

Abstract
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. Programmed death of neuronal cells plays a crucial role in acute and chronic neurodegeneration following TBI. The tumor suppressor protein p53, a transcription factor, has been recognized as an important regulator of apoptotic neuronal death. The p53 inactivator pifithrin-α (PFT-α) has been shown to be neuroprotective against stroke. A previous cellular study indicated that PFT-α oxygen analogue (PFT-α (O)) is more stable and active than PFT-α. We aimed to investigate whether inhibition of p53 using PFT-α or PFT-α (O) would be a potential neuroprotective strategy for TBI. To evaluate whether these drugs protect against excitotoxicity in vitro, primary rat cortical cultures were challenged with glutamate (50mM) in the presence or absence of various concentrations of the p53 inhibitors PFT-α or PFT-α (O). Cell viability was estimated by LDH assay. In vivo, adult Sprague Dawley rats were subjected to controlled cortical impact (CCI, with 4m/s velocity, 2 mm deformation). Five hours after injury, PFT-α or PFT-α (O) (2 mg/kg, i.v.) was administered to animals. Sensory and motor functions were evaluated by behavioral tests at 24 h after TBI. Apoptotic cells and p53-positive neurons were identified by double staining with cell-specific markers. Levels of mRNA encoding for p53-regulated genes (BAX, PUMA, Bcl-2 and p21) were measured by reverse transcription followed by real time-PCR from TBI animals without or with PFT- α/PFT- α (O) treatment. We found that PFT-α (O) (10uM) enhanced neuronal survival against glutamate-induced cytotoxicity in vitro more effectively than PFT-α (10uM). In vivo PFT-α (O) treatment enhanced functional recovery and decreased contusion volume at 24 h post-injury. Neuroprotection by PFT-α (O) treatment also reduced p53-positive neurons in the cortical contusion region. In addition, p53-regulated PUMA mRNA levels at 8h were significantly reduced by PFT-α (O) administration after TBI. PFT-α (O) treatment also decreased phospho-p53 positive neurons in the cortical contusion region. Our data suggest that PFT-α (O) provided a significant reduction of cortical cell death and protected neurons from glutamate-induced excitotoxicity in vitro, as well as improved neurological functional outcome and reduced brain injury in vivo via anti-apoptotic mechanisms. The inhibition of p53-induced apoptosis by PFT-α (O) provides a useful tool to evaluate reversible apoptotic mechanisms and may develop into a novel therapeutic strategy for TBI.
Keywords: Traumatic brain injury (TBI), p53, pifithrin-α (PFT-α), PFT-α oxygen analogue, apoptosis, controlled cortical impact
Introduction
Traumatic brain injury (TBI) is one of the most common causes of death and disability worldwide. The estimated incidence and mortality after TBI is approximately 200 cases and 20 cases for every 100,000 people, respectively (Reilly, 2007). TBI can be caused by traffic accidents, falls – particularly in the elderly - or violence, and represents a significant health concern within the US. Owing to their rapid industrial and economic growth, developing countries such as Taiwan have faced an enormous increase in the number of motorcycles, which has subsequently caused a rapid increase in motorcycle-related TBI. It is necessary to understand the mechanisms that contribute to dysfunction and death following serious trauma, and at the same time to devise appropriate therapeutic ways to improve patient prognosis.
The pathophysiology of TBI consists of two main phases: a primary (mechanical impact) phase of damage, and a secondary (delayed) damage phase. A great deal of effort has focused on various biochemical mechanisms involved in the secondary injury processes that follow TBI (Andrews et al., 1990; Miller, 1986). Primary damage occurs at the moment of insult, and includes contusion and laceration, diffuse axonal injury and intracranial hemorrhage (LaPlaca et al., 2007; Spaethling et al., 2007). Secondary damage includes altered neurochemical mechanisms, activation of degradative enzymes, swelling (edema) and ischemia. Ischemia has been suggested as one of the most important mechanisms underlying secondary brain damage following TBI, especially in severely injured patients (Maas et al., 2000; van den Brink et al., 2000; van Santbrink et al., 2002; Verweij et al., 2007).
In neurons, the transcription factor p53 is activated and results in delayed cell death after TBI. At 6 h following TBI, a robust increase in p53-labeled cells was evident within the site of maximal injury (contusion) in the cortex (Napieralski et al., 1999; Plesnila et al., 2007). A stable p53-inhibitor, Pifithrin-α (PFT-α) has been identified, which prevents neuronal cell death by inhibiting p53 transcriptional activity, mitochondrial damage, and caspase activation (Pietrancosta et al., 2006; Sohn et al., 2009). PFT α compounds have proven effective in preventing neuronal cell loss in models of stroke, PD and AD in tissue culture, and in animals (Culmsee et al., 2001; Duan et al., 2002; Leker et al., 2004). For example, in the middle cerebral artery occlusion (MCAO) murine stroke model, PFTα compounds decreased infarct volume by up to 50% in a concentration- and time-dependent manner, with reductions occurring in the ischemic penumbra as opposed to the central core. Reduced infarct size was associated with lower behavioral disability scores and was achieved within a therapeutic window extending to 4 h. These actions were associated with lowered levels of both induced- and phosphorylated p53, a reduced expression of its targeted genes, p21WAF and Bax, and reduced activation of caspases in the infarct tissue. Immunohistochemical staining for p53 and p21WAF within ischemic penumbra of treated vs. control mice suggested that PFTα agents act at the level of p53 translocation into the nucleus to prevent binding to specific DNA sites (Leker et al., 2004). In a regenerative stroke paradigm, these p53 inhibitors proved effective in improving behavioral outcomes of animals when administered a full 6 days post stroke (Luo et al., 2009). This was mediated via actions on neurogenesis, which although stimulated by trauma, is a highly inefficient process. p53 inhibition not only increased the number of neural progenitor cells, but augmented their survival, migration to areas of damage, and differentiation into neuronal phenotypes that integrated effectively into the remaining neural network (Luo et al., 2009), thereby suggesting therapeutic promise of p53 inactivators as both a protective as well as a regenerative treatment strategy. Administration of these PFTα p53 inhibitors in cellular and animal models of PD supported this potential by showing neuroprotective activity in substantia nigra from MPTP-induced cell death (Duan et al., 2002), and also providing neuroregenerative efficacy (Chou et al., 2011).
In this study, we attempted to interrupt the apoptotic cascade upstream of caspase activation in an experimental model of TBI. In this regard, an important regulatory step in apoptosis occurs at mitochondrial membranes where members of the Bcl-2 family of proteins either promote (PUMA, Bax and Bad) or prevent (Bcl-2 and Bcl-XL) membrane permeability transition (Polster and Fiskum, 2004; Yuan and Yankner, 2000). p53 may regulate delayed neuronal death through transactivation of these pro-apoptotic genes after TBI.
Previous studies indicated that PFT-α oxygen analogue (PFT-α (O)) is more stable and active in cellular studies than PFT-α (Greig et al., 2004; Zhu et al., 2002). We hypothesized that post-injury treatment with PFT-α or PFT-α (O) has therapeutic effect against TBI through inhibiting p53 transcriptional activity. In the present study, the efficacy and therapeutic window of these two p53 inhibitors were compared in TBI animal models.
Materials and methods
Synthesis of P53 inhibitors
Pifithrin-α (PFT-α: 1-(4-methylphenyl)-2-(4,5,6,7-tetrahydro-2-imino-3 (2H)-benzothiazolyl)ethanone) was synthesized as its HBr salt, according to the route of Zhu et al. (Fig. 1A). In contrast to sulfur containing PFT-α, the oxygen containing close analogue (PFT-α (O): 1-(4-methylphenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzooxazolyl)ethanone) was prepared from 2-hydroxycyclohexanone dimer and cyanamide. Specifically, 2-hydroxycyclohexanone dimer was reacted with cyanamide in a 95 °C oil bath to provide 2-amino-4,5,6,7-tetrahydrobenzoxazole, which was separated and characterized. This 2-amino-4,5,6,7-tetrahydrobenzoxazole was then stirred with α-bromomethyl ketone at room temperature for 2 days, and the precipitate was collected by filtration and recrystallized from MeOH/EtOAC to provide PFT-α (O), which was then prepared as a HBr salt. Chemical characterization confirmed the structures of the desired compounds in high purity (>99%), which were dissolved in 100% dimethyl sulfoxide (DMSO) for cell culture studies. In brief, neuronal cultures were challenged with excess glutamate (Sigma, St Louis, MO) to evaluate PFT-α and PFT-α (O) mediated neuroprotective actions (see below).
Fig. 1. Post-injury administration of PFT-α or PFT-α (O) at 5 h after TBI significantly reduced contusion volume at 24 h.
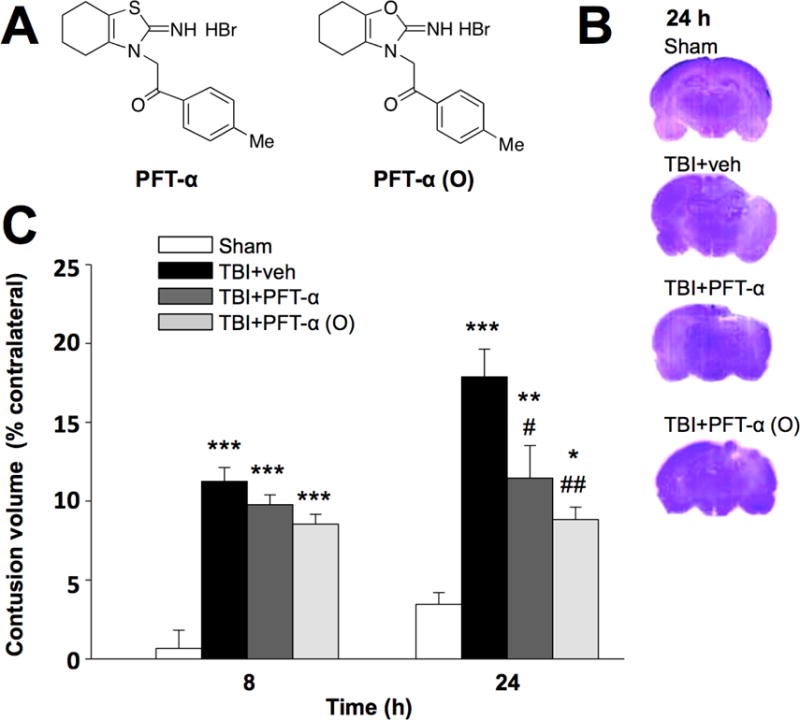
(A) Chemical structures of PFT-α and PFT-α (O). (B) Coronal brain sections (cresyl violet stained) from sham and TBI challenged rats treated with vehicle, PFT-α or PFT-α (O) at 24 h. (C) Contusion volumes induced by TBI at 8 h and 24 h (associated with the primary mechanical injury and secondary apoptotic phases, respectively). All TBI challenged groups were significantly different from the sham group at 8 and 24 h. Whereas the contusion volume of TBI groups were not different from one another at 8 h, by 24 h it was significantly reduced by PFT-α or PFT-α (O) (2mg/kg) treatment compared to the TBI vehicle group. Data are expressed as means ±SEM. * p <0.05, ** p <0.01, ***p<0.001 versus sham group; # p <0.05; ## p <0.01, versus TBI + veh group (n= 5 for each group).
Animal model of traumatic brain injury (TBI)
All animals were treated in accordance with the International Guidelines for animal research, and the study design was approved by the animal ethics committee of Taipei Medical University. Animals were housed in groups in a temperaturee- (21–25°C) and humidity (45–50%)-controlled room with a 12-h light/dark cycle and ad libitum access to pellet chow and water. All animals were randomized into 4 groups (sham injury, CCI+vehicle, CCI+PFT-α and CCI+ PFT-α (O)). Male Sprague-Dawley rats (250–300 g, body weight) were anaesthetized and placed in a stereotaxic frame. A 5-mm craniotomy was performed over the left parietal cortex, centered on the coronal suture and 3.5 mm lateral to the sagittal suture. Injury was made using a controlled cortical impact (CCI) instrument with a rounded metal tip (5 mm diameter). A velocity of 4 m/s and a deformation depth 2 mm below the dura were used as we previously described (Chen et al., 2008). Sham animals received anesthesia and crainiotomy but no CCI. Body temperature was monitored throughout the surgery by a rectal probe; temperature was maintained at 37.0±0.5°C using a heated pad. Rats were placed in a heated cage to maintain body temperature, while recovering from anaesthesia.
Drug Administration
CCI rats received vehicle (10% DMSO in saline), PFT-α or PFT-α (O) injections (2 mg/kg, i.v.) at 5-hr (n = 5) or 7-hr (n = 5) post-injury. Notably, body temperature was evaluated in a group of parallel TBI animals following vehicle, PFT-α or PFT-α (O) treatment over a period of 3 h, and no changes in body temperature were evident. Furthermore, no temperature changes were seen after PFT α or PFT α (O) in normal or sham animals as well. All behavioral testing and histological analysis were carried out by experimenters blinded to treatment conditions.
Primary rat cortical neuronal/glial cocultures
Primary neuronal/glial cocultures were isolated from cerebral cortex of 1-day-old neonatal Sprague–Dawley rats as previously described (Huang et al., 2009). These procedures were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Taipei Medical University, Taipei, Taiwan. Rat pup brains were removed immediately after sacrificed and the cerebral cortex were placed in ice-cold Hank’s solution (without Ca2+ or Mg2+). Cortical cells were dissociated and suspended in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL, Grand Island, NY, USA) with 10% fetal bovine serum (FBS). Thereafter, cells were plated at a density of 5×105 cells/ml, and then incubated at 37 °C in a humidified incubator (5% CO2, 95% air). All experiments were performed in 13–14-day after the plating. We used immunostaining to determine the percentage of the cell composition that was additionally followed by cell counting. Under our culture conditions, the neuronal/glial cocultures consisted of approximately 40% to 42% neurons, 40% to 45% astrocytes, and 8% to10% microglia, as previously described (Huang et al., 2009).
Neuronal/glial cocultures were treated with glutamate (Sigma-Aldrich) to mimic TBI-induced excitotoxicity in vitro. Different concentrations of glutamate were assessed to induce excitotoxicity. The concentration of 50 mM glutamate, which was sufficient to cause a mild cellular loss was selected for the following experiment. To evaluate neuroprotection against excitotoxicity, different concentrations of PFT-α, PFT-α (O) or vehicle (0.1% DMSO in PBS) was added at 30 min after glutamate challenge (50 mM) and cytotoxicity was appraised at 24 h by lactate dehydrogenase (LDH) assay, as previously described (Huang et al., 2009). LDH activity was determined by measuring optical absorbance at 340 nm wavelength, and scaled to the value of maximal death (100%) measured after freeze-thaw treatment of sister cultures. LDH release was expressed as the percentage of values relative to vehicle-treated control cultures.
Behavioral evaluation of neurological outcome
Behavioral testing was performed before CCI and at 24 h after CCI. The evaluation consisted of a modified neurological severity score (mNSS) assessment, a tactile adhesive removal test, an elevated body swing test and a beam walk test. All were performed by an observer blinded to the experimental groups.
To compare neurological deficit severity in TBI rats, a mNSS which is a composite of motor, sensory, reflex and balance tests was performed (Chen et al., 2008). One point was scored for the inability to perform the test or for the lack of a tested reflex; thus, the higher the score, the more severe the injury. Neurological function was graded on a scale of 0–18 (normal score, 0; maximal deficit score, 18).
A tactile removal test was used to evaluate somatosensory function. Two small adhesive-backed stickers (each 113.1 mm2) were used as bilateral tactile stimuli that were placed on the distal–radial region on the wrist of each forelimb (Chen et al., 2008). Rats were pre-trained daily for 3 days before CCI. The time required (not to exceed 180 s) for the rat to remove the sticker from the forelimb was recorded for five trials at 24 h after CCI.
Body asymmetry was quantitatively analyzed with the use of the elevated body swing test (EBST). Briefly, rats were examined for lateral movement/turning when their bodies were suspended 10 cm above the testing table by lifting their tails. A left/right swing was counted when the head/torso of the animal moved more than 10° from the vertical axis after elevation by their tail. As CCI was performed on the left side, a resulting weakness (hemiparesis) develops on the right side. The frequency of left/right swings was scored across 20 consecutive trials and expressed as a percentage [calculated by: (number of right-biased swings / the total number of swings) × 100%]. An uninjured animal showed an equal frequency to swing to either the left or right side.
The beam walk test was used to assess the fine motor coordination and balance (Feeney et al., 1982). Each rat was placed on a bright light platform and was allowed to walk along a narrow beam (2.5 × 122.0 cm) to reach a darkened goal box at the end of the beam (Chen et al., 2008). The beam-walk latency for a rat to reach the goal box was recorded (not to exceed 60 s). Five trials were recorded 1 h before CCI (baseline) and at 24 h after CCI. The mean values of latency were then quantified.
Fluoro Jade C staining
Fluoro Jade C (FJC) is a polyanionic fluorescein derivative that selectively binds to degenerating g neuronal cell, and is more specific and sensitive than Fluoro Jade B (Schmued et al., 2005). We used Fluoro-Jade C ready-to-dilute staining kit for identifying degenerating neurons (Biosensis, TR-100-FJ), with some modification. Tissue sections were de-paraffinized, rehydrated, incubated in distilled water for 2 min. They were then incubated in a solution of potassium permanganate (1:15) for 10 min, rinsed in distilled water for 2 min and incubated in FJC solution (1:25) for 30 min. The slides were then washed and mounted on coverslips with Vecta-shield mounting medium (Vector). All sections were observed and photographed under a fluorescence microscope with a blue (450–490 nm) excitation light.
Contusion volume
To measure contusion volume, cresyl violet-stained sections were digitized and analyzed using a 1× objective and computer image analysis system. Contusion volume measurement was performed as previously described (Chen et al., 2007). Contusion area was calculated from all images of cresyl violet-stained sections that contained contused brain; volume was computed by summation of areas multiplied by inter-slice distance (500 μm). Hemisphere tissue loss was expressed as a percentage calculated by [(contralateral hemispheric volume-ipsilateral hemispheric volume) / contralateral hemispheric volume) × 100%], as previously reported (Zhang et al., 1998).
Immunohistochemistry
All sections were dried, rehydrated in phosphate buffered saline (PBS), and rinsed in PBS. Sections were blocked for 60 min in 5% BSA (PBS containing 5% BSA and 0.2% Triton X-100; Sigma) and incubated with the appropriate primary antibodies, either mouse monoclonal anti-NeuN antibody (Millpore; 1:500, NeuN is a neuronal marker)/ anti-p53 antibody (GeneTex ; 1:500) / anti-annexin V antibody (Abcam ; 1:500) / anti-p-p53 antibody (Cell signaling ; 1:1000) / or anti-PUMA antibody (Abcam ; 1:200) at 4°C overnight and with secondary antibodies (Alexa Fluor® 488 goat anti-rabbit IgG (1:200, Jackson ImmunoResearch, West Grove, PA); Alexa Fluor® 594 anti-mouse IgG (1:200 dilution, Jackson ImmunoResearch, West Grove, PA)) at room temperature for 2 h. Sections were mounted with Mounting Medium H-1000 (Vector Laboratories, Burlingame, CA, USA). The numbers of NeuN-, p53-, annexin V-, p-p53-, and PUMA–positive cells were counted in 5 randomly selected fields by means of SPOT image analysis software (Diagnostic Instruments, Sterling Heights, MI)
Quantitative measurement of messenger RNA encoding for p53-regulated pro-apoptotic genes
Total RNA of tissue (approximately 50 mg)/cells isolated from neuronal/glial co-cultures was extracted by TRIzol reagent (Invitrogen, Life technologies, Carlsbad, CA, USA), as recommended by the manufacturer. The concentration and purity of RNA were determined by measuring absorbance at 260 and 280 nm wavelength. For reverse transcriptase (RT) and cDNA synthesis, 3 μg RNA was performed using a total reaction volume of 20 μl with ReverTra Ace set (no. PU-TRT-100; Purigo, Taipei, Taiwan). The RT products were chilled on ice as templates in quantitative (real-time) PCR or were stored at −20 °C. For quantification of mRNA levels, we performed qPCR by using QuantiFast SYBR Green PCR kit (Qiagen) with primers in a Rotor-Gene Q 2plex HRM Platform (Qiagen). The primers were designed using published cDNA sequences: PUMA, 5′-CATGGGACTCCTCCCCTTAC-3′ (forward) and 5′-CACCTAGTTGGGCTCCATTT-3′ (reverse); Bax, 5′-GTGAGCGGCTGCTTGTCT-3′ (forward) and 5′-GTGGGGGTCCCGAAGTAG-3′ (reverse); Bcl-2, 5′-GTACCTGAACCGGCATCTG-3′ (forward) and 5′-GGGGCCATATAGTTCCACAA-3′ (reverse); Apaf1, 5′-CATGAGCTGAAGGGC CATA-3′ (forward) and 5′-GGCCATCTGAGACATTCCAT-3′ (reverse); p21, 5′-ACATCTCAGGGCCGAAAAC-3′ (forward) and 5′-GCGCTTGGAGTGATAGAAAT-3′ (reverse); β-actin, 5′-GACCCAGATCATGTTTGAGACCTTC-3′ (forward) and 5′-GGTGACCGTAACACTACCTGAG-3′ (reverse). Reaction conditions were carried out for 35-40 cycles (5 min at 95 °C, 10 s at 95°C and 30 s at 60 °C). Finally, relative transcript expression of p53-regulated pro-apoptotic mRNA were normalized to β-actin, which was used as an internal control, and were expressed as values relative to the control group using the comparative cycle threshold (Ct) method.
Statistical analysis
Comparisons between four groups were conducted using one-way ANOVA with a post hoc test; Bonferonni correction was used for repeated measures. All statistical analyses and bar graph displays were performed using Sigma Plot and Stat version 2.0 from Jandel Scientific, San Diego, CA. Data are presented as mean ± standard error of the mean (SEM) values.
Results
Post-injury treatment with PTF-α and a PFT-α (O) analogue significantly reduced neuronal injury
PTF-α and PFT-α (O) were both well tolerated across all groups of treated rats, and no changes in body temperature (evaluated over a period of 3 h following administration) were evident – in line with our prior evaluation (Leker et al., 2004) in which, additionally, there were no changes in the physiological parameters of blood pressure, heart rate and oxygen saturation that were monitored for up to 1 h. Animals were euthanized after their functional evaluation by transcardial perfusion under anesthesia, and brains were removed and sectioned to measure brain contusion volume by cresyl violet staining at 8 and 24 h after CCI. CCI resulted in a loss of cortical tissue in the ipsilateral (left) parietal cortex, as reflected by gross reductions in cresyl violet staining intensity. In contrast, the cytoarchitecture of the cortex remained normal on the contralateral (right) cortex, as reflected by gross reductions in cresyl violet staining intensity (Fig. 1B). The volume of contusion estimated from cresyl violet-stained sections after vehicle treatment in CCI rats was significantly greater than the corresponding volume in sham animals. Post-injury treatment of PFT-α or PFT-α (O) significantly reduced contusion volume at 1 day after CCI (Fig. 1C; P <0.05). However, there was no significant difference in contusion volume between the PFT-α and PFT-α (O) treatment. These changes were further evaluated quantitatively. Specifically, we chose to appraise 8 and 24 h time points, based on the literature of when apoptotic mechanisms would be expected to influence contusion volume and given the well documented expression of caspase-3 in rat brain injury models.
As explained in the ‘Materials and methods’, the hemisphere tissue loss was expressed as a percentage calculated by [(contralateral hemispheric volume-ipsilateral hemispheric volume) / contralateral hemispheric volume) ×100%]. The average contusion volume percentages were 0.66%±1.16% for the sham group at 8 h, and 11.25%±0.88%, 9.78%±0.62%, and 8.54%±0.63% in CCI+vehicle, CCI+PFT-α, and CCI+PFT-α (O) groups, respectively (n= 5 in each group). The average contusion volume percentage at 24 h after the CCI were 17.88%±1.75%, 11.45%±2.97%, and 8.83%±0.79% in CCI+vehicle, CCI+PFT-α, and CCI+PFT-α (O) groups, respectively (n= 5 in each group) (Fig. 1C).
Analysis by 2-way Anova indicated that at 8 h (mechanical injury) there was no difference between treatment groups after injury, but all groups differed significantly from sham (p<0.001). At 24 h, when the apoptotic process would be manifested, injury volume was significantly influenced by treatment. The CCI+vehicle group was again significantly different than the sham (p<0.001). At 24 h after CCI both the PFTα and PFTα (O) groups were significantly different from the vehicle group (p<0.001). Indeed, there were no differences between 8 h and 24 h contusion volumes for both the TBI challenged PFT-α and PFT-α (O) treated groups (p=0.327 and p=0.864) respectively.
By subtracting the 8 h from the 24 h contusion volume data one can estimate that due to the secondary apoptotic phase - which is a volume of 6.63% for CCI alone (vehicle), 1.68% for CCI+PFT-α, and 0.29% for CCI+PFT-α (O). One can then express the two latter as a percent of the CCI vehicle value to gain an approximation of inhibition of the secondary phase by the two treatments. In this regard, PFT-α inhibits this secondary phase by 74.7% and PFT-α (O) inhibits it by 95.6%.
FJC-positive cells with neuronal morphology were evident 24 h after CCI in the cortical contusion margin (Fig. 2A) but not in the contralateral hemisphere. A representative HE-stained coronal section is shown in Figure 2B. PFT-α or PFT-α (O) significantly reduced (~50%) the number of FJC-positive cells compared with vehicle treatment (Fig. 2C; P<0.001).
Fig. 2. Post-injury administration of PFT-α (O) at 5 h after TBI significantly decreased FJC positive cells in the cortical contusion region at 24 h.
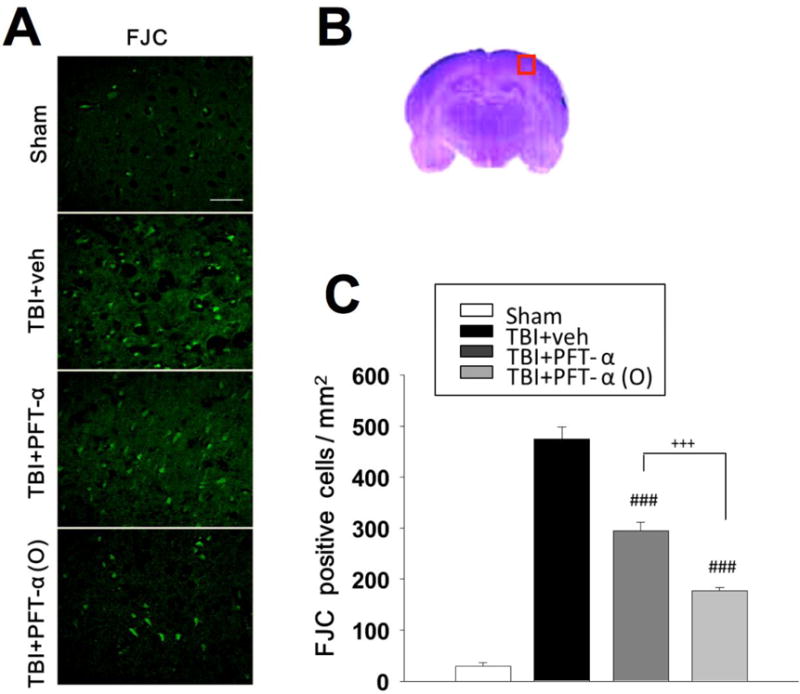
(A) Photomicrographs of FJC-stained regions of interest in sham group, TBI + veh group, TBI + PFT-α and TBI + PFT-α (O) groups. (B) The representative HE-stained coronal section showing the area as indicated by the red square box to compare the fluorescent signals between the 4 groups of rats (the brain section shown is from a sham control). (C) There was a significant decrease in the number of FJC-positive cells in both TBI +PFT- α and TBI +PFT- α (O) groups. The total number of FJC-positive cells was expressed as the mean number per field of view (0.087 mm2). Data are expressed as mean ± SEM. ### p <0.001 versus TBI+veh group; +++ p <0.001 versus TBI+PFT- α group, analyzed by one-way ANOVA. Bar= 50 μm. (n=5 in each group).
Post-injury treatment of PFT-α or PFT-α (O) at 5 but not 7 hr improved neurological functional deficits after TBI
Motor coordination impairment, measured by the beam walking test, after CCI was evident in vehicle-treated animals at 24 h (Fig. 3A). PFT-α or PFT-α (O) treated rats at 5 h showed significantly better performance in beam walking than vehicle-treated rats after injury. Neurological function was measured by mNSS scores at 24 h after CCI in sham and CCI rats indicated that the overall functional outcome caused by CCI was evident in the vehicle-treated group. Post-injury treatment with PFT-α or PFT-α (O) given at 5 h improved functional deficits after CCI as revealed by mNSS scores (Fig. 3B; P<0.001). Functional evaluation measured by the elevated body swing test (EBST) at 24h after CCI in sham and CCI rats indicated that impaired functional outcome, as measured by motor asymmetry, caused by CCI was evident in the vehicle-treated group (Fig 3C). PFT-α (O) treated rats at 5 h improved functional deficits after CCI as revealed by reduction in contralateral swings (Fig. 3C; P<0.05). Functional evaluation measured by tactile adhesive-removal at 24h after CCI in sham and CCI rats indicated that functional deficits caused by CCI was evident in the vehicle-treated group (Fig 3D) in which unilateral CCI resulted in a delay in the time needed to remove the patch. Post-injury treatment with PFT-α or PFT-α (O) given at 5 h improved functional deficits after CCI as revealed by tactile adhesive removal test (Fig. 3D; P<0.01). Delaying PFT treatment until 7 h post injury greatly reduced its effectiveness across all tests (Fig 3 A–D).
Fig. 3. Post-injury administration of PFT-α or PFT-α (O) given at 5h but not 7 h improved functional outcomes as revealed by behavioral evaluation.
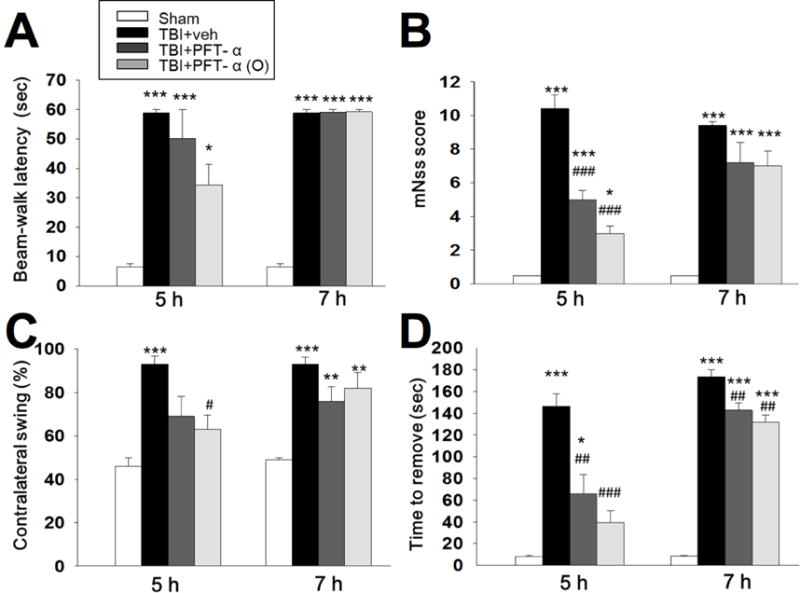
(A) Motor coordination measured by beam walking test. (B) Neurological function measured by mNSS. (C) Motor asymmetry measured by elevated body swing test (EBST) (D) Sensory-motor function measured by tactile adhesive removal test. Data represent the mean ±SEM. *P <0.05, ***P <0.001 versus sham group; #P<0.05, ###P<0.001 versus TBI +veh group. (n=5 in each group). Note that when PFT was given at 7h post injury, it was much less effective.
Post-injury PFT-α or PFT-α (O) treatment reduces p53-related protein and annexin V expression in neurons
To examine the effect of PFT-α or PFT-α (O) on p53 and apoptosis-related protein expression, immunohistochemistry was used to evaluate gene translation 8 h after injury in the cortical contusion region. Our results indicate that p53 positive cells were not colocalized in astrocytes (figure not shown). Therefore, we further evaluated whether the p53 protein was expressed in neurons (Fig. 4A). We found p53 was colocalized in neurons and p53 protein levels in the cortical contusion margin were significantly increased at 8 h after injury. Post-injury treatment with PFT-α or PFT-α (O) at 5 h significantly decreased the number of p53 positive neurons (Fig. 4B; P<0.001). We also used annexin V staining to examine apoptosis in neurons (Fig. 4C). Quantitative analysis revealed a 25% and 55% reduction in annexin V -a-positive neurons in PFT-α and PFT-α (O) treated groups, respectively. Our results indicated that post-injury treatment with PFT-α or PFT-α (O) significantly decreased the number of annexin V positive neurons (Fig. 4D; P<0.001). In addition, treatment with PFT-α (O) showed fewer apoptotic neurons than the PFT-α treated group (P <0.001).
Fig. 4. Post-injury administration of PFT-α or PFT-α (O) at 5 h after TBI significantly decreased p53 and Annexin V positive neurons in the cortical contusion region at 8 h.

(A) Co-immunohistochemistry of p53 and NeuN in cortical brain tissue. (C) Co-immunohistochemistry of Annexin V and NeuN in cortical brain tissue. p53 or annexin V immunoreactivity is shown in green, and NeuN (a marker for neurons) is shown in red. Yellow labelling indicates colocalization. (B, D) There was a significant decrease in the number of p53 and Annexin V positive neurons in TBI + PFT-α and TBI + PFT-α (O) group, respectively. Data represent the mean ±SEM. **p<0.01; ***p<0.001 versus the sham group; ##p<0.001; ###p<0.001 versus the TBI + veh group; ++p<0.01; +++p<0.001 versus the TBI + PFT-α group. Scale bar=100 μm. (n=5 for each group).
Post-injury PFT-α (O) treatment downregulated apoptosis-related mRNA expression induced by TBI
To examine whether PFT-α or PFT-α (O) would inhibit transcriptional activity of p53, we compared p53-regulated genes mRNA levels (BAX, PUMA, Bcl-2 Apaf1 and p21) in sham, vehicle treated, and PFT-α or PFT-α (O) groups by reverse transcription followed by real-time PCR (quantitative RT-PCR). Our results indicated that post-injury treatment with PFT-α (O) reduced PUMA mRNA expression in rat cortex following CCI. However, PFT-α (O) did not change the level of Bax, Bcl-2 Apaf1 and p21 genes (Fig. 5).
Fig. 5. Expression of p53-regulated mRNAs after CCI injury at 8 h, assessed by RT-PCR.
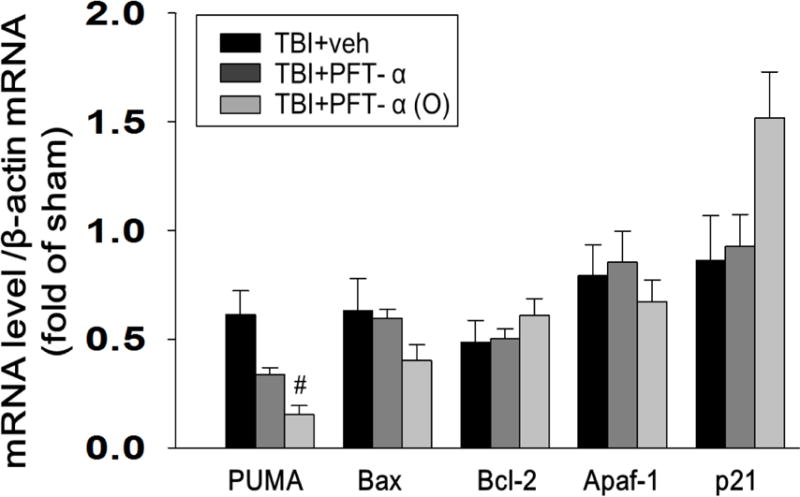
The mRNA levels of PUMA, Bax, Bcl-2, Apaf1, and p21 in brain tissue from sham, vehicle treated, and PFT-α or PFT-α (O) groups were analyzed by RT-PCR. Fold change relative to that of sham animals. PFT-α (O) significantly reduced TBI-induced PUMA mRNA expression in the ipsilateral hemisphere compared with vehicle-treated rats. Data are expressed as means±SEM. #p<0.05 vs. TBI + veh group (n= 5 in each group).
Post-injury PFT-α or PFT-α (O) treatment reduces phospho-p53 and PUMA expression in neurons
To examine the effect of PFT-α or PFT-α (O) on the inhibition of p53 transcriptional activity, immunohistochemistry was used to evaluate phospho-p53 expression in the cortical contusion region. We found p-p53 was colocalized in neurons following CCI (Fig. 6A) and that CCI-induced p-p53 protein levels within the cortical contusion margin were significantly decreased in the PFT-α and particularly in the PFT-α (O) treatment groups when administered 5 h following CCI (Fig. 6B; P<0.001). In addition, we evaluated PUMA staining to confirm that PUMA was suppressed by PFT-α (O) (Fig. 6C). These results indicated that CCI-induced PUMA expression was significantly decreased in the PFT-α (O) treated group (Fig. 6D; P <0.001), more substantially than that achieved by PFT-α.
Fig. 6. Post-injury administration PFT-α (O) at 5 h after TBI significantly decreased p-p53 and PUMA positive neurons in the cortical contusion region at 8 h.
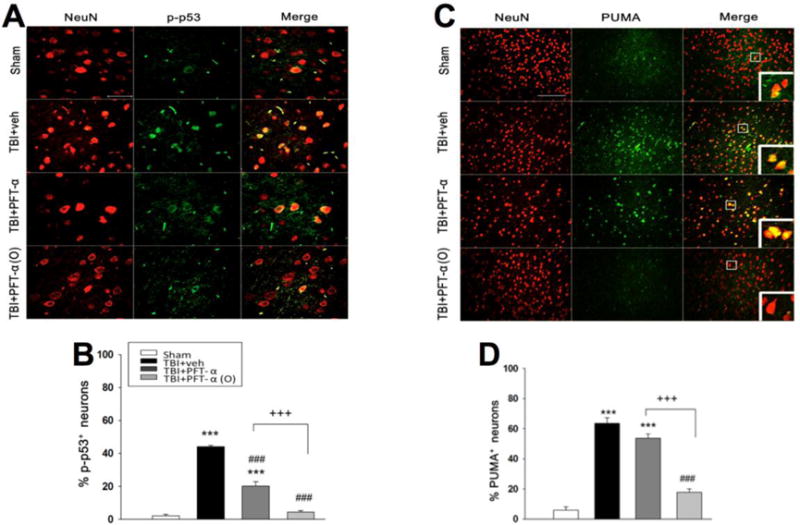
(A) Co-immunohistochemistry of p-p53 and NeuN in cortical brain tissue. (C) Co-immunohistochemistry of PUMA and NeuN in cortical brain tissue. Phospho-p53 or PUMA immunoreactivity is shown in green, and NeuN is shown in red. Yellow labelling indicates colocalization. (B, D) There was a significantly decrease in the number of p-p53 and PUMA positive neurons in TBI + PFT-α (O) group, respectively. Data represent the mean ±SEM. **p<0.01; ***p<0.001 versus the sham group; ##p<0.001; ###p<0.001 versus the TBI + veh group; ++p<0.01; +++p<0.001 versus the TBI + PFT-α group. Scale bar=100 μm. (n=5 for each group).
In Vitro Studies in primary cultured neurons/glia
To further examine mechanism of neuroprotection, PFT- α and PFT- α (O) were studied in primary neuronal/glial cultures in vitro. Exposure of primary cultures of neurons/glial to various concentrations of glutamate (1, 3, 10, 30, 50, 75 and 100 mM) for 24 h caused significant reductions in cell viability (Fig. 7A). As a substantial but submaximal toxicity was elicited by 50 mM glutamate, this concentration was used to further study the p53 inhibitors.
Fig. 7. PFT-α and PFT-α (O) reduce glutamate-induced excitotoxicity in cortical cultures.
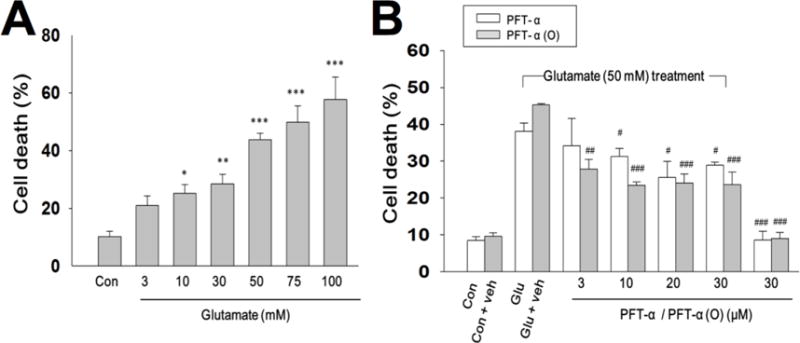
(A) Cultured cells were exposed to various concentrations of glutamate (3, 10, 50, 75 and 100 mM) for 24 h. Cell death (%) was measured by LDH release. 50 mM glutamate was chosen as the concentration for testing PFT analogues. (B) PFT- α or PFT- α (O) (3, 10, 20, 30 μM) was added 30 min after cells were exposed glutamate (50 mM) for 24 hrs in primary cultures of neuron/glia. Cell death was measured by LDH activity in culture media which was scaled to the value of maximal death (100%) measured after freeze-thaw treatment of sister cultures. Data are expressed as means±SEM. *p<0.05; **p<0.01; ***p<0.001 compared with the control group; #p<0.05; ##p<0.01; ###p<0.001 compared with the glutamate treatment (n=3-5 in each group).
PFT-α or PFT-α (O) (3, 10, 20 and 30 μM) concentration-dependently attenuated cell loss induced by glutamate (Fig. 7B). Immunocytochemical studies revealed that PFT-α and PFT-α (O) protected neurons from glutamate-induced excitotoxicity (Fig. 8 A, B). Moreover, microglial activation, as measured by ED1, and which was elevated by glutamate, was also reduced by PFT-α and PFT-α (O) (Fig. 8 A, C). For both the reductions in neuron loss and microglial activation, PFT-α (O) was more potent than PFT-α. Finally, PFT-α (O) significantly reduced the glutamate-induced elevation of PUMA mRNA, an important gene in the apoptotic pathway (Fig. 9).
Fig. 8. PFT-α or PFT-α(O) reduces glutamate-induced neuronal loss and activation of microglia.
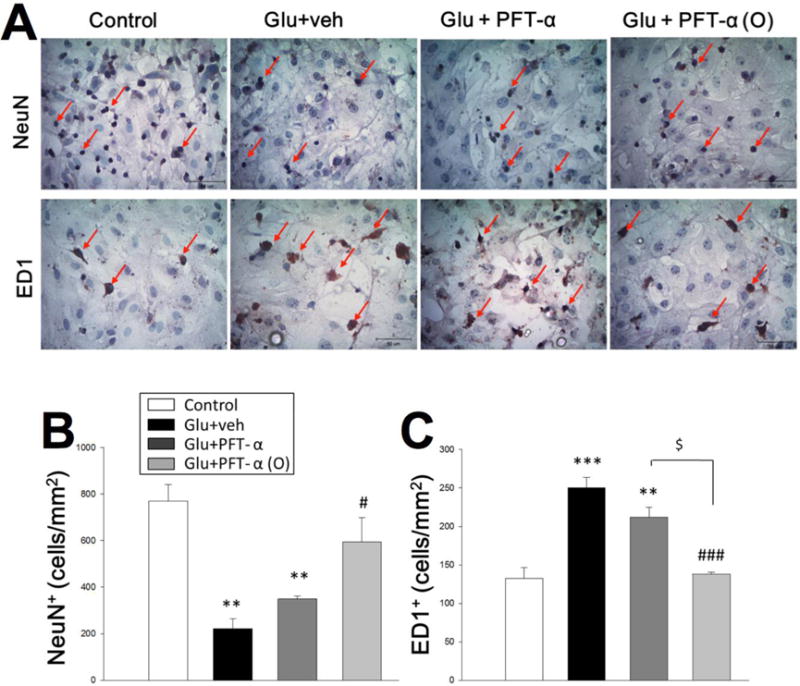
(A) Photomicrograph showing nuclear structure revealed by immunocytochemical staining with NeuN and ED1 in control cultures, cultures treated with glutamate (50mM), cultures treated with glutamate (50 mM) and PFT-α (10μM), and cultures treated with glutamate (50 mM) and PFT-α (O) (10μM) at 24 h after glutamate-induced excitotoxicity. Scale bar, 50 μ m. (B, C) Quantitative comparison of NeuN-positive (NeuN+) / ED1-positive (ED1+) cells in control cultures, glutamate cultures, glutamate cultures treated with PFT-α, and glutamate cultures treated with PFT-α (O). Data are expressed as means±SEM. ***P <0.001 versus control cultures; ###P<0.001 versus glutamate cultures. Bar= 50 μm. (n= 5 in each group).
Fig. 9. PFT-α (O) suppresses glutamate-induced the expression of PUMA in cortical cells.
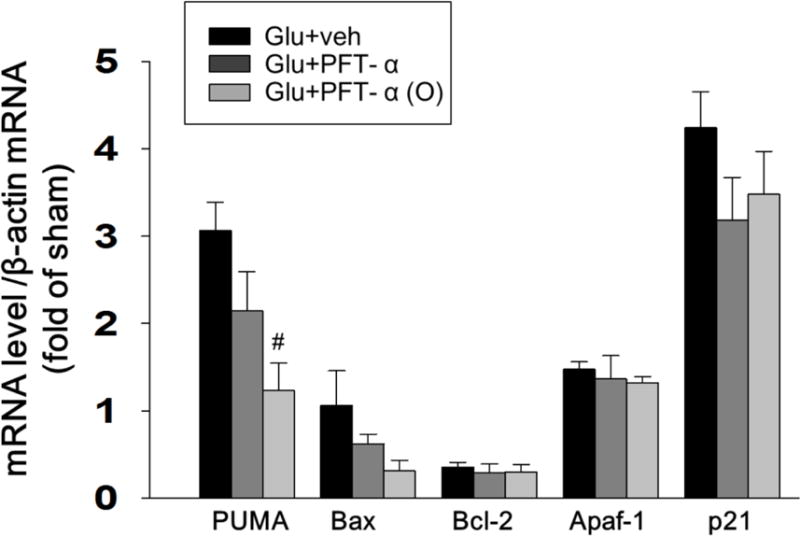
The mRNA levels of PUMA, Bax, Bcl-2, Apaf1and p21 in glutamate-treated (50mM) cortical cultures with PFT-α or PFT-α (O) (10 μM) were analyzed by qRT-PCR. Fold changes relative to that of control cultures are shown. Reduction of the elevated messenger RNA (mRNA) levels of PUMA in primary cultures by PFT-α (O) treatment at 24 hr after glutamate-induced excitotoxicity. PFT-α (O) did not change the level of Bax, Bcl2 Apaf1and p21 genes. Data are expressed as means±SEM. #p<0.05 vs. glutamate group (n= 5 in each group).
Discussion
Our data demonstrates that post-administration of imino-tetrahydrobenzothiazole- based p53 inhibitors PFT-α or PFT-α (O) significantly improved neurological functional deficits and reduced contusion volume following CCI. PFT-α or PFT-α (O) also reduced injury-induced elevations of p53 and annexinV expression in the neurons. CCI resulted in substantial upregulation of PUMA mRNA and protein, and PFT-α or PFT-α (O) attenuated PUMA mRNA transcription and protein synthesis. There was no effect of either PFT derivative in any animal group on body temperature, indicating hypothermia is not a confound to these p53 inhibitor effects.
Our results in rats support two previous studies in mice (Plesnila et al., 2007; Rachmany et al., 2013), evaluating different types of injury and examining other behavioral tests and neurochemical markers, that demonstrate the delayed (secondary) neuronal death associated with TBI is p53-dependent and can be pharmacologically blocked to mitigate functional impairments. In this regard, the p53 inactivator PFT-α has also been reported to be effective in preventing neuronal cell loss in models of stroke, PD, AD and Huntington’s disease models in vitro and in animals (Bae et al., 2005; Culmsee et al., 2001; Duan et al., 2002; Endo et al., 2006; Leker et al., 2004; Zhang et al., 2009; Zhu et al., 2002). This is the first study showing that the neuroprotective effects of PFT-α (O) are significantly better than that of PFT-α in animals, which is in line with actions in cell culture where their 50% effective neuroprotective concentration (EC50) values were 70 nM and 250 nM, respectively (Zhu et al., 2002). Results from the present study also demonstrated a therapeutic window in TBI of between 5 to 7 h for reversal of neuronal demise and behavioral abnormalities.
Extensive studies have demonstrated ischemia-induced p53-dependent apoptosis to have a key role in delayed neuronal cell dysfunction and death occurring in cortical infarction and stroke (Covini et al., 1999; Li et al., 1997; Li et al., 1994; Watanabe et al., 1999). A prior study, evaluating PFT-α in a MCAO murine stroke model, reported a time- and concentration-dependent decrease in infarct volume of up to 50%, with reductions evident within the ischemic penumbra rather than the central core. Diminished infarct size was accompanied by lower behavioral disability scores and attained within an optimal therapeutic window of some 4 h. These actions were associated with lowered levels of both induced-and phosphorylated p53 (Leker et al., 2004). Such p53 involvement in ischemic neuron damage and its amelioration by PFT-α is in line with a reduced infarct volume evaluated in p53 deficient mice (Crumrine et al., 1994). In these animals, a 50% decline in p53 expression in heterozygous p53 knockout animals reduced stroke volume more effectively than homozygous knockout mice, and suggests that partial rather than complete inactivation of p53 is desirable. Notably, ischemic preconditioning that, similar to PFT-α, provides neurons resistance to subsequent ischemia is associated with lowered p53 levels (Tomasevic et al., 1999).
In an open head controlled cortical impact (CCI) model of TBI in mice that causes a cerebral contusion similar to the present study, levels of p53 were reported raised within brain at 15 min following TBI (Plesnila et al., 2007). They were further elevated at 3, 6, and 12 h, and sustained for some 24 h as evaluated at the rim and center of the contusion (Plesnila et al., 2007). This rapid upsurge in p53 protein levels preceded neuronal cell death, and was strongly correlated with the secondary expansion of contusion volume (Plesnila et al., 2007). This is in accord with early heightened p53 expression following fluid percussion TBI injury in the rat (Napieralski et al., 1999) and a time-dependent rise in caspase-3 activity within the hippocampus of rats subjected to TBI-induced concussion and contusion (Nakajima et al., 2010). Of interest, p53 levels within areas of contralateral brain not directly associated with CCI were reported as not different from sham animals (Plesnila et al., 2007), albeit that a diffuse neuron loss throughout the brain has been described across a number of TBI animal models (Greig et al., 2014; Tashlykov et al., 2007; Tashlykov et al., 2009). The administration of PFT-α (6 mg/kg, but particularly 8 mg/kg, i.p.) either before or up to 6 h following CCI, 3 h was optimal, markedly decreased the rise in p53 expression and mitigated the secondary brain tissue loss. Our results in rat are consistent with this prior study in relation to the therapeutic window of opportunity, but were achieved with a lower dose by use of a more potent p53 inactivator, PFT-α (O), and are in line with the PFT-α dose effectively used in a mild concussive TBI model (2 mg/kg) (Rachmany et al., 2013). They are additionally in line with cerebral ischemia studies, in which PFT-α (2 mg/kg, i.p.) mitigated the rise in p53 levels, reduced the infarct size and improved behavioral outcome measures with a therapeutic window of 3 to 4 h (Leker et al., 2004; Zhu et al., 2002). In the present study we found that administration of PFT-α (O) (2 mg/kg, i.v.) at 5 h post-injury reduced p53 translocation. Furthermore, the neuroprotective actions of PFT-α and PFT-α (O) in our primary cultures challenged with glutamate are in line with, and extend, prior studies mitigating excitotoxic and chemical-induced cell death (Culmsee et al., 2003; Culmsee et al., 2001). Our present studies additionally demonstrated that PFT-α and, in particular, PFT-α (O) mitigated microglial cell activation, a marker of neuroinflammation (Felts et al., 2005) that is known to occur following a TBI insult (Tobinick et al., 2012; Greig et al., 2014; Sharp et al., 2014; Baratz et al., 2015). This action on microglial cells may have been a direct anti-inflammatory one, but more likely was a consequence of neuronal neuroprotection - thereby limiting any induced neuroinflammation – and, again, in this in vitro model temperature change is not a confound.
In large part, these effects are also in accord with rodent models of PD, in which both p53 knockout mice and mice pretreated with PFT-α analogues were protected from MPTP-induced dopaminergic neuron loss within the substantia nigra of the brain (Duan et al., 2002; Trimmer et al., 1996). A similar protection was afforded by PFT-α against 6-hydroxydopamine-induced neuron loss in cellular and animal PD models (Biswas et al., 2005; Liang et al., 2007), as well as against neuron loss after proteasome inhibitors (Nair et al., 2006) and paraquat (Yang and Tiffany-Castiglioni, 2008). Our studies, additionally, are in line with PFT-α induced protection against mitochondria-associated cellular dysfunction caused by the mutant huntingtin protein, mHtt, that upregulates p53 levels in Huntington’s disease mHtt-transgenic mice (Bae et al., 2005), as well as PFT-α induced motor neuron protection in spinal cord in a mouse model of amyotrophic lateral sclerosis associated with spinal cord cell loss (Eve et al., 2007). Moreover, p53 is present in synaptosomes and is critical in synapse loss that, likewise, can be protected by p53 inhibitors (Gilman et al., 2003). This is relevant to axonal shearing consequent to TBI (Marklund and Hillered, 2011; Whiting et al., 2006).
The mechanism underpinning the protective actions of PFT-α are reported mediated by inhibiting interaction of p53 with the transcriptional activator p300, with the preservation or enhancement of NF-κB binding to p300 (Culmsee and Mattson, 2005; Culmsee et al., 2003; Gudkov and Komarova, 2010). In such a scenario, proapoptotic cascades are impeded and prosurvival ones augmented, which is largely in accord with our cellular and in vivo data. In light of extensive studies by Gudkov and Komarova (2010), we therefore propose that acutely lowering p53 translocation may provide neuroprotection and neuroregenerative efficacy after a TBI insult without acute or long-term toxicity.
As our data suggest that both PFT-α analogues protect neurons from glutamate-induced excitotoxicity in vitro and improve neurobehavioral outcomes and reduce brain injury in vivo with a therapeutic window between 5 h to 7 h after TBI. Our studies reinforce the usefulness of these agents as pharmacological tools to understand mechanisms underlying delayed neuronal death and cognitive impairments induced by mTBI. In this study, we also demonstrated that PFT-α (O) suppressed the mRNA expression of the pro-apoptotic gene PUMA both in cellular and animal models of TBI. PUMA plays an important role in mediating neuronal apoptosis (Jeffers et al., 2003). Indeed, the initial report of PUMA in an animal neurologic injury model involved global ischemia in rats, where it was found upregulated (Reimertz et al, 2003). This PUMA elevation has been confirmed in models of global cerebral ischemia (Niizuma et al, 2008) and focal cerebral ischemia (Luo et al, 2009; Kuroki et al, 2009). PFT-α mitigated the upregulation of PUMA in these models, supporting p53 involvement (Luo et al, 2009; Niizuma et al, 2008). Likewise, Toth et al. reported that PUMA was up-regulated by ischemia-reperfusion induced hypoxia in rat brain (Toth et al., 2006), which can lead to caspase activation (Puthalakath and Strasser, 2002), with the deletion of PUMA mitigating cell death and improving physiological function after ischemia-reperfusion injury (Toth et al., 2006). Hence, p53 and PUMA appear to be key regulators of neuronal cell death in stroke and across multiple physiologically relevant insults (Engel et al., 2011), and their role in TBI, albeit less well investigated, appears to likewise be important and could be further investigated in knockout models. In conclusion and clearly evident herein, the neuroprotective effects of PFT-α (O) appear to be significantly better than those of PFT-α (with the former providing some 95.6% inhibition of the secondary apoptotic phase of cell death triggered by CCI, reductions in apoptotic markers and improved behavioral outcome). Our studies support the notion that a substantial neural population is amenable to rescue following a TBI, and add to the increasing weight of evidence that inhibition of p53-induced apoptosis by a well-tolerated PFT-α analogue may have the potential to develop into a novel therapeutic strategy for TBI and possibly other tissue injuries of different causes.
Acknowledgments
This study was supported in part by a grant from the Ministry of Science and Technology (MOST103-2321-B-038-002 to JY Wang) and by the Intramural Research Program, National Institute on Aging, NIH.
Footnotes
Financial Disclosure
No competing financial interest exists.
Conflict of interest
The authors have no conflicts of interest relevant to this article to disclose.
References
- Andrews PJ, Piper IR, Dearden NM, Miller JD. Secondary insults during intrahospital transport of head-injured patients. Lancet. 1990;335:327–330. doi: 10.1016/0140-6736(90)90614-b. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Wang JY, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J Neuroinflamm. 2015 doi: 10.1186/s12974-015-0237-4. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Ryu E, Park C, Malagelada C, Greene LA. Puma and p53 play required roles in death evoked in a cellular model of Parkinson disease. Neurochemical research. 2005;30:839–845. doi: 10.1007/s11064-005-6877-5. [DOI] [PubMed] [Google Scholar]
- Chen SF, Hsu CW, Huang WH, Wang JY. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. British journal of pharmacology. 2008;155:1279–1296. doi: 10.1038/bjp.2008.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SF, Hung TH, Chen CC, Lin KH, Huang YN, Tsai HC, Wang JY. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life sciences. 2007;81:288–298. doi: 10.1016/j.lfs.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Chou J, Greig NH, Reiner D, Hoffer BJ, Wang Y. Enhanced survival of dopaminergic neuronal transplants in hemiparkinsonian rats by the p53 inactivator PFT-alpha. Cell transplantation. 2011;20:1351–1359. doi: 10.3727/096368910X557173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covini N, Tamburin M, Consalez G, Salvati P, Benatti L. ZFM1/SF1 mRNA in rat and gerbil brain after global ischaemia. The European journal of neuroscience. 1999;11:781–787. doi: 10.1046/j.1460-9568.1999.00485.x. [DOI] [PubMed] [Google Scholar]
- Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1994;14:887–891. doi: 10.1038/jcbfm.1994.119. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochemical and biophysical research communications. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. Journal of neurochemistry. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Annals of neurology. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- Endo H, Saito A, Chan PH. Mitochondrial translocation of p53 underlies the selective death of hippocampal CA1 neurons after global cerebral ischaemia. Biochemical Society transactions. 2006;34:1283–1286. doi: 10.1042/BST0341283. [DOI] [PubMed] [Google Scholar]
- Engel T, Plesnila N, Prehn JH, Henshall DC. In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:1196–210. doi: 10.1038/jcbfm.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eve DJ, Dennis JS, Citron BA. Transcription factor p53 in degenerating spinal cords. Brain Research. 2007;1150:174–181. doi: 10.1016/j.brainres.2007.02.088. [DOI] [PubMed] [Google Scholar]
- Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217:855–857. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- Felts PA, Woolston AM, Fernando HB, Asquith S, Gregson NA, Mizzi OJ, Smith KJ. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain. 2005;128:1649–66. doi: 10.1093/brain/awh516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman CP, Chan SL, Guo Z, Zhu X, Greig N, Mattson MP. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Medicine. 2003;3:159–172. doi: 10.1385/NMM:3:3:159. [DOI] [PubMed] [Google Scholar]
- Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Annals of the New York Academy of Sciences. 2004;1035:290–315. doi: 10.1196/annals.1332.018. [DOI] [PubMed] [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri DK, Sambamurti K, Becker RE, Pick CG. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2014;10:S62–75. doi: 10.1016/j.jalz.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA. Pathologies associated with the p53 response. Cold Spring Harbor perspectives in biology. 2010;2:a001180. doi: 10.1101/cshperspect.a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YN, Wu CH, Lin TC, Wang JY. Methamphetamine induces heme oxygenase-1 expression in cortical neurons and glia to prevent its toxicity. Toxicology and applied pharmacology. 2009;240:315–326. doi: 10.1016/j.taap.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kuroki K, Virard I, Concannon CG, Engel T, Woods I, Taki W, Plesnila N, Henshall DC, Prehn JH. Effects of transient focal cerebral ischemia in mice deficient in puma. Neurosci Lett. 2009;451:237–240. doi: 10.1016/j.neulet.2009.01.019. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Simon CM, Prado GR, Cullen DK. CNS injury biomechanics and experimental models. Progress in brain research. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Experimental neurology. 2004;187:478–486. doi: 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M, Powers C, Jiang N. Apoptosis and protein expression after focal cerebral ischemia in rat. Brain research. 1997;765:301–312. doi: 10.1016/s0006-8993(97)00524-6. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S. p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke; a journal of cerebral circulation. 1994;25:849–855. doi: 10.1161/01.str.25.4.849. discussion 855–846. [DOI] [PubMed] [Google Scholar]
- Liang ZQ, Li YL, Zhao XL, Han R, Wang XX, Wang Y, Chase TN, Bennett MC, Qin ZH. NF-kappaB contributes to 6-hydroxydopamine-induced apoptosis of nigral dopaminergic neurons through p53. Brain research. 2007;1145:190–203. doi: 10.1016/j.brainres.2007.01.130. [DOI] [PubMed] [Google Scholar]
- Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, Wang Y. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Annals of neurology. 2009;65:520–530. doi: 10.1002/ana.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Dearden M, Servadei F, Stocchetti N, Unterberg A. Current recommendations for neurotrauma. Current opinion in critical care. 2000;6:281–292. [PubMed] [Google Scholar]
- Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? British journal of pharmacology. 2011;164:1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD. Minor, moderate and severe head injury. Neurosurgical review. 1986;9:135–139. doi: 10.1007/BF01743065. [DOI] [PubMed] [Google Scholar]
- Nair VD, McNaught KS, Gonzalez-Maeso J, Sealfon SC, Olanow CW. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. The Journal of biological chemistry. 2006;281:39550–39560. doi: 10.1074/jbc.M603950200. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Horiuchi Y, Kamata H, Yukawa M, Kuwabara M, Tsubokawa T. Distinct time courses of secondary brain damage in the hippocampus following brain concussion and contusion in rats. The Tohoku journal of experimental medicine. 2010;221:229–235. doi: 10.1620/tjem.221.229. [DOI] [PubMed] [Google Scholar]
- Napieralski JA, Raghupathi R, McIntosh TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain research. Molecular brain research. 1999;71:78–86. doi: 10.1016/s0169-328x(99)00155-2. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2008;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrancosta N, Moumen A, Dono R, Lingor P, Planchamp V, Lamballe F, Bahr M, Kraus JL, Maina F. Imino-tetrahydro-benzothiazole derivatives as p53 inhibitors: discovery of a highly potent in vivo inhibitor and its action mechanism. Journal of medicinal chemistry. 2006;49:3645–3652. doi: 10.1021/jm060318n. [DOI] [PubMed] [Google Scholar]
- Plesnila N, von Baumgarten L, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell death and differentiation. 2007;14:1529–1541. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. Journal of neurochemistry. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell death and differentiation. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Yu QS, Li Y, Wang JY, Pick CG, Greig NH. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53-dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT-alpha. PloS one. 2013;8:e79837. doi: 10.1371/journal.pone.0079837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly P. The impact of neurotrauma on society: an international perspective. Progress in brain research. 2007;161:3–9. doi: 10.1016/S0079-6123(06)61001-7. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain research. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol. 2014;210:156–66. doi: 10.1038/nrneurol.2014.15. [DOI] [PubMed] [Google Scholar]
- Sohn D, Graupner V, Neise D, Essmann F, Schulze-Osthoff K, Janicke RU. Pifithrin-alpha protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell death and differentiation. 2009;16:869–878. doi: 10.1038/cdd.2009.17. [DOI] [PubMed] [Google Scholar]
- Spaethling JM, Geddes-Klein DM, Miller WJ, von Reyn CR, Singh P, Mesfin M, Bernstein SJ, Meaney DF. Linking impact to cellular and molecular sequelae of CNS injury: modeling in vivo complexity with in vitro simplicity. Progress in brain research. 2007;161:27–39. doi: 10.1016/S0079-6123(06)61003-0. [DOI] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Gazit V, Zohar O, Schreiber S, Pick CG. Apoptotic changes in the cortex and hippocampus following minimal brain trauma in mice. Brain research. 2007;1130:197–205. doi: 10.1016/j.brainres.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Volkov A, Gazit V, Schreiber S, Zohar O, Pick CG. Minimal traumatic brain injury induce apoptotic cell death in mice. Journal of molecular neuroscience : MN. 2009;37:16–24. doi: 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26:1051–70. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- Tomasevic G, Shamloo M, Israeli D, Wieloch T. Activation of p53 and its target genes p21(WAF1/Cip1) and PAG608/Wig-1 in ischemic preconditioning. Brain research. Molecular brain research. 1999;70:304–313. doi: 10.1016/s0169-328x(99)00146-1. [DOI] [PubMed] [Google Scholar]
- Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. American journal of physiology. Heart and circulatory physiology. 2006;291:H52–60. doi: 10.1152/ajpheart.01046.2005. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Smith TS, Jung AB, Bennett JP., Jr Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration : a journal for neurodegenerative disorders, neuroprotection, and neuroregeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ, Suazo JA, Hogesteeger C, Jansen WJ, Kloos LM, Vermeulen J, Maas AI. Brain oxygen tension in severe head injury. Neurosurgery. 2000;46:868–876. doi: 10.1097/00006123-200004000-00018. discussion 876–868. [DOI] [PubMed] [Google Scholar]
- van Santbrink H, Schouten JW, Steyerberg EW, Avezaat CJ, Maas AI. Serial transcranial Doppler measurements in traumatic brain injury with special focus on the early posttraumatic period. Acta neurochirurgica. 2002;144:1141–1149. doi: 10.1007/s00701-002-1012-8. [DOI] [PubMed] [Google Scholar]
- Verweij BH, Amelink GJ, Muizelaar JP. Current concepts of cerebral oxygen transport and energy metabolism after severe traumatic brain injury. Progress in brain research. 2007;161:111–124. doi: 10.1016/S0079-6123(06)61008-X. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Ohta S, Kumon Y, Sakaki S, Sakanaka M. Increase in p53 protein expression following cortical infarction in the spontaneously hypertensive rat. Brain research. 1999;837:38–45. doi: 10.1016/s0006-8993(99)01652-2. [DOI] [PubMed] [Google Scholar]
- Whiting MD, Baranova AI, Hamm RJ. In: Cognitive Impairment following Traumatic Brain Injury. Levin ED, Buccafusco JJ, editors. Animal Models of Cognitive Impairment; Boca Raton (FL): 2006. [PubMed] [Google Scholar]
- Yang W, Tiffany-Castiglioni E. Paraquat-induced apoptosis in human neuroblastoma SH-SY5Y cells: involvement of p53 and mitochondria. Journal of toxicology and environmental health. Part A. 2008;71:289–299. doi: 10.1080/15287390701738467. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhang C, Raghupathi R, Saatman KE, Smith DH, Stutzmann JM, Wahl F, McIntosh TK. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. Journal of neuroscience research. 1998;52:342–349. doi: 10.1002/(SICI)1097-4547(19980501)52:3<342::AID-JNR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Wang Y, Wang Y, Zhang X, Han R, Wu JC, Liang ZQ, Gu ZL, Han F, Fukunaga K, Qin ZH. p53 mediates mitochondria dysfunction- triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5:339–350. doi: 10.4161/auto.5.3.8174. [DOI] [PubMed] [Google Scholar]
- Zhu X, Yu QS, Cutler RG, Culmsee CW, Holloway HW, Lahiri DK, Mattson MP, Greig NH. Novel p53 inactivators with neuroprotective action: syntheses and pharmacological evaluation of 2-imino-2,3,4,5,6,7- hexahydrobenzothiazole and 2-imino-2,3,4,5,6,7-hexahydrobenzoxazole derivatives. Journal of medicinal chemistry. 2002;45:5090–5097. doi: 10.1021/jm020044d. [DOI] [PubMed] [Google Scholar]