

ABSTRACT
Non-small cell lung cancer (NSCLC) still constitutes the most common cancer-related cause of death worldwide. All efforts to introduce suitable treatment options using chemotherapeutics or targeted therapies have, up to this point, failed to exhibit a substantial effect on the 5-year-survival rate. The involvement of epigenetic alterations in the evolution of different cancers has led to the development of epigenetics-based therapies, mainly targeting DNA methyltransferases (DNMTs) and histone-modifying enzymes. So far, their greatest success stories have been registered in hematologic neoplasias. As the effects of epigenetic single agent treatment of solid tumors have been limited, the investigative focus now lies on combination therapies of epigenetically active agents with conventional chemotherapy, immunotherapy, or kinase inhibitors. This review includes a short overview of the most important preclinical approaches as well as an extensive discussion of clinical trials using epigenetic combination therapies in NSCLC, including ongoing trials. Thus, we are providing an overview of what lies ahead in the field of epigenetic combinatory therapies of NSCLC in the coming years.
KEYWORDS: Chromatin, DNA methylation; DNMT, epigenetics, histone modifications, LSD1, lung cancer, NSCLC
Abbreviations
- AML
Acute myeloid leukemia
- ATRA
All-trans retinoic acid
- AZA
Azacitidine
- CR
Complete remission
- DLTs
Dose limiting toxicities
- DNMTi
DNA methyltransferase inhibitors
- DNMTs
DNA methyltransferases
- EGFR
Epidermal growth factor receptor
- EMA
European Medicine Agency
- EZH2
Enhancer of zeste homolog 2
- FDA
Food and Drug Administration
- HATs
Histone acetyltransferases
- HDAC
Histone deacetylases
- HDACi
HDAC inhibitors
- KDMs
Histone lysine demethylases
- KMTs
Histone lysine methyltransferases
- MDS
Myelodysplastic syndrome
- MTD
Maximum tolerated dose
- NSCLC
Non-small cell lung cancer
- OS
Overall survival
- P2RD
Phase 2 recommended dose
- PD-1
Programmed cell death receptor 1
- PD-L1
Programmed cell death receptor ligand 1
- PFS
Progression-free survival
- PR
Partial remission
- RFS
Relapse-free survival
- SAM
S-adenosyl methionine
- SCLC
Small cell lung cancer
- SD
Stable disease
- TC
Treatment choice
- TKIs
Tyrosine kinase inhibitors
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide. Despite continuous research and development of new therapeutic regimens, the 5-year overall survival (OS) rate of non-small cell lung cancer (NSCLC) remains at a mere 15%.1 Epigenetic therapy approaches offer novel, innovative treatment options that may improve this troubling statistic, namely with DNA methyltransferase inhibitors (DNMTi) and histone-modifying agents. These classes of compounds have been clinically tested as single agents and in combination with chemotherapeutics, small-molecule inhibitor drugs, and differentiating agents. Combination strategies often are used with the rationale to epigenetically “prime” the cancer cells by treatment with epigenetically active agents to the activity of the subsequently administered second agent.2
DNA methylation usually occurs by transfer of a methyl group to the cytosine of a cytosine-guanine dinucleotide (CpG), e.g., of gene promoters. This allows the binding of different proteins that ultimately prohibit the RNA polymerase to access this area and can therefore silence the respective gene.3
Histones are nuclear proteins around which the DNA is wrapped. Posttranslational modifications, such as addition or removal of methyl or acetyl groups to amino acids within the histones, can lead to a change of conformation and therefore facilitate or hinder access of the transcription factor machinery to the DNA.4,5
Reversing the aberrant epigenetic patterns of cancer cells can re-sensitize them to established treatment, e.g., chemotherapeutics or radiation therapy. In this review, we provide an overview of published and ongoing clinical combination trials using epigenetic drugs in NSCLC.
DNA methylation
DNA methyltransferases (DNMTs) transfer methyl groups to cytosines by employing S-adenosyl methionine (SAM) as their methyl donor. Both DNA hypo- and hyper-methylation are found in cancer cells, the latter can lead to silencing of tumor suppressor genes6 or of genes that are involved in, e.g., metastasis, angiogenesis, invasion, or immune response by T-cell recognition.7 Table 1 provides a list of currently investigated DNMTi.
Table 1.
Currently available DNA methyltransferase inhibitors. This table lists the most important DNMTi used in research. Nucleoside analogs resemble nucleosides, but lead to a chain termination when they are incorporated in the DNA. Antisense oligonucleotide inhibitors hybridize with their complementary mRNAs, prevent their translation and thereby the biosynthesis of certain proteins.99
| DNA Methyltransferase Inhibitors | |
|---|---|
| Substance Group | Substance Name |
| Nucleoside analogs | Decitabine (5-aza-2′-deoxycytidine, Dacogen®, DAC)*+ |
| Azacitidine (5-azacytidine, Vidaza®)*+ | |
| 5-aza-fluoro-2′-deoxycytidine (FCdR) | |
| CC486 (oral azacitidine) | |
| Guadecitabine (SGI-110) | |
| Sinefungin | |
| Zebularine | |
| Antisense oligonucleotide (ASO) inhibitors | DNMT1 ASO |
| MG98 | |
| Others | 1-Hydrazinophthalazine |
| CBC12 | |
| Epigallocatechin gallate (EGCG) | |
| Procainamide | |
| Psammaplin A | |
| RG 108 | |
| SGI-1027 | |
| Thioguanine |
Food and Drug Administration (FDA)-approved drugs +European Medicine Agency (EMA)−approved drugs.
Azacitidine (AZA) and 5-aza-2′-deoxycytidine (decitabine), the 2 clinically approved DNMT inhibitors (DNMTi), are generally considered the “flagships” of epigenetic therapy. After a lengthy development period, they have finally become accepted as new, non-intensive frontline treatment standards for (mostly elderly) patients with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). In these 2 related myeloid neoplasias, remission rates, improvements in survival, and quality of life are very encouraging. Their role in the treatment of solid tumors is much less defined, necessitating active investigations, including their mechanisms of action.
Histone modifications: Enzymes and their inhibitors
Histone modifications are dynamic processes that are regulated by so-called “writer” and “eraser” enzymes. These writers and erasers either deposit or remove specific posttranslational modifications from histones, which in turn are recognized by “readers.” The 2 most broadly studied chemical modifications of histones, on which we focus in this review, are histone acetylation and histone methylation. Whether a histone modification acts as a repressive or active mark depends on the chemical group itself as well as its position within the histone. Fig. 1 depicts the most relevant modifications at histone H3, including their function, the respective modifying enzymes, as well as prototypical pharmacological inhibitors of these enzymes.
Figure 1.
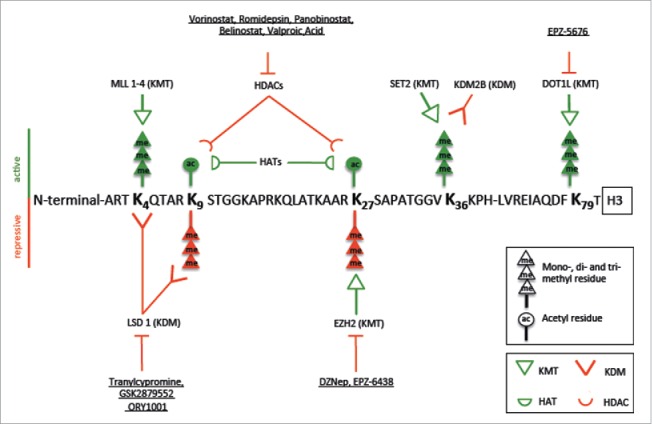
Amino acid sequence of histone H3 with active (green) and repressive (red) marks attached to lysine residues (K). Histone marks, such as acetyl and methyl groups, are regulated by writers (e.g., EZH2) and erasers (e.g., LSD1, HDACs). Depending on their position, they can either lead to an active or an inactive chromatin conformation. The finely tuned regulation is often altered in human cancers. Epigenetic drugs, such as tranylcypromine or DZNep, inhibit the chromatin modifying enzymes and may thereby influence gene expression and revert aberrant gene silencing. (KMT: histone lysine methyltransferase, KDM: histone demethylase, HDAC: histone deacetylase, HAT: histone acetyltransferase).
Histone acetyltransferases (HATs) add acetyl groups to lysines and lead to an open conformation of the chromatin by altering its charge, which supports active transcription.
The four different classes of histone deacetylases (HDAC I-IV), on the other hand, are global mediators of transcriptional repression. Like HATs, they also target non-histone proteins. HDACs are overexpressed, or their recruitment is altered, in a large variety of cancers, including NSCLC.8 In vitro, HDAC inhibitors (HDACi) exhibit antineoplastic activity in cancer cells by inhibiting proliferation and angiogenesis. Additionally, they induce apoptosis by regulating both pro- and anti-apoptotic genes. Six different structural categories of HDACi have been described, and drugs representing 3 of them have been approved for cancer treatment: Vorinostat, belinostat, chidamide, and romidepsin for cutaneous and peripheral T-cell lymphoma,9,10,11 and panobinostat for multiple myeloma.12 Table 2 provides a list of currently investigated HDAC inhibitors.
Table 2.
Currently investigated histone deacetylase inhibitors. This table depicts the most important HDACi that are being used in research. They can be categorized into 6 groups: hydroxamic acid based compounds, cyclic tetrapeptides, short-chain and aromatic fatty acids, benzamides, electrophilic ketone, and others.
| Histone Deacetylase Inhibitors8,100–106 | ||
|---|---|---|
| Substance Group | Substance Name | Targeted HDACs |
| Hydroxamic acid based compounds | Givinostat (ITF2357) | Class I and II |
| Panobinostat (LBH 589)*+ | Class I and II | |
| Dacinostat (NVP-LAQ 824) | Class I and II | |
| Oxamflatin | Class I and II | |
| Abexinostat (PCI-24781) | Class I and II | |
| Pracinostat (SB939) | Class I, II and IV | |
| Belinostat (PXD101)*+ | Class I and II | |
| Quisinostat | Class I and II | |
| Resminostat | Class I and II | |
| Rocilinostat | Class IIb | |
| Suberoylanilide hydroxamic acid (SAHA, Vorinostat)* | Class I and II | |
| Trichostatin A (TSA) | Class I and II | |
| Cyclic peptides | Apicidin | Class I and III |
| Trapoxin A and B | Class I and IIa | |
| Romidepsin (FK228, Depsipeptide)* | Class I | |
| Short-chain and aromatic fatty acids and their salts | (Sodium) 4-phenylbutyrate | Class I and II |
| Sodium butyrate | Class I and IIa | |
| Valproic acid/Valproate (VPA) | Class I and IIa | |
| Benzamides | Chidamide’ | Class I |
| Tacedinaline (CI-994) | Class I | |
| Mocetinostat (MGCD0103) | Class I | |
| Entinostat (MS-275) | Class I | |
| Electrophilic ketone | Trifluoromethylketone | Class II |
| Others | Psammaplin A | Class I |
*FDA-approved drugs +EMA-approved drugs 'Chinese FDA-approved drugs.
Histone methylation marks are deposited by histone lysine methyltransferases (KMTs), which can be divided into non-SET and SET-domain-containing KMTs (e.g., MLL1-5, SET1A/B, SET 7/9). They can be removed by histone lysine demethylases (KDMs), such as LSD-1 and -2, or JmJD-domain-containing histone demethylases.13,14
LSD1 demethylates mono- and di-methylated lysines 4 and 9 of histone H3. It is frequently overexpressed in NSCLC and promotes proliferation and invasion.15 Again, the efficacy of LSD1 inhibition is deduced from its success in hematology: LSD1 inhibition was shown to remove the differentiation block in AML cells16 and re-sensitize AML cells to all-trans retinoic acid (ATRA).17 The first compound clinically used as an LSD1 inhibitor is tranylcypromine, a monoamine oxidase inhibitor approved more than 50 y ago for treatment-refractory depression. More potent and specific LSD1 inhibitors are presently under preclinical and early clinical development.18
The methylation of lysine 27 of histone H3, H3K27, is regulated by the enhancer of zeste homolog 2 (EZH2), the catalytic domain of the polycomb repressive complex 2 (PRC2). Trimethylation of H3K27 by EZH2 leads to silencing of PRC2 target genes that are involved in stem cell differentiation and embryonic development. EZH2 is overexpressed in a variety of cancers, including NSCLC. 3-Deazaneplanocin A (DZNep) is an EZH2 inhibitor that leads to reduced trimethylated H3K27 levels in breast cancer cells and the de-repression of aberrantly silenced genes.19 Table 3 provides a list of currently investigated histone methylation modifiers.
Table 3.
Currently available histone methylation modifiers. This table depicts the most important histone methyltransferase and demethylase inhibitors that are being used in research. EZH2 catalyzes the addition of methyl groups to histone H3 at lysine 27. LSD1 demethylates di- and tri-methylated H3K4.
| Histone Methyltransferase and Demethylase Inhibitors | |
|---|---|
| Group | Substance name |
| EZH2 inhibition | 3-deazaneplanocin A (DZnep) |
| Tazemetostat (EPZ-6438) | |
| GSK126 | |
| GSK2816126 | |
| GSK343 | |
| UNC-1999 | |
| LSD1 inhibition | Tranylcypromine (2-PCPA) |
| GSK LSD1 | |
| GSK2879552 | |
| ORY-1001 | |
| Pargyline | |
| RN 1 dihydrochloride | |
| SP2509 | |
| Dual HDAC-LSD1 Inhibition | 4SC-202 |
| DOT1L inhibition | EPZ-5676 |
Epigenetic therapy in myeloid neoplasias and multiple myeloma
Until now, epigenetic therapy has been most effective in hematologic neoplasias. In a randomized phase 3 study in older patients with mostly higher-risk MDS comparing azacitidine (administered over 7 d every 4 weeks) to standard of care, the 2-year survival was 51% in the azacitidine arm compared to 26% in the standard of care arm.20 In another randomized phase 3 trial for newly diagnosed AML patients of 65 y and older and more than 30% bone marrow blasts, azacitidine (given at the same dose and schedule as for MDS) increased median overall survival from 6.5 months in the conventional treatment arm to 10.4 months in the azacitidine arm.21
In a phase 3 trial with 170 MDS patients, decitabine led to a higher response rate (17% vs. 0%) and a trend to longer progression-free survival (PFS) (12.1 vs. 7.8 months) when compared to best supportive care.22 The EORTC Intergroup trial 06011 with 233 higher-risk MDS-patients of 60 y and older comparing low-dose decitabine with sole best supportive care resulted in improvement of PFS, AML transformation, and quality of life.23 Decitabine is also active in older AML patients. Particularly, when administered over 10 d decitabine induced response rates almost comparable to standard induction chemotherapy.24,25In a phase III trial comparing decitabine with treatment choice (TC) in 485 newly diagnosed AML patients of 65 y and older with poor to intermediate risk cytogenetics, decitabine led to an increase in complete remission rate (17.8% with decitabine vs. 7.8% with TC) and longer OS.26
Last but not least, the HDAC inhibitor panobinostat significantly increased PFS and complete remission rate in multiple myeloma when combined with bortezomib and dexamethasone.27
Potential for epigenetic therapy in lung cancer
Epigenetic changes are an important feature in NSCLC development, making them viable targets in lung cancer therapy. Aberrant promoter methylation of genes like CDKN2A 28, MLH1 and MSH2 29, APC 30, RARB 31, MGMT,32 and many others33 has been described in lung cancer. Furthermore, different chromatin modifications can be used as prognostic markers. For example, overexpression of class I HDACs34 and globally elevated H3 and H4 methylation are associated with a poor prognosis35, whereas high dimethyl H3K4 levels and low acetylated H3K9ac36 appeared to confer a better prognosis.
Two groups of lung cancer patients might obtain particular benefit from epigenetically active drugs: patients that are not fit enough for aggressive chemotherapy and high-risk NSCLC patients. The first group, i.e., patients not eligible for chemotherapy, might still be fit enough for the—usually less energy draining—epigenetic therapy. Rather than being cytotoxic like conventional chemotherapeutics, epigenetic therapies are thought to induce apoptosis and/or differentiation by reversing aberrant silencing or activation of genes. Elegantly enough, this should in principle eliminate only cancer cells, leaving normal cells untouched, resulting in fewer and less intense side effects. The second group comprises high-risk NSCLC patients, i.e., those with shorter relapse-free survival (RFS), who seem to be prone to relevant epigenetic alterations, e.g., harboring aberrant DNA methylation of HIST1H4F, PCDHGB6, NPBWR1, ALX1, and HOXA9.37
Combination treatment including epigenetically active agents: Preclinical studies
DNMTi38, HDACi39, inhibitors of EZH2,40 and LSD115 have all been demonstrated to have anticancer effects in in vitro NSCLC studies. However, single-agent clinical trials of these groups of inhibitors in lung cancer patients showed mostly limited or transient effects, or high toxicity41,42, which is why combination therapies seem favorable. Epigenetic agents have been shown to be able to “prime” cancer cells to standard chemotherapy, possibly by reactivation of tumor suppressor genes or DNA repair pathways.43 They can also be used to re-sensitize cancer cells after the development of resistance, e.g., to tyrosine kinase inhibitors (TKIs).44 The following paragraphs provide an overview of some interesting preclinical studies using epigenetic drugs in NSCLC.
Preclinical combination therapy with DNMTi
Azacitidine was shown to act synergistically with cytarabine and etoposide in NSCLC cell lines.45 This combination led to a further hypomethylation of CpG sites located within 2 tumor suppressor genes (MGMT and THRB).
Li et al. observed a correlation between DNA promoter methylation of the EGFR gene and TKI resistance in NSCLC cell lines. DNMT inhibition using decitabine enhanced or even restored sensitivity to gefitinib, resulting in growth inhibition and apoptosis, as well as reduced EGFR protein expression.46
Several proven or potential DNMTi (azacitidine, decitabine, zebularine, hydralazine, epigallocatechin gallate, and psammaplin A) caused radiosensitization in the NSCLC cell line A549.47
Preclinical combination therapy with HDACi
Two novel HDAC inhibitors (ST2782 and ST3595) showed a synergistic effect with taxanes, which act by stabilizing the microtubuli in the spindle apparatus and disrupting mitosis. Combination therapy was followed by an increase in growth inhibition, apoptosis, and cell cycle delay at the G2/M-transition in different cancer cell lines, among them, the NSCLC cells H460 and A549. This might be caused by a supportive effect of acetylation on microtubular stabilization.48,49
HDACi also led to downregulation of thymidylate synthase, an enzyme involved in the folate cycle and a target of cytostatic agents such as pemetrexed. High thymidylate synthase levels correlate with pemetrexed resistance. When sequentially treated with pemetrexed followed by ITF2357, a pan-HDACi, several NSCLC cell lines showed a synergistic effect on growth inhibition and apoptosis. Results were confirmed in xenograft models derived from the adenocarcinoma cell line H1650.50
The HDACi romidepsin was able to enhance the anti-tumor effect of erlotinib in 9 NSCLC lines of different histology and mutation status, including EGFR-, KRAS-mutant, and wild type cell lines, as well as reduce tumor burden in NCI-H1299 xenograft models.51 HDAC inhibition using entinostat (MS-275) re-sensitized different TKI-resistant NSCLC cell lines to gefitinib, probably by restoring E-Cadherin expression.52
The pan-HDAC-inhibitor Panobinostat did not only make TKI-resistant A549-cells available to the antineoplastic activity of Erlotinib, it also led to an increase in mono-, di- and trimethylation of histone H3 lysine 4 (H3K4), an indicator for a crosstalk between HDAC inhibitor and LSD1.53 The same pan-HDACi, panobinostat, was also able to prime NSCLC cell lines to the differentiating effect of ATRA54 and, furthermore, combining ATRA with the novel HDACi SL142 or SL325 suppressed colony formation, induced apoptosis via Bax expression, and increased caspase-3 activity in NSCLC cell lines.55
The HDACi sodium valproate also enhanced the anti-tumoral effect of cisplatinum-vinorelbine-based chemoradiation in NSCLC cell lines.56 Trichostatin A (TSA) was also able to radiosensitize NSCLC cell lines, promoting apoptosis and G2/M-cell-cycle arrest.57
Preclinical combination treatment approaches targeting aberrant histone methylation
Aberrant histone methylation is a relatively recently discovered feature in NSCLC, which is reflected in the scarceness of studies using agents affecting histone methylation.
Fillmore et al. could show that EZH2 knockdown as well as indirect EZH2 inhibition using 3-deazaneplanocin A (DZNep) were both able to prime NSCLC cell lines to the effect of the topoisomerase inhibitor etoposide.58
Even less research has until now been conducted concerning aberrant histone demethylation in NSCLC. LSD1 knockdown as well as LSD1 inhibition using pargyline suppressed invasion, migration, and proliferation in lung cancer specimens;15 crosstalk between LSD1i and HDACi is known to play a role in breast cancer.59 To our knowledge, there are as yet no studies investigating combination therapies for lung cancer using LSD1 inhibitors.
Preclinical epigenetic combination therapy
There is substantial crosstalk between the different epigenetic regulator enzymes, e.g., between LSD1 and class I, II and IV HDACs in breast cancer,59 as well as DNA methylation and histone methylation.60 Different histone modifications even influence each other in yeast cells.61 These findings have led to a number of epigenetic combination studies, but only few targeting lung cancer.
The combination of the HDACi SAHA and the non-specific EZH2 inhibitor DZNep decreased phosphorylation of proteins essential for the EGFR pathway, increased apoptosis in NSCLC cell lines and reduced tumor burden in H1975 xenograft mice.62
Combining the DNMTi azacitidine with the HDACi entinostat led to the reduction of tumor burden, increased expression of pro-apoptotic genes and genes involved in cell cycle regulation in an orthotopic lung cancer model.63
These encouraging results stand in contrast to a series of experiments on NSCLC cell lines and xenografts using azacitidine and entinostat as priming agents to chemotherapeutics published by Vendetti et al. in 2014. The group found no difference in the response to cisplatin, docetaxel, 17-AAG (an antitumor antibiotic), or gemcitabine after epigenetic priming in A549, H358, H838, or H1229 cells. And, although epigenetic priming enhanced the response of A549 xenografts to irinotecan, no effect on A549 or H460 xenografts was seen when using cisplatin or docetaxel. Priming even diminished the effect of irinotecan on H460 cells. Application of azacitidine and entinostat did however enhance the response to repeat treatment of irinotecan in a patient derived adenocarcinoma xenograft. All in all, the results of Vendetti et al. did not generally strengthen the hypothesis of epigenetic chemosensitization. The authors themselves note that efficacy might be context- and host-dependent and encourage further investigation.64
Clinical trials
Single-agent clinical trials using chromatin-modifying drugs
The Canadian scientist Richard Momparler is one of the pioneers in the epigenetic treatment of NSCLC patients. In a trial in the 1990s using decitabine in 15 stage IV NSCLC patients with no prior treatment, one patient survived more than 81 months.65 After this astonishing observation, Momparler et al. suggested a delayed mode of action of decitabine, which had almost been dismissed as a treatment choice in NSCLC, and thus sparked a new interest in the field. Since then, numerous clinical trials have studied the single-agent effects of DNMTi38,66 or HDACi67,68 in NSCLC. As mentioned earlier, however, clinical activity was often limited due to transient effects or toxicity. Combining epigenetic substances with established therapies might increase efficacy and reduce side effects. Table 4 depicts ongoing and published trials using epigenetic combination therapies in NSCLC.
Table 4.
Ongoing and published trials using epigenetic combination therapies in NSCLC. In this table, ongoing and finished trials using epigenetic combination therapies in NSCLC are displayed, including their phase, number of recruited patients, dose limiting toxicity, response rate respectively median survival, study population, and their status (ongoing or published).
| Drugs | Phase | Patient number | Dose-Limiting Toxicities | Response rate, PFS, OS (months) | Study population | Reference |
|---|---|---|---|---|---|---|
| DNMTi combination therapy | ||||||
| Azacitidine + erlotinib | 1 | 30 | Conjunctivitis, infusion reaction in 2 of 5 cohorts | PR in 2, SD in 11 patients; Median PFS 2 months | Advanced solid tumors, among them 2 NSCLC patients, who had already received standard therapy | 68 |
| Oral azacitidine + pembrolizumab vs. placebo + pembrolizumab* | 1 | 90 | — | — | Patients with squamous or non-squamous stage IIIB or IV NSCLC pretreated with only 1 prior systemic platinum-based chemotherapy | 95* |
| 5-Fluoro-2-deoxycytidine + tetrahydrouridine* | 2 | Ca. 185 | — | — | Advanced NSCLC, breast cancer, bladder cancer, or head or neck cancer that had progressed after standard treatment or for whom no effective therapy exists | 78* |
| Decitabine + cisplatin | 1/2 | 21/14 | — | No objective response; OS 3,7 months | Phase 1: Patients with histopathologically confirmed diagnosis of malignancy and progressive disease; Phase 2: stage IIIB and IV NSCLC | 69 |
| Decitabine + genistein* | 2 | Ca. 48 | — | — | Phase 1: Non-estrogen dependent advanced solid malignancy that has failed standard therapies and/or for which no curative therapeutic option exists, Phase 2a: NSCLC of stage IIIb or IV that has failed or is ineligible to standard therapies |
77* |
| HDAC Combination therapy | ||||||
| Belinostat, carboplatin, paclitaxel* and bevacizumab* | 1/2 | 7 | — | — | Advanced NSCLC (stage IV), not previously treated with any chemotherapy regimen (prior adjuvant chemotherapy and/or chemotherapy/radiation for stage III allowed) | 81* |
| Belinostat + Erlotinib* | 1/2 | 5 | — | — | Dose escalation phase: NSCLC patients suitable for treatment with Erlotinib; MTD expansion phase: patients with NSCLC rated suitable for treatment with Erlotinib and with measurable disease | 82* |
| Entinostat + Pembrolizumab* | 1b/2 | 158 | — | — | Patients with recurrent/metastatic NSCLC tested for anaplastic lymphoma kinase (ALK) rearrangements and epidermal growth factor receptor (EGFR) mutations and, if positive, have been treated with prior EGFR or ALK therapy; at least 1 chemotherapeutic regimen; progressive disease; no prior treatment with a PD-1/PD-L1-blocking antibody | 96* |
| Erlotinib +/− entinostat | 2 | 132 | — | Only patients with high E-cadherin levels at time of diagnosis benefit from combination | Stage III or stage IV NSCLC patents that had received one or 2 previous chemotherapy or chemoradiotherapy regimens for advanced NSCLC and their disease had progressed based on radiologic evidence | 74 |
| Paclitaxel + carboplatin +/− vorinostat* | 2/3 | 253 | — | — | NSCLC patients with no prior systemic treatment for lung cancer except patients at least 12 months from prior adjuvant therapy | 80* |
| Panobinostat + erlotinib | 1 | 33 | 2 grade 3 DLTs of nausea and grade 3 prolonged QTc | 7 EGFR mutant patients: 3 PR, 3 SD, 1 progressed; PFS was 4.7 in EGFR-mutant vs. One.9 months in EGFR wild-type patients, OS est. 41 vs. Five.2 months | Patients with advanced/metastatic NSCLC or head and neck cancer, who had failed at least one line of systemic therapy | 72 |
| Vorinostat + bortezomib + Surgery | 1 | 21 | 2 grade III DLTs of fatigue and hypophosphatemia | >60% histologic necrosis of tumor in 6 of 20 patients | NSCLC patient that had no clinical or pathologic evidence of N2, N3, or M1 disease | 70 |
| Vorinostat or placebo + Carboplatin + paclitaxel | 2 | 94 | — | PFS 6.0 vs. Four.1 months | Stage IIIB (with malignant pleural effusion) or IV NSCLC patients with no prior therapy for advanced-stage disease | 75 |
| Vorinostat + erlotinib | 1/2 | 1 DLT of grade 3 diarrhea | No objective response; PFS 8 weeks, OS 10.3 months | NSCLC patients with advanced disease and EGFR mutations in exons 19 or 21, who had been treated with full doses of erlotinib for a minimum of 3 months | 73 | |
| Vorinostat + gemcitabine + platinum* | 1 | 61 | — | — | Metastatic or locally advanced NSCLC patients that have not been previously treated with systemic chemotherapy or have received non-platinum and non- gemcitabine based neoadjuvant or adjuvant chemotherapy if the last dose was at least 6 months prior to study enrollment | 79* |
| Vorinostat + sorafenib | 1 | 35 | — | 1 PR, 8 SD; PFS 2.2 months | Locally advanced or metastatic solid tumors refractory to established forms of therapy or for which sorafenib alone would be considered as appropriate therapy, among them 15 NSCLC patients | 71 |
| DNMTi+HDACi combination therapy | ||||||
| Azacitidine (s.c.) + oral entinostat followed by chemotherapy* | 2 | Ca. 165 | — | — | NSCLC patients that have received exactly one prior therapy; patients with epidermal growth factor receptor (EGFR) mutations in exon 19 or 21 and patients with detected anaplastic lymphoma kinase (ALK) translocation may have had 2 prior therapies if one was a tyrosine kinase inhibitor specific to their mutation | 83* |
| Azacitidine (s.c.) + oral entinostat or oral azacitidine alone prior to nivolumab* | 2 | Ca. 120 | — | — | Stage IIIB, IV or recurrent NSCLC patients with at least one platinum based chemotherapy, and not more than 3 prior therapies for stage IIIB/IV disease | 93* |
| Decitabine + valproic acid | 1 | 8 | 2 grade 3 DLTs of neurotoxicity | No objective response; | NSCLC patients | 76 |
| Azacitidine + entinostat | 1/2 | 10/42 | 0 | 1 CR, 1 PR for 24 months | Metastatic NSCLC patients with disease progression after at least one prior anti-cancer regimen for metastatic disease; any number of prior therapies was allowed; patients with treated brain metastases were included | 42 |
| Azacitidine + entinostat* | 1/2 | 162 | — | — | Metastatic or unresectable NSCLC patients that have failed at least one previous chemotherapy regimen | 84* |
| LSD1-inhibition (single treatment in SCLC) | ||||||
| GSK2879552* | 1 | 100 | — | — | SCLC patients with recurrent or refractory disease after receiving at least one prior standard/approved platinum-containing chemotherapy regimen, or where standard therapy is refused | 86* |
CR = Complete response, DLT = Dose limiting toxicity, OS = overall survival, PR = partial response, PFS = progression free survival, SD = stable disease, − = Information was not provided
= Ongoing
Combining DNMTi with erlotinib or cisplatinum
In a phase I trial with 30 patients with different tumor entities including 2 patients with NSCLC, Bauman et al. showed that azacitidine plus erlotinib was well tolerated. The phase 2 recommended dose (P2RD) was 150 mg erlotinib daily and 75 mg/m2 azacitidine daily on days 1–4 and 15–18 of a 28-day cycle.69
Schwartsmann et al. conducted a phase I trial with 21 patients with different tumor entities including 8 NSCLC patients and a consecutive phase II trial with 14 inoperable, non-pretreated stage III and IV NSCLC patients using decitabine and cisplatin. Apart from dose finding, they measured adverse effects, response rate, and median survival. The dose-limiting toxicity was myelosuppression, median survival was 15 weeks—a result comparable or slightly inferior to cisplatin alone. The disappointing result might be partially explained by a predominance of stage IV cancer patients.70
Combination therapy of NSCLC using HDACi
After conducting a phase I study with 21 NSCLC patients, Jones et al. concluded that HDAC inhibition using vorinostat combined with proteasome inhibition using bortezomib followed by surgery was a feasible treatment option.71
Another phase I study with 17 patients with different cancers, including 3 NSCLC patients, determined the phase 2 recommended dose (P2RD) of vorinostat and the receptor tyrosine kinase inhibitor (TKI) sorafenib to be vorinostat 300 mg on days 1 to 14 and sorafenib 400 mg daily during a 21-day cycle. Using this dosing scheme, 12 additional NSCLC patients were further evaluated. While the entire patient group tolerated the drugs well, the NSCLC group showed one case of grade V hemoptysis and one coronary event, the majority was not able to finish 2 cycles.72
In a phase 1 trial, panobinostat plus erlotinib in 33 patients with NSCLC and head and neck cancer was overall well tolerated, dose-limiting toxicities (DLTs) occurred in 2 patients (one grade 3 nausea and grade 3 prolonged QTc), but resolved without interference. The combination also resulted in an OS of 41 (estimated) vs. 5.2 months in patients harboring an epidermal growth factor receptor (EGFR) mutation vs. wild type EGFR patients. The trial therefore identified EGFR mutated patients as especially susceptible to this combination treatment.73
Although it could be safely administered, vorinostat combined with erlotinib only prevented progression in 28 percent of the patients at 12 weeks after treatment in a phase I/II study with 33 NSCLC EGFR-mutant patients, who had progressed after erlotinib treatment.74
In a phase II trial with 132 stage III and IV NSCLC patients who had advanced after prior treatment (one or 2 previous chemotherapy or chemoradiotherapy regimens), entinostat with the TKI erlotinib did not exhibit a beneficial effect on the study population. However, for those patients showing high E-cadherin levels at enrolment, the OS was longer compared to erlotinib alone.75
Ninety-four patients with advanced NSCLC were enrolled in the randomized, double blind, placebo-controlled phase II trial by Ramalingam et al. comparing cisplatin/paclitaxel combined either with vorinostat or placebo. The combination therapy was superior regarding response rate (34% vs. 12.5%, P = 0.02), but not median PFS (6.0 vs. 4.1 months, P = 0.48) or OS (13.0 vs. 9.7 months, P = 0.17).76
Therapy approaches combining different epigenetically effective drugs
The combination of decitabine with valproic acid in a phase I trial with 8 NSCLC patients led to an increase of fetal hemoglobin, which indicated reactivation of initially silenced β-globin genes by hypomethylation. However, a phase II study was not recommended because of grade 3 neurological toxicities in 2 patients including disorientation, lethargy, memory loss, and ataxia at dose level 1.77
In 10 extensively pretreated, refractory advanced NSCLC patients in a phase I/II trial, the combination of azacitidine and entinostat was well tolerated. Following epigenetic treatment, a complete remission was observed in one patient and a partial remission in another, who remained with stable disease for about 2 y after ending the study.42 Median survival was 6.4 months. However, a subset of 10 “methylation signature”-positive patients was identified, whose median survival amounted to 10.4 months. These patients showed demethylation of at least 2 of 4 initially silenced NSCLC signature genes (APC, RASSF1A, CDH13, CDKN2A) following treatment. Eight of these “methylation signature”-positive patients had stable disease or an objective response. Of the 16 identified “methylation signature”-negative patients, only 4 showed stable disease and no objective responses were observed. Additionally, the epigenetic treatment improved the response rate to subsequent anti-cancer treatments, with 4 of 19 patients enjoying major objective responses.42
Ongoing combination clinical trials
A number of researchers are currently investigating epigenetic combination therapies in NSCLC. The following section provides a summary of ongoing trials, including those that had to be terminated early.
In a phase I study researchers will determine the phase 2 recommended dose (P2RD) of the DNMTi decitabine administered together with the phytoestrogen genistein in patients with solid tumors. The phase II part of this study aims to monitor safety and preliminary efficacy in 48 stage III and IV NSCLC patients.78
Progression-free survival (PFS) and response rate will be evaluated in a currently recruiting phase II study using the DNMTi 5-fluoro-2-deoxycytidine and the competitive cytidine deaminase inhibitor tetrahydrouridine, with a target of 185 NSCLC patients.79
There are several ongoing trials using HDAC inhibitors. To assess DLTs and MTD of vorinostat combined with gemcitabine plus either cis- or carbo-platin is the goal of a phase I study monitoring 61 chemo-naive patients with advanced NSCLC.80
Several studies had to be terminated early: OS and PFS were planned to be measured in a phase II trial using vorinostat in combination with paclitaxel and carboplatin in 253 stage III and IV NSCLC patients. The study had to be terminated as the endpoint had not been achieved.81 Another phase I/II study was terminated for logistic reasons. It had aimed to determine maximum tolerated dose (MTD) of belinostat combined with standard chemotherapy (carboplatin, paclitaxel, and bevacizumab induction) as well as OS and long-term safety in 7 stage IV NSCLC patients.82 Another phase I study investigated belinostat in combination with erlotinib in NSCLC patients; it was terminated when the MTD was exceeded.83
As expected, the current interest in HDAC inhibitors combined with DNMT inhibitors is reflected in a number of ongoing trials using both agent types. A phase II study will test the priming ability of azacitidine combined with entinostat followed by a variety of chemotherapeutic agents (docetaxel, gemcitabine, irinotecan, pemetrexed) and is planning to include 165 advanced NSCLC patients.84
A hundred and sixty-two advanced NSCLC patients have enrolled in an all-epigenetic phase II study, in which MTD and response rate of azacitidine combined with entinostat will be determined.85
Importantly, another study (neither using a combination therapy nor including NSCLC patients) has recently been initiated: the first clinical trial in lung cancer (in this case small cell lung cancer, SCLC) studying a novel LSD1 inhibitor, GSK2879552. This irreversible selective inhibitor of LSD1 led to growth inhibition in a number of SCLC cell lines. The authors could show that certain cell lines harboring a specific hypomethylation signature were more susceptible to LSD1 inhibition than those lacking this signature. The anti-tumor effect was confirmed in SCLC xenograft mice.86 One hundred SCLC patients with recurrent or refractory disease after platinum-based chemotherapy regimen will be enrolled in a phase-1 study examining the safety, including adverse events and DLTs, and disease control rate of GSK2879552.87
New immunotherapeutic approaches using epigenetic therapies
One of the most interesting recent discoveries in hemato-oncology is the induction of immune responses in cancer cells by hypomethylating agents. In a follow-up observation of patients with refractory NSCLC that the group of Wrangle had treated with low-dose azacitidine and the HDAC inhibitor entinostat in 2013, they noticed that a number of these patients had above-average responses to subsequent therapies. Among those, impressively robust remissions were induced by anti-PD1-antibodies. It seemed that azacitidine exhibited a priming effect to other treatment regimen in these patients.42 Encouraged by this observation and to examine the underlying molecular changes, they treated NSCLC cells with 500 nM of azacitidine for 72 h. Azacitidine induced the upregulation of different immune-related pathways as well as the expression of cancer/testis antigens and transcripts of HLA Class I antigens, which are important for tumor recognition and their destruction by cytotoxic T-cells. It also led to the upregulation of programmed cell death receptor ligand 1 (PD-L1), a key ligand-mediator of immune tolerance. Usually the interaction of PD-L1 with its receptor programmed cell death receptor 1 (PD-1) acts as a checkpoint for immunological responses to inflammation. When cancer cells express PD-L1, it can enable them to evade recognition and degradation by cytotoxic T-cells.88 Anti-PD1 antibodies have already been demonstrated to induce complete or partial remissions in NSCLC, e.g., in 5 of 49 NSCLC patients in a phase I study including different tumor types.89 However, a subset of NSCLC patients could be identified who did not respond to the treatment due to low PD-L1 expression.90 The authors then suggested a priming effect of DNA hypomethylating agents to anti-PD-L1-antibodies, especially in those patients with low PD-L1 expression. Two research groups have investigated the effect of azacitidine or decitabine on colorectal and ovarian cancer cells. Very impressive evidence suggests that the anti-tumor activity of these DNMTi is at least partially based on the activation of endogenous retroviral sequences, which express double-stranded RNAs and thus trigger interferon responses that result in elimination of the tumor cells.91,92
The new angle on DNMTi has already led to the set-up of a large phase II study, in which up to 120 patients with recurrent, metastatic NSCLC will be recruited using azacitidine and entinostat or orally administered azacitidine to prime the tumors to the monoclonal anti-PD1 antibody nivolumab.93 Nivolumab has been approved for NSCLC treatment in the EU in 2015 following the CheckMate-017-study.94 Two similar studies exist using the PD-1 inhibitor pembrolizumab, approved for NSCLC and melanoma treatment in the USA. A phase 1 study including up to 90 patients with stage IIIB and IV NSCLC will test the safety and efficacy of oral azacitidine administration in combination with pembrolizumab vs. pembrolizumab and placebo.95 Up to 158 patients will be enrolled in a phase 1b/2 dose escalation study using the HDAC inhibitor entinostat combined with pembrolizumab in patients with NSCLC and an expansion cohort with NSCLC and melanoma patients.96
Summary and conclusions
In this review we describe present developments in the field of epigenetic combination therapies. Combining epigenetic agents with standard treatment may increase their efficacy. Epigenetic drugs as single agents or combined with biologicals might result in treatment options that are available to patients too unfit for aggressive chemotherapy, due to age, reduced performance status, and comorbidities. Efficacy of combining epigenetic drugs with standard chemotherapy has been demonstrated in a plethora of preclinical studies. Only in recent years researchers started to translate these findings into clinical testing, which is why the majority of studies are still phase I and II trials. Different approaches have been taken, mainly combining DNMTi or HDACi with, e.g., chemotherapy, monoclonal antibodies, or TKIs. Especially interesting is the involvement of hypomethylating agents in immunological pathways and their priming effect to anti-PD1-antibodies.
So far, epigenetic combination therapies have not brought the desired breakthrough in NSCLC treatment. A couple of studies had to be terminated due to intolerable toxicities or limited results. However, some trials were able to identify special subgroups (e.g., patients with high E-cadherin levels) that would benefit from a certain combination treatment. Others showed promising results altogether. For instance, the synergistic effect that has been observed when combining HDACi and DNMTi97,98 has sparked a special interest and is reflected in a number of trials using both epigenetic drugs together in combination with standard therapy.
Also, new epigenetic drugs are constantly emerging, as demonstrated by the recent discovery of histone methylation and demethylation and agents modifying these marks, such as EZH2 and LSD1 inhibitors. Combination therapies using these inhibitors are already being tested in a preclinical setting and it seems it is only a matter of time until clinical trials will be designed. In conclusion, epigenetic therapies may yield great opportunities in the treatment of NSCLC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Christoph Plass, Roland Schüle, and Justus Duyster for helpful discussions.
Funding
Supported by the DFG, CRC992 Medep (A04, C04) and DKTK.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer Statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.Miller CP, Singh MM, Rivera-Del Valle N, Manton CA, Chandra J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J Biomed Biotechnol 2011; 2011:514261; PMID:21765634; http://dx.doi.org/ 10.1155/2011/514261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baylin SB, Jones PA. The next 10 years — Timeline: A decade of exploring the cancer epigenome — biological and translational implications. Nat Rev Cancer 2011; 11:726-34; http://dx.doi.org/ 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin Modifications and Their Function. Cell 2007; 128:693-705; PMID:17320507; http://dx.doi.org/ 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 5.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381-95; PMID:21321607; http://dx.doi.org/ 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herman JG, Baylin SB. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N Engl J Med 2003; 349:2042-54; PMID:14627790; http://dx.doi.org/ 10.1056/NEJMra023075 [DOI] [PubMed] [Google Scholar]
- 7.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al.. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527:1-16; http://dx.doi.org/ 10.1038/527S1a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5:769-84; PMID:16955068; http://dx.doi.org/ 10.1038/nrd2133 [DOI] [PubMed] [Google Scholar]
- 9.Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Morschhauser F, Wilhelm M, Pinter-Brown L, et al.. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol 2014; 7:11; PMID:24456586; http://dx.doi.org/ 10.1186/1756-8722-7-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, Yu L, Ke X, Huang H, Shen Z, et al.. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol 2015; 26:1766-71; http://dx.doi.org/ 10.1093/annonc/mdv237. [DOI] [PubMed] [Google Scholar]
- 11.Campbell P, Thomas CM. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J Oncol Pharm Pr 2016; http://dx.doi.org/ 10.1177/1078155216634178. [DOI] [PubMed] [Google Scholar]
- 12.Mu S, Tajima T, Corrado C, Suzuki K, Hino M, Kuroda Y, Shibayama H, Lin R, Waldron E, Binlich F. Panobinostat PK/PD in combination with bortezomib and dexamethasone in patients withrelapsed and relapsed/ refractory multiple myeloma (RRMM). Clin Pharmacol Ther 2015; 97:S49; http://dx.doi.org/ 10.1007/s00228-015-1967-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann I, Roatsch M, Schmitt ML, Carlino L, Pippel M, Sippl W, Jung M. The role of histone demethylases in cancer therapy. Mol Oncol 2012; 6:683-703; PMID:22902149; http://dx.doi.org/ 10.1016/j.molonc.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morera L, Lübbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenet 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One 2012; 7:e35065; PMID:22493729; http://dx.doi.org/ 10.1371/journal.pone.0035065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al.. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012; 21:473-87; PMID:22464800; http://dx.doi.org/ 10.1016/j.ccr.2012.03.014 [DOI] [PubMed] [Google Scholar]
- 17.Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al.. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 2012; 18:605-11; PMID:22406747; http://dx.doi.org/ 10.1038/nm.2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM. Reversible Inhibitors of LSD1 as Therapeutic Agents in Acute Myeloid Leukemia: Clinical Significance and Progress to Date. Med Res Rev 2014; 35:586-618; http://dx.doi.org/ 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi J Takashina T, Kinoshita I, Kikuchi E, Shimizu Y, Sakakibara-Konishi J, Oizumi S, Marquez VE, Nishimura M, Dosaka-Akita H. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer 2012; 29:997-1003; http://dx.doi.org/ 10.1016/j.lungcan.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, et al.. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10:223-32; PMID:19230772; http://dx.doi.org/ 10.1016/S1470-2045(09)70003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, Kumar R, Cavenagh J, Schuh AC, Candoni A, et al.. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015; 126:291-300; PMID:25987659; http://dx.doi.org/ 10.1182/blood-2015-01-621664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kantarjian H, Issa J-PJ, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, et al.. Decitabine improves patient outcomes in myelodysplastic syndromes. Cancer [Internet] 2006; 106:1794-803; PMID:16532500; http://dx.doi.org/ 10.1002/cncr.21792 [DOI] [PubMed] [Google Scholar]
- 23.Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, Selleslag D, Labar B, Germing U, Salih HR, et al.. Low-Dose Decitabine Versus Best Supportive Care in Elderly Patients With Intermediate- or High-Risk Myelodysplastic Syndrome (MDS) Ineligible for Intensive Chemotherapy: Final Results of the Randomized Phase III Study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS. J Clin Oncol 2011; 29:1987-96; PMID:21483003; http://dx.doi.org/ 10.1200/JCO.2010.30.9245 [DOI] [PubMed] [Google Scholar]
- 24.Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, Liu S, Havelange V, Becker H, Schaaf L, et al.. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A 2010; 107:7473-8; PMID:20368434; http://dx.doi.org/ 10.1073/pnas.1002650107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruijsen M, Lübbert M, Wijermans P, Huls G. Clinical Results of Hypomethylating Agents in AML Treatment. J Clin Med 2014; 4:1-17; PMID:26237015; http://dx.doi.org/ 10.3390/jcm4010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, Gau JP, Chou WC, Buckstein R, Cermak J, et al.. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, With Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients With Newly Diagnosed Acute Myeloid Leukemia. J Clin Oncol [Internet] 2012; 30:1-10; http://dx.doi.org/ 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A, Nakorn TN, Siritanaratkul N, et al.. Panobinostat plus bortezomib and dexamethasone vs. placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014; 15:1195-206; PMID:25242045; http://dx.doi.org/ 10.1016/S1470-2045(14)70440-1 [DOI] [PubMed] [Google Scholar]
- 28.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995; 1:686-92; PMID:7585152; http://dx.doi.org/ 10.1038/nm0795-686 [DOI] [PubMed] [Google Scholar]
- 29.Gomes A, Reis-Silva M, Alarcão A, Couceiro P, Sousa V, Carvalho L. Promoter hypermethylation of DNA repair genes MLH1 and MSH2 in adenocarcinomas and squamous cell carcinomas of the lung. Rev Port Pneumol 2014; 20:20-30; PMID:24360395; http://dx.doi.org/ 10.1016/j.rppneu.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 30.Grote HJ, Schmiemann V, Kiel S, Böcking A, Kappes R, Gabbert HE, Sarbia M. Aberrant methylation of the adenomatous polyposis coli promoter 1A in bronchial aspirates from patients with suspected lung cancer. Int J Cancer 2004; 110:751-5; PMID:15146565; http://dx.doi.org/ 10.1002/ijc.20196 [DOI] [PubMed] [Google Scholar]
- 31.Virmani AK, Rathi A, Zöchbauer-Müller S, Sacchi N, Fukuyama Y, Bryant D, Maitra A, Heda S, Fong KM, Thunnissen F, et al.. Promoter methylation and silencing of the retinoic acid receptor-β gene in lung carcinomas. J Natl Cancer Inst 2000; 92:1303-7; PMID:10944551; http://dx.doi.org/ 10.1093/jnci/92.16.1303 [DOI] [PubMed] [Google Scholar]
- 32.Do H, Wong NC, Murone C, John T, Solomon B, Mitchell PL, Dobrovic A. A critical re-assessment of DNA repair gene promoter methylation in non-small cell lung carcinoma. Sci Rep 2014; 4:4186; PMID:24569633; http://dx.doi.org/ 10.1038/srep04186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belinsky SA. Unmasking the Lung Cancer Epigenome. Annu Rev Physiol 2015; 77:453-74; PMID:25668024; http://dx.doi.org/ 10.1146/annurev-physiol-021014-072018 [DOI] [PubMed] [Google Scholar]
- 34.Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, Motoyama S, Ogawa J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011; 74:300-4; PMID:21466904; http://dx.doi.org/ 10.1016/j.lungcan.2011.02.019 [DOI] [PubMed] [Google Scholar]
- 35.Song JS, Kim YS, Kim DK, Park S, Il Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int 2012; 62:182-90; PMID:22360506; http://dx.doi.org/ 10.1111/j.1440-1827.2011.02776.x [DOI] [PubMed] [Google Scholar]
- 36.Barlési F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA. Global histone modifications predict prognosis of resected non-small-cell lung cancer. J Clin Oncol 2007; 25:4358-64; http://dx.doi.org/ 10.1200/JCO.2007.11.2599 [DOI] [PubMed] [Google Scholar]
- 37.Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A, et al.. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol 2013; 31:4140-7; PMID:24081945; http://dx.doi.org/ 10.1200/JCO.2012.48.5516 [DOI] [PubMed] [Google Scholar]
- 38.Momparler RL. Epigenetic therapy of non-small cell lung cancer using decitabine (5-aza-2′-deoxycytidine). Front Oncol 2013; 3:188; PMID:23908969; http://dx.doi.org/ 10.3389/fonc.2013.00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gridelli C, Rossi A, Maione P. The potential role of histone deacetylase inhibitors in the treatment of non-small-cell lung cancer. Crit Rev Oncol Hematol 2008; 68:29-36; PMID:18424067; http://dx.doi.org/ 10.1016/j.critrevonc.2008.03.002 [DOI] [PubMed] [Google Scholar]
- 40.Kikuchi J, Takashina T, Kinoshita I, Kikuchi E, Shimizu Y, Sakakibara-Konishi J, Oizumi S, Marquez VE, Nishimura M, Dosaka-Akita H. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer 2012; 78:138-43; PMID:22925699; http://dx.doi.org/ 10.1016/j.lungcan.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoeffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs 2008; 26:483-8; PMID:18425418; http://dx.doi.org/ 10.1007/s10637-008-9131-6 [DOI] [PubMed] [Google Scholar]
- 42.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al.. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011; 1:598-607; PMID:22586682; http://dx.doi.org/ 10.1158/2159-8290.CD-11-0214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5:37-50; PMID:16485345; http://dx.doi.org/ 10.1038/nrd1930 [DOI] [PubMed] [Google Scholar]
- 44.Tang J, Salama R, Gadgeel SM, Sarkar FH, Ahmad A. Erlotinib resistance in lung cancer: current progress and future perspectives. Front Pharmacol 2013; 4:15; PMID:23407898; http://dx.doi.org/ 10.3389/fphar.2013.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Füller M, Klein M, Schmidt E, Rohde C, Göllner S, Schulze I, Qianli J, Berdel W, Edemir B, Müller-Tidow C, et al.. 5-Azacytidine enhances efficacy of multiple chemotherapy drugs in AML and lung cancer with modulation of CpG methylation. Int J Oncol 2014; 46:1192-204; PMID:25501798; http://dx.doi.org/ 10.3892/ijo.2014.2792. [DOI] [PubMed] [Google Scholar]
- 46.Li XY, Wu JZ, Cao HX, Ma R, Wu JQ, Zhong YJ, Feng JF. Blockade of DNA methylation enhances the therapeutic effect of gefitinib in non-small cell lung cancer cells. Oncol Rep 2013; 29:1975-82; PMID:23440266; http://dx.doi.org/ 10.3892/or.2013.2298. [DOI] [PubMed] [Google Scholar]
- 47.Kim HJ, Kim JH, Chie EK, Park DY, Kim IA, Kim IH. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat Oncol 2012; 7:39; PMID:22429326; http://dx.doi.org/ 10.1186/1748-717X-7-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuco V, de Cesare M, Cincinelli R, Nannei R, Pisano C, Zaffaroni N, Zunino F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS One 2011; 6:1-12; http://dx.doi.org/ 10.1371/journal.pone.0029085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q, Jiang S, Zhang S, Ma X. Histone Deacetylase Inhibitor Trichostatin A Enhances Anti- tumor Effects of Docetaxel or Erlotinib in A549 Cell Line. 2012; 13:3471-6; PMID:22994780; http://dx.doi.org/ 10.7314/APJCP.2012.13.7.3471. [DOI] [PubMed] [Google Scholar]
- 50.Del Bufalo D, Desideri M, De Luca T, Di Martile M, Gabellini C, Monica V, Busso S, Eramo A, De Maria R, Milella M, et al.. Histone deacetylase inhibition synergistically enhances pemetrexed cytotoxicity through induction of apoptosis and autophagy in non-small cell lung cancer. Mol Cancer 2014; 13:230; http://dx.doi.org/ 10.1186/1476-4598-13-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frenkel EP. Histone Deacetylase Inhibitor Romidepsin Enhances Anti-Tumor Effect of Erlotinib in Non-small Cell Lung Cancer (NSCLC). Cell Lines 2009; 4:161-6; http://dx.doi.org/ 10.1097/JTO.0b013e318194fae7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, et al.. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006; 66:944-50; PMID:16424029; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1988 [DOI] [PubMed] [Google Scholar]
- 53.Greve G, Schiffmann I, Pfeifer D, Pantic M, Schüler J, Lübbert M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer. 2015;15(1):947; http://dx.doi.org/ 10.1186/s12885-015-1967-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greve G, Schiffmann I, Lübbert M. Epigenetic priming of non-small cell lung cancer cell lines to the antiproliferative and differentiating effects of all-trans retinoic acid. J Cancer Res Clin Oncol 2015; 141:2171-80; PMID:26008188; http://dx.doi.org/ 10.1007/s00432-015-1987-1. [DOI] [PubMed] [Google Scholar]
- 55.Han S, Fukazawa T, Yamatsuji T, Matsuoka J, Miyachi H, Maeda Y, Durbin M, Naomoto Y. Anti-tumor effect in human lung cancer by a combination treatment of novel histone deacetylase inhibitors: SL142 or SL325 and retinoic acids. PLoS One 2010; 5:e13834; PMID:21079797; http://dx.doi.org/ 10.1371/journal.pone.0013834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gavrilov V, Lavrenkov K, Ariad S. Sodium Valproate, a Histone Deacetylase Inhibitor, Enhances the Efficacy of Vinorelbine-Cisplatin- based Chemoradiation in Non-small Cell Lung Cancer Cells. Anticancer Res 2014; 34:6572. [PubMed] [Google Scholar]
- 57.Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, Mei QB. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther 2009; 8:823-31; PMID:19270532; http://dx.doi.org/ 10.4161/cbt.8.9.8143 [DOI] [PubMed] [Google Scholar]
- 58.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK, et al.. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015; 520:239-42; PMID:25629630; http://dx.doi.org/ 10.1038/nature14122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vasilatos SN, Katz TA, Oesterreich S, Wan Y, Davidson NE, Huang Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013; 34:1196-207; PMID:23354309; http://dx.doi.org/ 10.1093/carcin/bgt033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med J 2009; 50:455-63; PMID:19718392; http://dx.doi.org/ 10.3349/ymj.2009.50.4.455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guillemette B, Drogaris P, Lin HH, Armstrong H, Hiragami-Hamada K, Imhof A, Bonneil E, Thibault P, Verreault A, Festenstein RJ. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet 2011; 7:e1001354; http://dx.doi.org/ 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takashina T, Kinoshita I, Kikuchi J, Shimizu Y, Konishi JS, Oizumi S, Nishimura M, Akita HD. Abstract 5162: Effects of combined epigenetic therapy with histone methyltransferase EZH2 inhibitor 3-deazaneplanocin and histone deacetylase inhibitor SAHA on non-small cell lung cancer cells. Cancer Res 2014; 74:5162-2; http://dx.doi.org/ 10.1158/1538-7445.AM2014-5162 [DOI] [Google Scholar]
- 63.Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, Channell MM, Liu Y, Casero RA, Baylin SB, et al.. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res 2011; 71:454-62; PMID:21224363; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vendetti FP, Topper M, Huang P, Dobromilskaya I, Easwaran H, Wrangle J, Baylin SB, Poirier JT, Rudin CM. Evaluation of azacitidine and entinostat as sensitization agents to cytotoxic chemotherapy in preclinical models of non-small cell lung cancer. Oncotarget 2015; 6:56-70; PMID:25474141; http://dx.doi.org/ 10.18632/oncotarget.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Momparler RL. Pilot phase I-II study on 5-aza-2′deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anti-Cander Drugs 1997; 53:358-68; PMID:9074840; http://dx.doi.org/ 10.1097/00001813-199704000-00008 [DOI] [PubMed] [Google Scholar]
- 66.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, et al.. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res 2006; 12:5777-85; PMID:17020984; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0669 [DOI] [PubMed] [Google Scholar]
- 67.Traynor AM, Dubey S, Eickhoff JC, Kolesar JM, Schell K, Huie MS, Groteluschen DL, Marcotte SM, Hallahan CM, Weeks HR, et al.. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol 2009; 4:522-6; PMID:19347984; http://dx.doi.org/ 10.1097/JTO.0b013e3181952478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Otterson GA, Hodgson L, Pang H, Vokes EE. Phase II study of the HDAC inhibitor romidepsin in relapsed small cell lung cancer. Changes 2012; 29:997-1003; http://dx.doi.org/ 10.1016/j.biotechadv.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bauman J, Verschraegen C, Belinsky S, Muller C, Rutledge T, Fekrazad M, Ravindranathan M, Lee SJ, Jones D. A phase I study of 5-azacytidine and erlotinib in advanced solid tumor malignancies. Cancer Chemother Pharmacol 2012; 69:547-54; PMID:21901396; http://dx.doi.org/ 10.1007/s00280-011-1729-2 [DOI] [PubMed] [Google Scholar]
- 70.Schwartsmann G, Schunemann H, Gorini CN, Filho AF, Garbino C, Sabini G, Muse I, DiLeone L, Mans DR. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer. Invest New Drugs 2000; 18:83-91; PMID:10830142; http://dx.doi.org/ 10.1023/A:1006388031954 [DOI] [PubMed] [Google Scholar]
- 71.Jones DR, Moskaluk CA, Gillenwater HH, Petroni R, Burks SG, Philips J, Rehm PK, Kozower BD, Bao Y. Phase I Trial of Induction Histone Deacetylase and Proteasome Inhibition Followed by Surgery in Non-small Cell Lung Cancer. J Thorac Oncol 2012; 7:1683-90; http://dx.doi.org/ 10.1097/JTO.0b013e318267928d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dasari A, Gore L, Messersmith WA, Diab S, Jimeno A, Weekes CD, Lewis KD, Drabkin HA, Flaig TW, Camidge DR. A phase i study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Invest New Drugs 2013; 31:115-25; PMID:22415798; http://dx.doi.org/ 10.1007/s10637-012-9812-z [DOI] [PubMed] [Google Scholar]
- 73.Gray J, Haura EB, Chiappori A, Tanvetyanon T, Williams CC, Pinder-Schenk M, Kish JA, Kreahling JM, Lush RM, Neuger AM, et al.. A phase I, pharmacokinetic and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin Cancer Res [Internet] 2014. [cited 2014 Feb 5]; 20:1644-55; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, Moran T, Rolfo C, Pallarès MC, Insa A, et al.. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 2014; 84:161-7; PMID:24636848; http://dx.doi.org/ 10.1016/j.lungcan.2014.02.011 [DOI] [PubMed] [Google Scholar]
- 75.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, Varella-Garcia M, Bunn PA, Hirsch FR. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol 2012; 30:2248-55; PMID:22508830; http://dx.doi.org/ 10.1200/JCO.2011.38.9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Thomas S, Espinoza-Delgado I, Vokes EE, Gandara DR, et al.. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol 2010; 28:56-62; PMID:19933908; http://dx.doi.org/ 10.1200/JCO.2009.24.9094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chu BF, Karpenko MJ, Liu Z, Aimiuwu J, Villalona-Calero MA, Chan KK, Grever MR, Otterson GA. Phase i study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol 2013; 71:115-21; PMID:23053268; http://dx.doi.org/ 10.1007/s00280-012-1986-8 [DOI] [PubMed] [Google Scholar]
- 78. http://clinicaltrials.gov/show/NCT01628471 [Google Scholar]
- 79. http://clinicaltrials.gov/show/NCT00978250 [Google Scholar]
- 80. http://clinicaltrials.gov/show/NCT00423449 [Google Scholar]
- 81. https://clinicaltrials.gov/ct2/show/record/NCT00473889 [Google Scholar]
- 82. http://clinicaltrials.gov/show/NCT01090830 [Google Scholar]
- 83. http://clinicaltrials.gov/show/NCT01188707 [Google Scholar]
- 84. https://clinicaltrials.gov/ct2/show/record/NCT01935947 [Google Scholar]
- 85. http://clinicaltrials.gov/show/NCT00387465 [Google Scholar]
- 86.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al.. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015; 28:57-69; PMID:26175415; http://dx.doi.org/ 10.1016/j.ccell.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 87. https://clinicaltrials.gov/ct2/show/NCT02034123 [Google Scholar]
- 88.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, et al.. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget [Internet] 2013; 4:2067-79; PMID:24162015; http://dx.doi.org/ 10.18632/oncotarget.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med [Internet] 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N Engl J Med 2010; 362:2251-9; PMID:20525993; http://dx.doi.org/ 10.1056/NEJMoa0912614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al.. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell [Internet] 2015; 162:974-86. Available from: http://dx.doi.org/ 10.1016/j.cell.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al.. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell [Internet] 2015; 162:961-73. Available from: http://dx.doi.org/10.1016/j.cell.2015.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. https://clinicaltrials.gov/ct2/show/NCT01928576 [Google Scholar]
- 94.Brahmer J, Reckamp KL, Baas P, Crin∫ L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al.. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med 2015; 373:1-13; http://dx.doi.org/ 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. https://clinicaltrials.gov/ct2/show/NCT02546986 [Google Scholar]
- 96. https://clinicaltrials.gov/show/NCT02437136 [Google Scholar]
- 97.Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999; 21:103-7; PMID:9916800; http://dx.doi.org/ 10.1038/5047 [DOI] [PubMed] [Google Scholar]
- 98.Huang Y, Vasilatos SN, Boric L, Shaw PG, Davidson NE. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res Treat 2012; 131:777-89; PMID:21452019; http://dx.doi.org/ 10.1007/s10549-011-1480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fahy J, Jeltsch A, Arimondo PB. DNA methyltransferase inhibitors in cancer: a chemical and therapeutic patent overview and selected clinical studies. Expert Opin Ther Pat 2012; 22:1-16; PMID:22214283; http://dx.doi.org/ 10.1517/13543776.2012.729579 [DOI] [PubMed] [Google Scholar]
- 100.Ono T, Li C, Mizutani E, Terashita Y, Yamagata K, Wakayama T. Inhibition of class IIb histone deacetylase significantly improves cloning efficiency in mice. Biol Reprod [Internet] 2010; 83:929-37; http://dx.doi.org/ 10.1095/biolreprod.110.085282. [DOI] [PubMed] [Google Scholar]
- 101.Ali A, Bluteau O, Messaoudi K, Palazzo A, Boukour S, Lordier L, Lecluse Y, Rameau P, Kraus-Berthier L, Jacquet-Bescond A, et al.. Thrombocytopenia induced by the histone deacetylase inhibitor abexinostat involves p53-dependent and -independent mechanisms. Cell Death Dis [Internet] 2013; 4:e738; PMID:23887629; http://dx.doi.org/ 10.1038/cddis.2013.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, Merchant TE, Boulad F, Cheung NK, Bonilla MA, et al.. Desmoplastic small round-cell tumor: Prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 1996; 14:1526-31; PMID:8622067; http://dx.doi.org/ 10.1002/pbc. [DOI] [PubMed] [Google Scholar]
- 103.Carol H, Gorlick R, Kolb EA, Morton CL, Manesh DM, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak A, et al.. Initial Testing (Stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2010; 199:1442-8; http://dx.doi.org/ 10.1086/597422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mandl-Weber S, Meinel FG, Jankowsky R, Oduncu F, Schmidmaier R, Baumann P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol 2010; 149:518-28; PMID:20201941; http://dx.doi.org/ 10.1111/j.1365-2141.2010.08124.x [DOI] [PubMed] [Google Scholar]
- 105.Kusaczuk M, Krętowski R, Bartoszewicz M, Cechowska-Pasko M. Phenylbutyrate - a pan-HDAC inhibitor that suppresses proliferation of glioblastoma LN-229 cell line. Tumor Biol 2016; 37:931-42; http://dx.doi.org/ 10.1007/s13277-015-3781-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim DH, Shin J, Kwon HJ. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp Mol Med [Internet] 2007; 39:47-55. PMID:17334228; http://dx.doi.org/ 10.1038/emm.2007.6 [DOI] [PubMed] [Google Scholar]