study from Taiwan followed 80,000 patients with peptic ulcer for 10 years after H. pylori eradication therapy35). The patients were assigned to an early eradication group (patients underwent H. pylori eradication therapy at the time of diagnosis) or a late eradication group (patients underwent H. pylori eradication therapy 1 year after diagnosis). The incidence of gastric cancer was markedly lower in the early eradication group than in the late eradication group suggesting that, while the effect of H. pylori eradication therapy in reducing the incidence of gastric cancer is obvious, the earlier eradication the better.
Mass eradication of H. pylori was started in Taiwan in 2004 and initially included 4,121 subjects. Compared to the 5 year period before H. pylori therapy, the effectiveness of H. pylori eradication therapy in reducing the incidence of gastric cancer was estimated to be 25% (rate ratio 0.753, 95% CI 0.372 to 1.525)36). Identification of a significant effect of eradication on cancer prevention will often depend on the duration of follow-up duration (i.e., whether the duration is sufficiently long to allow the difference to become evident)37). This is shown in the Shangdong intervention trial which failed to find a difference in gastric cancer incidence after 7.3 years but after 14.7 years the incidence of gastric cancer was significantly reduced among those who had received H. pylori eradication therapy38).
Metachronous gastric cancer
The phenotype of the individual at risk for gastric cancer includes the presence of extensive atrophic gastritis. The entire gastric mucosa is exposed to H. pylori and intraluminal carcinogens (i.e., field carcinogenesis) and thus it is not surprising that gastric cancer is often multifocal. For example, careful examination of stomachs resected for one cancer report that 4% to 15% will contain an identifiable foci of cancer separate from the lesion which prompted the resection39-41). It is thus unclear how many metachronous lesions are new lesions vs. appearance of missed or previously inapparent synchronous lesions.
The surprising finding following H. pylori eradiation after removal of an early gastric cancer was that H. pylori eradication reduced the rate of metachronous lesions approximately 3-fold. This was originally reported by Uemura et al. in a prospective nonrandomized study and confirmed by Fukase et al. in a multicenter randomized trial42, 43). Since that time there have been a relatively large number of studies and a meta-analysis confirming that H. pylori eradication is associated with a reduction in the incidence of metachronous gastric cancer44). While the pathogenesis of this phenomenon remains unclear, it is clear that the natural history of the disease is altered following H. pylori eradication. H. pylori eradication alters or greatly reduces the direct H. pylori-associated H. pylori-induced inflammation (Figure 1). In addition, there are likely significant changes the interactions of the host's immune system and residual premalignant and as yet inapparent areas of tumorigenesis. For example, regression of acute inflammatory cells and cytokines may also reduce immune mediated tumor-promoting activity45, 46). Removal of the H. pylori also reduces or eliminates ongoing H. pylori-induced direct and epigenetic genetic damage and abnormal expression of miRNA's47). Finally, improvement in acid secretion would also be expected to reduce non-H. pylori bacterial overgrowth and its potentially deleterious effects1).
Risk of development of a metachronous gastric cancer
Little is known regarding the relation of a metachronous lesion and its relation to the original early gastric cancer in terms of similarities in genetic signatures, expression of tumor-specific antigens, or natural history. Table 1 lists risk factors that have been suggested as indicative of an increased risk of developing a metachronous lesion. The majority of these have been identified in retrospective analyses and not yet by prospective trials. Most are also the same risk factors that are associated with development of the original cancer such as extent and severity of atrophy and age. Prospective research is needed to identify reliable biomarkers predictive of development of metachronous cancer as well as to define surveillance methods, intervals, and duration.
Table 1.
Risk factors suggested to be indicative of an increased risk of metachronous gastric cancer.
|
Prospective studies also must stratify patients in relation to suspected risk factors in that the incidence of metachronous cancers is low (typically less than 5% per year) making randomization critical to reduce bias. Some candidate examples of prognostic or predictive biomarkers for the prediction of subsequent risk include the presence or absence, titers, and changes in anti-H. pylori IgG and or anti-CagA antibodies, serum pepsinogens, gastrin, and miRNA's. The location, extent, and severity of histologic changes before and after H. pylori eradication, the effect on acid secretion and gastric microbiome may also be candidate biomarkers.
CD44 (CD44v9) has been identified as one of the cell surface markers associated with cancer stem-like cells48) (CD44v9) and is associated with suppression of the production of reactive oxygen species resulting in subsequent therapeutic resistance, recurrence, and metastasis of tumors48-51). A recent report demonstrated that the recurrence rate of early gastric cancer was significantly higher in the CD44v9-positive group than in the CD44v9-negative group (hazard ratio (HR), 21.8; 95% confidence interval (CI), 5.71–83.1).52) suggesting the presence of CD44v9 as a possible marker of more rapid recurrence and a poor outcome.
Possible approaches to reducing risk
Clinical trials might include randomized trials of whether the use of adjuvants in addition to H. pylori eradication can further reduce risk (i.e., co-therapy with anti-inflammatory agents, gastroprotectives, antioxidants, or demethylating agents)53-55). The gastric microbiome both luminal as well as mucosal is likely to be greatly influenced by H. pylori eradication therapy and deserves detailed study. As noted above, the stomach of a patient with an endoscopically removable early gastric cancer likely contains other lesions. Decades ago the congo red-methylene blue test was proposed to assist in identifying such lesions56). Since that time there have been tremendous improvements in imaging technology and these advances should be explored for their utility.
If patients at increased risk could be reliably identified they would seem to be ideal candidates for targeted therapy or for cancer immunotherapy to eliminate residual disease before it becomes evident. It is becoming possible to culture gastric cells as “organoids” such it may be possible to culture the early gastric cancer initially removed to provide the antigens needed for patient-specific vaccination or other immunotherapy57, 58). In addition, targeted chemotherapy based on markers is beginning to show promise and may be useful for patients at especially high risk. For example, Sox2 is a member of the SRY-related HMG-box (SOX) family of transcription factors and well known as an essential embryonic stem cell gene. SOX2 is aberrantly expressed in many solid tumors and is a potential immunogenic antigen. SOX2 actives AKT signaling and has shown neoplastic transformation effect in gastric cancer cell lines and has been identified as an oncogene in gastric cancer59). Cells overexpressing SOX2 also showed significant insensitivity to therapeutic antibody against human epidermal growth factor receptor 2 (HER2)59). HER2 belongs to a family of four receptors (EGFR/HER1, HER2, HER3, HER4). HER regulate cell growth, survival and differentiation through interlinked signal transduction involving activation of the PI3K/ Akt and the Ras/Raf/MEK/MAPK pathways60). The monoclonal antibody against HER2 (trastuzumab) is approved as adjuvant treatment specifically for patients with HER2-positive early stage breast cancer and trastuzumab plus chemotherapy showed a survival benefit for advanced gastric cancer patients61, 62). Risk stratification may allow appropriate early use of targeted therapy.
Reversal of gastric mucosal atrophy
There are data in humans regarding the use of steroid therapy to promote recovery of normal mucosal histology in atrophy gastritis/gastric atrophy63-67). Such studies were typically done long before H. pylori was recognized and included both autoimmune and H. pylori gastritis. More recently, patients and animals taking tamoxifen have been shown to have regression in intestinal metaplasia, ADP ribosylation inhibitors such as Olaparib™ and prostaglandin E2 have been shown in animal studies to partial reverse intestinal metaplasia suggesting that it may be possible to at least partially reverse these changes68-72). More studies are needed especially those coupled with estimates of the changes in cancer risk as well as whether genetic instability has been improved. Possibly these changes are largely cosmetic and relate to changes in transdifferention and thus have little effect with regards to modifying underlying cancer risk73).
References
- 1.Shiotani A, Cen P, Graham DY. Eradication of gastric cancer is now both possible and practical. Semin Cancer Biol. 2013;23:492–501. doi: 10.1016/j.semcancer.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Hanada K, Graham DY. Helicobacter pylori and the molecular pathogenesis of intestinal-type gastric carcinoma. Expert Rev Anticancer Ther. 2014;14:947–54. doi: 10.1586/14737140.2014.911092. [DOI] [PubMed] [Google Scholar]
- 3.Wang C, Weber A, Graham DY. Age-, period- and cohort effects on gastric cancer mortality. Dig Dis Sci. 2014 doi: 10.1007/s10620-014-3359-0. in press. [DOI] [PubMed] [Google Scholar]
- 4.Graham DY. History of Helicobacter pylori, duodenal ulcer, gastric ulcer and gastric cancer. World J Gastroenterol. 2014;20:5191–204. doi: 10.3748/wjg.v20.i18.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JL. The role of gastric acid in preventing foodborne disease and how bacteria overcome acid conditions. J Food Prot. 2003;66:1292–1303. doi: 10.4315/0362-028x-66.7.1292. [DOI] [PubMed] [Google Scholar]
- 6.Peterson WL, Mackowiak PA, Barnett CC, et al. The human gastric bactericidal barrier: mechanisms of action, relative antibacterial activity, and dietary influences. J Infect Dis. 1989;159:979–83. doi: 10.1093/infdis/159.5.979. [DOI] [PubMed] [Google Scholar]
- 7.Correa P, Cuello C, Gordillo G, et al. The gastric microenvironment in populations at high risk to stomach cancer. Natl Cancer Inst Monogr. 1979:167–70. [PubMed] [Google Scholar]
- 8.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–68. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.Foulds L. The development of tumors. Philadelphia: Lippincott; 1961. p. 85. [Google Scholar]
- 10.Bockus HL. Carcinoma of the stomach. In: Bockus HL, editor. Gastroenterology. 2nd. Vol. 1. Philadelphia: W.B. Saunders Co.; 1963. p. 748. [Google Scholar]
- 11.Graham DY, Asaka M. Eradication of gastric cancer and more efficient gastric cancer surveillance in Japan: two peas in a pod. J Gastroenterol. 2010;45:1–8. doi: 10.1007/s00535-009-0117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faber K. Gastritis and its consequences. Paris: Oxford University Press; 1935. [Google Scholar]
- 13.El-Zimaity HM, Ota H, Graham DY, et al. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer. 2002;94:1428–36. doi: 10.1002/cncr.10375. [DOI] [PubMed] [Google Scholar]
- 14.Halldorsdottir AM, Sigurdardottrir M, Jonasson JG, et al. Spasmolytic polypeptide-expressing metaplasia (SPEM) associated with gastric cancer in Iceland. Dig Dis Sci. 2003;48:431–41. doi: 10.1023/a:1022564027468. [DOI] [PubMed] [Google Scholar]
- 15.Hanby AM, Poulsom R, Singh S, et al. Spasmolytic polypeptide is a major antral peptide: distribution of the trefoil peptides human spasmolytic polypeptide and pS2 in the stomach. Gastroenterology. 1993;105:1110–16. doi: 10.1016/0016-5085(93)90956-d. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt PH, Lee JR, Joshi V, et al. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest. 1999;79:639–46. [PubMed] [Google Scholar]
- 17.Goldenring JR, Nam KT. Oxyntic atrophy, metaplasia, and gastric cancer. Prog Mol Biol Transl Sci. 2010;96:117–31. doi: 10.1016/B978-0-12-381280-3.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer. 2009;12:189–97. doi: 10.1007/s10120-009-0527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatematsu M, Furihata C, Katsuyama T, et al. Independent induction of intestinal metaplasia and gastric cancer in rats treated with N-methyl-N′-nitro-N-nitrosoguanidine. Cancer Res. 1983;43:1335–41. [PubMed] [Google Scholar]
- 20.Tatematsu M, Tsukamoto T, Inada K. Stem cells and gastric cancer: role of gastric and intestinal mixed intestinal metaplasia. Cancer Sci. 2003;94:135–41. doi: 10.1111/j.1349-7006.2003.tb01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaoka Y, Graham DY. Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 2014;10:1487–500. doi: 10.2217/fon.14.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin WC, Tsai HF, Kuo SH, et al. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010;70:5740–48. doi: 10.1158/0008-5472.CAN-09-4690. [DOI] [PubMed] [Google Scholar]
- 23.Smyth MJ, Cretney E, Kershaw MH, et al. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202:275–93. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 24.Chang WJ, Du Y, Zhao X, et al. Inflammation-related factors predicting prognosis of gastric cancer. World J Gastroenterol. 2014;20:4586–96. doi: 10.3748/wjg.v20.i16.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim R, Emi M, Tanabe K, et al. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006;66:5527–36. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- 26.Matsueda S, Graham DY. Immunotherapy in gastric cancer. World J Gastroenterol. 2014;20:1657–66. doi: 10.3748/wjg.v20.i7.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan AO, Lam SK, Wong BC, et al. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–6. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perri F, Cotugno R, Piepoli A, et al. Aberrant DNA methylation in non-neoplastic gastric mucosa of H. pylori infected patients and effect of eradication. Am J Gastroenterol. 2007;102:1361–71. doi: 10.1111/j.1572-0241.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- 29.Chan AO, Peng JZ, Lam SK, et al. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut. 2006;55:463–68. doi: 10.1136/gut.2005.077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung WK, Man EP, Yu J, et al. Effects of Helicobacter pylori eradication on methylation status of E-cadherin gene in noncancerous stomach. Clin Cancer Res. 2006;12:3216–21. doi: 10.1158/1078-0432.CCR-05-2442. [DOI] [PubMed] [Google Scholar]
- 31.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–76. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu T, Marusawa H, Endo Y, et al. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012;103:1201–6. doi: 10.1111/j.1349-7006.2012.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham DY, Shiotani A, El-Zimaity HM. Chromoendoscopy points the way to understanding recovery of gastric function after Helicobacter pylori eradication. Gastrointest Endosc. 2006;64:686–90. doi: 10.1016/j.gie.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 35.Wu CY, Kuo KN, Wu MS, et al. Early Helicobacter pylori eradication decreases risk of gastric cancer in patients with peptic ulcer disease. Gastroenterology. 2009;137:1641–48. doi: 10.1053/j.gastro.2009.07.060. [DOI] [PubMed] [Google Scholar]
- 36.Lee YC, Chen TH, Chiu HM, et al. The benefit of mass eradication of Helicobacter pylori infection: a community-based study of gastric cancer prevention. Gut. 2013;62:676–82. doi: 10.1136/gutjnl-2012-302240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham DY, Uemura N. Natural history of gastric cancer after Helicobacter pylori eradication in Japan: after endoscopic resection, after treatment of the general population, and naturally. Helicobacter. 2006;11:139–43. doi: 10.1111/j.1523-5378.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- 38.Ma JL, Zhang L, Brown LM, et al. Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. J Natl Cancer Inst. 2012;104:488–92. doi: 10.1093/jnci/djs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng J, Wang Y. Epidemiology, pathology and clinical management of multiple gastric cancers: a mini-review. Surg Oncol. 2010;19:e110–14. doi: 10.1016/j.suronc.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Hattori T. Development of adenocarcinomas in the stomach. Cancer. 1986;57:1528–34. doi: 10.1002/1097-0142(19860415)57:8<1528::aid-cncr2820570815>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Kodama M, Tur GE, Shiozawa N, et al. Clinicopathological features of multiple primary gastric carcinoma. J Surg Oncol. 1996;62:57–61. doi: 10.1002/(SICI)1096-9098(199605)62:1<57::AID-JSO12>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 42.Takeda J, Toyonaga A, Koufuji K, et al. Resected early gastric cancer-clinicopathological studies on 610 cases. Kurume Med J. 1995;42:87–94. doi: 10.2739/kurumemedj.42.87. [DOI] [PubMed] [Google Scholar]
- 43.Uemura N, Okamoto S. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer in Japan. Gastroenterol Clin North Am. 2000;29:819–27. doi: 10.1016/s0889-8553(05)70149-7. [DOI] [PubMed] [Google Scholar]
- 44.Fukase K, Kato M, Kikuchi S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–97. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 45.Yoon SB, Park JM, Lim CH, et al. Effect of Helicobacter pylori eradication on metachronous gastric cancer after endoscopic resection of gastric tumors: A meta-analysis. Helicobacter. 2014;19:243–48. doi: 10.1111/hel.12146. [DOI] [PubMed] [Google Scholar]
- 46.Mantovani A, Cassatella MA, Costantini C, et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–31. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 47.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 48.Shiotani A, Uedo N, Iishi H, et al. H. pylori eradication did not improve dysregulation of specific oncogenic miRNAs in intestinal metaplastic glands. J Gastroenterol. 2012;47:988–98. doi: 10.1007/s00535-012-0562-7. [DOI] [PubMed] [Google Scholar]
- 49.Jang BI, Li Y, Graham DY, Cen P. The role of CD44 in the pathogenesis, diagnosis, and therapy of gastric cancer. Gut Liver. 2011;5:397–405. doi: 10.5009/gnl.2011.5.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 51.Tsugawa H, Suzuki H, Saya H, et al. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stemlike cells. Cell Host Microbe. 2012;12:764–77. doi: 10.1016/j.chom.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Yae T, Tsuchihashi K, Ishimoto T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- 53.Hirata K, Suzuki H, Imaeda H, et al. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br J Cancer. 2013;109:379–86. doi: 10.1038/bjc.2013.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong BC, Zhang L, Ma JL, et al. Effects of selective COX-2 inhibitor and Helicobacter pylori eradication on precancerous gastric lesions. Gut. 2012;61:812–18. doi: 10.1136/gutjnl-2011-300154. [DOI] [PubMed] [Google Scholar]
- 55.Nardone G, Rocco A. Chemoprevention of gastric cancer: role of COX-2 inhibitors and other agents. Dig Dis. 2004;22:320–26. doi: 10.1159/000083593. [DOI] [PubMed] [Google Scholar]
- 56.Graham DY, Shiotani A. The time to eradicate gastric cancer is now. Gut. 2005;54:735–38. doi: 10.1136/gut.2004.056549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iishi H, Tatsuta M, Okuda S. Diagnosis of simultaneous multiple gastric cancers by the endoscopic Congo red-methylene blue test. Endoscopy. 1988;20:78–82. doi: 10.1055/s-2007-1018137. [DOI] [PubMed] [Google Scholar]
- 58.Mahe MM, Aihara E, Schumacher MA, et al. Establishment of gastrointestinal epithelial organoids. Curr Protoe Mouse Biol. 2013;3:217–40. doi: 10.1002/9780470942390.mo130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, Nadauld L, Ootani A, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20:769–77. doi: 10.1038/nm.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian Y, Jia X, Wang S, et al. SOX2 oncogenes amplified and operate to activate AKT signaling in gastric cancer and predict immunotherapy responsiveness. J Cancer Res Clin Oncol. 2014;140:1117–24. doi: 10.1007/s00432-014-1660-0. [DOI] [PubMed] [Google Scholar]
- 61.Arteaga CL, Sliwkowski MX, Osborne CK, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 62.Van Cutsem E, Bang YJ, Feng-Yi F, et al. HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer. 2014 doi: 10.1007/sl0120-014-0402-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nordenstedt H, Graham DY, Kramer JR, et al. Helicobacter pylori-negative gastritis: prevalence and risk factors. Am J Gastroenterol. 2013;108:65–71. doi: 10.1038/ajg.2012.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeffries GH. Recovery of gastric mucosal structure and function in pernicious anemia during prednisolone therapy. Gastroenterology. 1965;48:371–78. [PubMed] [Google Scholar]
- 65.Jeffries GH, Todd JE, Sleisenger MH. The effect of prednisolone on gastric mucosal histology, gastric secretion, and vitamin B 12 absorption in patients with pernicious anemia. J Clin Invest. 1966;45:803–12. doi: 10.1172/JCI105395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baggett RT, Welsh JD. Observations on the effects of glucocorticoid administration in pernicious anemia. Am J Dig Dis. 1970;15:871–81. doi: 10.1007/BF02236052. [DOI] [PubMed] [Google Scholar]
- 67.Rodbro R, Dige-Petersen H, Schwartz M, et al. Effect of steroids on gastric mucosal structure and function in pernicious anemia. Acta Med Scand. 1967;181:445–52. doi: 10.1111/j.0954-6820.1967.tb07262.x. [DOI] [PubMed] [Google Scholar]
- 68.Ardeman S, Chanarin I. Steroids and addisonian pernicious anemia. N Engl J Med. 1965;273:1352–55. doi: 10.1056/NEJM196512162732502. [DOI] [PubMed] [Google Scholar]
- 69.Goldenring JR. Gastric intestinal metaplasia and tamoxifen: can we reverse the inevitable? Dig Dis Sci. 2014;59:1078–79. doi: 10.1007/s10620-014-3088-4. [DOI] [PubMed] [Google Scholar]
- 70.Toller IM, Hitzler I, Sayi A, et al. Prostaglandin E2 prevents Helicobacter-induced gastric preneoplasia and facilitates persistent infection in a mouse model. Gastroenterology. 2010;138:1455–67. doi: 10.1053/j.gastro.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Toller IM, Altmeyer M, Kohler E, et al. Inhibition of ADP ribosylation prevents and cures Helicobacter-induced gastric preneoplasia. Cancer Res. 2010;70:5912–22. doi: 10.1158/0008-5472.CAN-10-0528. [DOI] [PubMed] [Google Scholar]
- 72.Sayi A, Kohler E, Toller IM, et al. TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J Immunol. 2011;186:878–90. doi: 10.4049/jimmunol.1002269. [DOI] [PubMed] [Google Scholar]
- 73.Moon CM, Kim SH, Lee SK, et al. Chronic tamoxifen use is associated with a decreased risk of intestinal metaplasia in human gastric epithelium. Dig Dis Sci. 2014;59:1244–54. doi: 10.1007/s10620-013-2994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mesquita P, Raquel A, Nuno L, et al. Metaplasia-a trans-differentiation process that facilitates cancer development: the model of gastric intestinal metaplasia. Crit Rev Oncog. 2006;12:3–26. doi: 10.1615/critrevoncog.v12.i1-2.20. [DOI] [PubMed] [Google Scholar]