

Abstract
Products of the LMNA gene, primarily lamin A and C, are key components of the nuclear lamina, a proteinaceous meshwork that underlies the inner nuclear membrane and is essential for proper nuclear architecture. Alterations in lamin A and C that disrupt the integrity of the nuclear lamina affect a whole repertoire of nuclear functions, causing cellular decline. In humans, hundreds of mutations in the LMNA gene have been identified and correlated with over a dozen degenerative disorders, referred to as laminopathies. These diseases include neuropathies, muscular dystrophies, lipodystrophies, and premature aging diseases. This review focuses on one of the most severe laminopathies, Hutchinson-Gilford Progeria Syndrome (HGPS), which is caused by aberrant splicing of the LMNA gene and expression of a mutant product called progerin. Here, we discuss current views about the molecular mechanisms that contribute to the pathophysiology of this devastating disease, as well as the strategies being tested in vitro and in vivo to counteract progerin toxicity. In particular, progerin accumulation elicits nuclear morphological abnormalities, misregulated gene expression, defects in DNA repair, telomere shortening, and genomic instability, all of which limit cellular proliferative capacity. In patients harboring this mutation, a severe premature aging disease develops during childhood. Interestingly, progerin is also produced in senescent cells and cells from old individuals, suggesting that progerin accumulation might be a factor in physiological aging. Deciphering the molecular mechanisms whereby progerin expression leads to HGPS is an emergent area of research, which could bring us closer to understanding the pathology of aging.
Keywords: Laminopathies, nuclear lamina, progerin, genomic instability, HGPS pathology, HGPS treatment
1. Introduction
The nuclear lamina has emerged as a nuclear compartment with critical roles in the maintenance of nuclear architecture and stability, as well as genome organization and function (Burke and Stewart 2014, Gruenbaum and Foisner 2015, Gruenbaum and Medalia 2015). The nuclear lamina is composed of the type V intermediate filament proteins A-type lamins (primarily lamin A/C) and B-type lamins (lamin B1/B2), in addition to lamina-associated proteins. B-type lamins are expressed in all cells and found almost exclusively at the nuclear periphery. A-type lamins (lamin A/C) result from alternative splicing of the LMNA gene and are found at the nuclear lamina and throughout the nuclear interior, being expressed mainly in differentiated cells. The current view is that lamins serve a scaffolding role, anchoring chromatin and transcription factors to the nuclear periphery, providing the compartmentalization of the genome that is required for proper DNA transactions such as transcription, replication, and repair, as well as transducing signals from the cytoskeleton into the nucleus. In addition, lamin expression level is directly linked to mechanical stability of the nucleus, and tissue rigidity and plasticity (Swift, Ivanovska et al. 2013). The association of mutations in lamin proteins, primarily in the LMNA gene, with over a dozen degenerative disorders underscores the importance of nuclear lamins in health and disease. Lamin-associated diseases or laminopathies encompass a range of phenotypes with different tissue pathologies, including muscular dystrophy disorders (e.g., Emery-Dreyfus Muscular Dystrophy or EDMD), peripheral neuropathies (e.g. Charcot-Marie-Tooth-Disease type 2B1 or CMT2B), lipodystrophies, as well as premature aging syndromes such as Hutchinson Gilford Progeria Syndrome (HGPS), Atypical Werner Syndrome (AWS), and restrictive dermopathy (RD) (Worman, Fong et al. 2009, Gordon, Rothman et al. 2014, Gonzalo and Kreienkamp 2015, Vidak and Foisner 2016). Despite intensive research, the relationships between genotypes and phenotypes in laminopathies remain poorly understood (Smith, Kudlow et al. 2005, Bertrand, Chikhaoui et al. 2011). Some hotspot mutations in the LMNA gene have been identified and associated with specific types of laminopathies, such is the case of HGPS. However, different mutations throughout the LMNA gene can cause the same type of disorder, and different substitutions of the same base can cause different disorders. Furthermore, the same LMNA mutation can cause disease in some individuals and be asymptomatic in others, stressing the enormous phenotypic variability in laminopathies (Rodriguez and Eriksson 2011). This variability suggests that mechanisms other than LMNA mutation contribute to the development of the disease. Understanding the factors that determine disease severity in laminopathies is an active area of investigation.
Another intriguing question in the field has been why laminopathies affect only a single or a few tissues, when lamin A/C are ubiquitously expressed. Some models propose that lamins alterations impact the 3D organization of the genome, inducing changes in gene expression. These changes vary among tissues, providing tissue specificity of laminopathies. Other models propose that alterations in lamins impact the mechanotransduction properties of cells, being especially detrimental for tissues such as muscle that are exposed to strong mechanical tension. Here we review the knowledge about the molecular mechanisms whereby mutations in the LMNA gene cause cellular and organismal decline, as well as the pathophysiology of HGPS and current therapeutic strategies for ameliorating this devastating disease.
2. HGPS is caused by expression of a mutant lamin A protein “progerin”
The LMNA gene encodes four lamins (A, C, CΔ10, and C2) via alternative splicing, of which lamin A and C are the most ubiquitously expressed. Lamin A and C are identical up to residue 574. Lamin C possesses five unique C-terminal residues, and lamin A is synthesized as a 664-residue prelamin A precursor that after post-translational processing results in a mature lamin A protein of 646 residues. Prelamin A contains a C-terminal -CAAX motif that is farnesylated, followed by cleavage of the last three residues and carboxymethylation of the terminal cysteine (Figure 1A). These modifications facilitate the association of prelamin A with the nuclear envelope. Subsequently, the endoprotease Zmpste24 (FACE-1) cleaves the 15 C-terminal residues, rendering mature lamin A. To date, over 400 mutations in the LMNA gene have been identified and associated with disease (http://www.umd.be/LMNA/).
Figure 1. Abnormal processing of lamin A in HGPS.

(A) Mature lamin A is produced from prelamin A, which is farnesylated followed by trimming and methylation at the C-terminus. Finally, cleavage by the Zmpste24 protease between amino acid residues 646-647 removes the farnesylated C-terminal end. (B) In HGPS patients, a mutation in exon 11 activates a cryptic splice site leading to deletion of 50 amino acid residues from the precursor protein, including the final Zmpste24 cleavage site, and accumulation of farnesylated progerin. (C) Overview of cellular consequences of progerin expression as well as proteins, small compounds and processes that influence progerin levels and/or progerin-induced defects.
In 2003, two laboratories reported simultaneously the identification of a mutation in the LMNA gene that causes HGPS (De Sandre-Giovannoli, Bernard et al. 2003, Eriksson, Brown et al. 2003). This mutation is a de novo single-base substitution within the LMNA exon 11 (c.1824C>T) that activates a cryptic splice site, leading to an in-frame deletion of 50 amino acids near the C-terminus of prelamin A (Figure 1B). The abnormal protein produced, known as ‘progerin’, retains the –CAAX motif and is farnesylated and carboxymethylated. However, progerin lacks the second site for endoproteolytic cleavage, and thus remains permanently farnesylated and carboxymethylated. This mutant form of lamin A acts in a dominant fashion to induce a whole variety of abnormalities in nuclear processes, which eventually lead to cellular and organismal decline. In fact, a morpholino oligonucleotide inhibiting usage of the cryptic splice site decreases progerin production and significantly ameliorates cellular phenotypes (Scaffidi and Misteli 2006). Although c.1824C>T remains the most frequent mutation in HGPS patients, other mutations in the LMNA gene have been reported that result in increased usage of the cryptic splice site. Some of these mutations cause a more severe phenotype than classical HGPS. Several studies indicate that the amount of progerin relative to prelamin A dictates the severity of the disease (Moulson, Fong et al. 2007, Reunert, Wentzell et al. 2012).
Post-translational modifications of progerin seem to play a major role in the pathophysiology of disease (Toth, Yang et al. 2005, Yang, Bergo et al. 2005, Fong, Frost et al. 2006, Varela, Pereira et al. 2008, Ibrahim, Sayin et al. 2013). For instance, blocking farnesylation of progerin results in a marked improvement of nuclear abnormalities in fibroblasts from both HGPS patients and mouse models of progeria. In addition, treatment with prenylation inhibitors in vivo improves the aging-like phenotype of the mice and increases lifespan (Varela, Pereira et al. 2008). Similarly, a mouse model of progeria with a hypomorphic allele of isoprenylcysteine carboxyl methyltransferase (ICMT), the enzyme that methylates the C-terminal end of prelamin A/progerin, exhibits a marked improvement of phenotype and increased survival. ICMT inhibition also delays senescence in HGPS fibroblasts (Ibrahim, Sayin et al. 2013).
Other post-translational modifications of lamin A include phosphorylation and sumoylation. Phosphorylation of lamin A facilitates disassembly of lamin filaments and breakdown of the nuclear lamina in preparation for mitosis (Snider and Omary 2014). In HGPS cells, phosphorylation of a prominent site (S22) by CDK1 during interphase is reduced, and prenylation inhibitors can rescue this defect (Moiseeva, Lopes-Paciencia et al. 2016). Moreover, preventing phosphorylation of lamin A/progerin by CDK inhibitors accelerates senescence in HGPS fibroblasts. Sumoylation of lamin A at lysine K201 by the SUMO E2 enzyme Ubc9 has been reported, and shown to require the conserved E203 residue (Zhang and Sarge 2008). Sumoylation is important for the proper localization of lamin A within the nucleus. Expression of GFP-tagged lamin A mutant proteins K201R, E203G, or E203K, which all have reduced sumoylation, shows an altered localization pattern, with the mutant proteins accumulating at foci near the nuclear periphery instead of a more continuous pattern. This is accompanied by decreased cell viability. Moreover, fibroblasts from a patient with the E203K lamin A mutation show decreased lamin A sumoylation and increased cell death, supporting a role for sumoylation in lamin A localization and function. Importantly, HGPS patient-derived fibroblasts exhibit a reduction in the nuclear/cytoplasmic concentration of Ran GTPase, resulting in reduced nuclear levels of Ubc9, as well as TPR, a nucleoporin from the basket of the nuclear pore complex (Kelley, Datta et al. 2011). Ectopic expression of progerin in HeLa cells was sufficient to reduce nuclear levels of Ubc9, while forced localization of Ubc9 to the nucleus ameliorated these progerin-induced phenotypes. These data suggest that progerin impacts the function of Ran GTPase and sumoylation pathways. Overall, these studies demonstrate that lamin A undergoes a variety of post-translational modifications that are important for its proper localization and function. Alterations in these modifications with progerin contribute to progerin-induced phenotypes.
Progerin expression provokes cellular decline, eliciting nuclear morphological abnormalities, misregulated gene expression, chromatin changes, mitochondrial dysfunction, defects in DNA repair, alternate splicing, accelerated telomere shortening and premature senescence (Figure 1C) (Goldman, Shumaker et al. 2004, Prokocimer, Barkan et al. 2013, Gonzalo and Kreienkamp 2015, Gonzalo and Eissenberg 2016). Despite enormous progress in recent years identifying cellular processes altered by progerin, we still lack a clear picture of the molecular mechanisms whereby progerin expression causes all these cellular phenotypes.
3. Molecular mechanisms behind cellular decline in HGPS
3.1. Nuclear morphological abnormalities
The cytological hallmark of HGPS patient-derived fibroblasts is nuclear morphological abnormality (Figure 2). HGPS nuclei appear bigger and dysmorphic, with protrusions and chromatin herniations throughout, as well as thickening of the nuclear lamina and disorganization of nuclear pores and chromatin (Goldman, Shumaker et al. 2004). In addition, HGPS nuclei accumulate basal level of DNA damage owed to deficiencies in DNA repair mechanisms (Figure 3). These nuclear defects are exacerbated during proliferation in culture, concomitant with accumulation of progerin and immobilization of lamin A at the nuclear lamina. Recent studies have shown a dosage-dependent effect of progerin expression in normal fibroblasts inducing phenotypes characteristic of HGPS fibroblasts (Chojnowski, Ong et al. 2015). This suggests that reducing progerin levels under a threshold could be sufficient to reduce phenotype severity. In fact, Lee and colleagues have reported the effectiveness of a LMNA gene exon 11 antisense oligonucleotide (ASO) increasing lamin C production at the expense of lamin A in normal primary fibroblasts (Lee, Nobumori et al. 2016). These cells did not present a detectable phenotype, consistent with lamin C-only mice being disease free (Fong, Ng et al. 2006). This ASO was able to reduce progerin expression in HGPS fibroblasts. Importantly, administration of exon 11 ASO in a mouse model of progeria reduced significantly lamin A and progerin levels in mouse tissues and improved aortic pathology. This strategy to reduce progerin might be of therapeutic applicability. Mechanistically, the study showed that the serine/arginine-rich splicing factor 2 (SRSF2) is critical for alternative splicing of the LMNA gene, with SRSF2 loss of function shifting the output of splicing towards lamin C, with lower expression of all lamin A forms.
Figure 2. Expression of progerin alters nuclear organization and genome stability.

Cells from HGPS patients are characterized by a series of alterations including reduced expression of extracellular matrix (ECM) components, nuclear envelope blebs, clustering of nuclear pore complexes (NPC), loss of peripheral heterochromatin, and reorganized microtubules. Progerin expression also affects dynamics of nuclear envelope transmembrane proteins (NETs), including emerin, and their interactions with chromatin-associated proteins, such as BAF, transcription factors (TF) and chromatin modifiers. HGPS cells have higher levels of reactive oxygen species (ROS) and DNA damage, whereas LAP2α is downregulated.
Figure 3. Nuclear defects in HGPS cells.
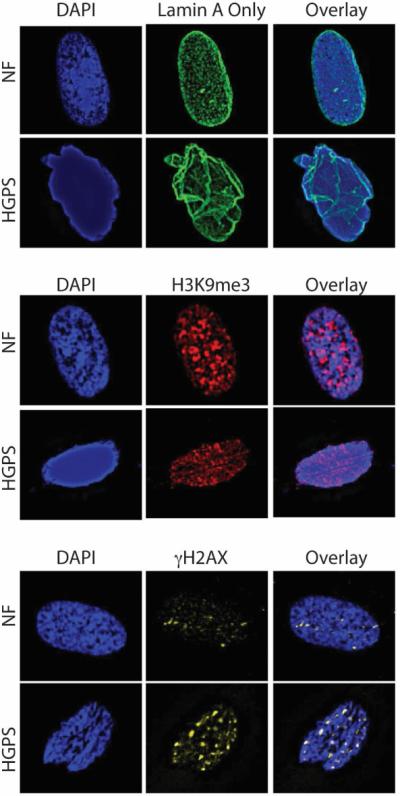
Immunofluorescence performed in primary normal human fibroblasts (NF) and HGPS patient derived fibroblasts (HGPS) with antibodies recognizing lamin A (green, top panels), histone modification H3K9me3 (red, medium panels), and γH2AX (yellow, bottom panels), a marker of DNA damage. DAPI staining was used to demarcate nuclei (blue staining in all panels). Note how HGPS patient derived fibroblasts exhibit nuclear morphological abnormalities, decreased levels of H3K9me3, and accumulation of basal levels of unrepaired DNA damage, when compared to normal fibroblasts.
Progerin also has different behavior than lamin A during mitosis. Progerin causes defects in chromosome segregation and nuclear envelope reassembly in addition to trapping lamina components and inner nuclear envelope proteins in the endoplasmic reticulum after mitosis (Eisch, Lu et al. 2016). Interestingly, progerin seems to displace the centromere protein F (CENP-F) from kinetochores, resulting in increased genomic instability, which in turn contributes to premature senescence.
Another characteristic of HGPS fibroblasts is their increased nuclear stiffness with increasing passage and their sensitivity to mechanical strain. While strain application in normal cells induces proliferative signals, HGPS cells lack this cell cycle activation response (Verstraeten, Ji et al. 2 008). In addition, polarization microscopy showed that lamins in HGPS nuclei have a reduced ability to rearrange under mechanical stress (Dahl, Scaffidi et al. 2006). These different responses to force may explain tissue specific effects of progerin. For instance, tissues with high levels of mechanical stress such as bone, skeletal muscle, heart, and blood vessels could be especially affected by progerin expression. Thus, impaired mechanotransduction could contribute to bone and vascular defects in HGPS patients (Prokocimer, Barkan et al. 2013).
3.2. Epigenetic changes
Alterations in histone modifications, DNA methylation, chromatin-modifying activities, and overall chromatin architecture have been observed in progerin-expressing cells (Arancio, Pizzolanti et al. 2014). In HGPS, there is loss of peripheral heterochromatin and reduced levels of repressive histone modifications (H3K9me3, H3K27me3)(Figure 3) and associated proteins (HP1), as well as increased transcription of pericentromeric satellite repeats (Scaffidi and Misteli 2006, Shumaker, Dechat et al. 2006, Dechat, Pfleghaar et al. 2008). Interestingly, these chromatin changes and expression of progerin are also observed in cells from old individuals, suggesting their implication in physiological aging (Scaffidi and Misteli 2006). Levels of H3K9me3 vary with changes of histone methyltransferase Suv39h1 expression in HGPS cells in culture (Liu, Wang et al. 2013). This enzyme seems to play a key role in progeria because knockout of Suv39h1 in a mouse model of progeria improves a variety of phenotypes. The changes in H3K27me3 correlate with upregulation of poorly expressed genes and downregulation of highly expressed genes, a trend that is observed in senescent HGPS cells (McCord, Nazario-Toole et al. 2013). Recently, deregulation of H3K27me3 in HGPS cells has been linked to impaired interaction of progerin with lamina-associated polypeptide-α (LAP2α). This protein binds tightly to lamin A, facilitating the interaction of lamin A with euchromatin (Gesson, Rescheneder et al. 2016). In fact, the chromatin immunoprecipitation (ChIP) profile of lamin A/C shows strong overlap with that of LAP2α (Gesson, Rescheneder et al. 2016). Consistent with this data, LAP2α-deficient cells exhibit reduced binding of lamin A/C to euchromatic domains. In contrast, progerin interacts weakly with LAP2α, and expression of progerin reduces dramatically the cellular levels of LAP2α (Vidak, Kubben et al. 2015). This is accompanied by reduced H3K27me3 levels and proliferation defects, which are rescued by increasing LAP2α levels (Chojnowski, Ong et al. 2015, Vidak, Kubben et al. 2015). Barrier-to-Autointegration Factor (BAF) is another lamina-associated protein recently proposed to mediate chromatin defects upon expression of prelamin A/progerin (Loi, Cenni et al. 2015). BAF is required for the changes in H3K9me3, HP1 and LAP2α observed in response to prelamin A accumulation. Progerin interacts with BAF and this study shows that the LAP2α nuclear localization defects observed in HGPS cells involves progerin-BAF interaction. These findings establish functional links between lamina-associated proteins and lamin A/C pathological forms.
HGPS patient cells also exhibit reduced expression of components of the NURD (Nucleosome Remodeling Deacetylase) complex, including RBBP1, RBBP7, MTA3, and HDCA1 subunits (Pegoraro, Kubben et al. 2009). Loss of NURD subunits RBBP1 and RBBP7 precedes DNA damage accumulation in progerin-expressing cells and depletion of these subunits in normal cells causes decreased methylation of histone H3 and DNA damage. In addition, the function of SIRT6, a member of the sirtuin family with both deacetylation and mono-ADP ribosylation activities, is significantly compromised in progerin-expressing cells. SIRT6 deficiency has been proposed to contribute to DNA repair defects and genomic instability in HGPS cells (Ghosh, Liu et al. 2015). Moreover, hypoacetylation of histone H4 has been reported in a mouse model of progeria, and linked to deficiency in the histone acetyltransferase MOF. Treatment of mouse progeria cells with a histone deacetylase inhibitor improved DNA repair and delayed entry into senescence (Krishnan, Chow et al. 2011).
In addition to histone modifications, DNA methylation patterns are altered in progeria cells. In particular, hypermethylation of ribosomal RNA genes and thus a reduction in their production has been reported in a mouse model of progeria (Osorio, Varela et al. 2010). HGPS cells also show a gain in methylation of CpG sites that tend to be hypomethylated in normal cells, and a decrease in methylation of CpG sites normally hypermethylated (Heyn, Moran et al. 2013). All these studies reveal that epigenetic changes are induced by expression of progerin and that strategies that reverse these changes could be investigated as a therapeutic possibility.
3.3. Misregulated gene expression
Genome-wide expression profiling of HGPS fibroblasts revealed a widespread transcriptional misregulation when compared to normal fibroblasts. Transcription factors (TFs) and extracellular matrix (ECM) proteins were the more prominent functional categories differentially expressed in HGPS cells (Csoka, English et al. 2004). The reported gene expression profile is consistent with disorders affecting mesodermal and mesenchymal cell lineages. In particular, HGPS fibroblasts exhibit high expression of ECM proteins and low expression of ECM remodelling enzymes, which can result in aberrant ECM deposition. In a mouse model carrying a progeric mutation within the Lmna gene, inhibition of the Wnt pathway was reported, leading to reduced function of the transcription factor LEF1, which regulates expression of ECM proteins (Hernandez, Roux et al. 2010). These studies suggest that deficiencies in Wnt signalling could cause changes in ECM composition, contributing to vascular stiffness in HGPS (Vidak and Foisner 2016).
In addition, HGPS fibroblasts exhibit defects in the retinoblastoma protein (pRb) signalling pathway and these defects are reversed by farnesyltransferase inhibitor (FTI) treatment (Marji, O'Donoghue et al. 2010). Members of the retinoblastoma family, which include pRb, p107 and p130, participate in multiple cellular processes such as cell cycle progression, differentiation, senescence, and apoptosis (Gonzalo and Blasco 2005). Rb proteins induce a repressive chromatin state around euchromatic promoters and also participate in the assembly of heterochromatin domains such as centromeres and telomeres (Gonzalo, Garcia-Cao et al. 2005). Thus, deficiency in Rb function in HGPS cells could contribute to proliferation defects, changes in gene expression, as well as alterations in the structure and function of heterochromatin domains.
Moreover, HGPS fibroblasts activate downstream effectors of the Notch signalling pathway (Scaffidi and Misteli 2008). This is owed to progerin releasing the sequestration of the Notch co-activator SKIP by wild-type lamins, resulting in transcriptional activation of downstream genes such as TEL1, HES1, and HES5. This phenotype is also observed upon induction of progerin expression in hMSCs (human mesenchymal stem cells), which in turn impacts their differentiation potential. These data provide a link between accelerated aging in HGPS patients and adult stem cell dysfunction. Further support for altered stem cell functions in HGPS came from cellular reprogramming of HGPS fibroblasts to induced pluripotent stem cells (iPSCs), followed by differentiation of HGPS-iPSCs into different lineages (Zhang, Lian et al. 2011). In particular, this study identified vascular smooth muscle and mesenchymal stem cell defects. Thus, shortage or exhaustion of stem cells needed for tissue replacement could represent a major cause of the pathology of HGPS.
Interestingly, the aberrant interaction of progerin with TFs that control adipogenesis has been proposed as a mechanism behind lipodystrophy in HGPS. For instance, SREBP1 binds with high affinity to progerin, resulting in SREBP1 sequestration at the nuclear periphery and reduction of its transcriptional activity (Duband-Goulet, Woerner et al. 2011). Similarly, impaired differentiation of human mesenchymal stem cells (hMSCs) into the adipose lineage was recently reported upon accumulation of prelamin A (Ruiz de Eguino, Infante et al. 2012). This effect is owed to sequestration of the transcription factor Sp1 by prelamin A, resulting in altered expression of ECM genes.
Sequestration of TFs seems to be a recurrent theme in laminopathies. Approaches on a global scale have attempted to map the interactome of lamin A, and to elucidate how progerin alters these interactions. A yeast two-hybrid screening identified 225 progerin-specific interactions, 51 proteins that interacted with both lamin A and progerin, and 61 interactions specific from lamin A (Dittmer, Sahni et al. 2014). The majority of progerin-specific interacting proteins were localized at the nuclear envelope (89%), while only half of lamin A-binding proteins accumulate at this location. Progerin-specific interactors were enriched for intrinsic membrane proteins with channel and transport function, including ER components and proteins involved in membrane organization, protein localization, and vesicle-mediated transport. In contrast, many lamina and nuclear envelope components were found among the lamin A-specific interacting proteins. It is tempting to speculate that gain or loss of tissue-specific lamin A/C-interactions could provide tissue specificity of laminopathies. Expression of progerin might either increase or decrease the sequestration of proteins at the nuclear periphery or in the nuclear interior, impacting their function.
A recent study has also shown that lamin A/C play a role in the compartmentalization and function of the Polycomb group (PcG) of proteins, key regulators of development and differentiation (Cesarini, Mozzetta et al. 2015). Depletion of A-type lamins leads to dispersion of PcG proteins, hindering their gene repressive functions. As a consequence, loss of lamin A/C expression accelerates the differentiation of myoblasts to myotubes (Cesarini, Mozzetta et al. 2015). These data indicate that through regulation of PcG group of factors, lamin A/C proteins can modulate the expression of a variety of genes. Further studies are needed to determine if progerin expression can impact the localization and function of PcG proteins, and if these processes contribute to the alterations in gene expression observed in HGPS patient-derived cells.
3.4. DNA repair defects and genomic instability
Fibroblasts from HGPS patients and from mouse models of progeria exhibit accumulation of DNA damage, chromosomal instability, increased sensitivity to DNA-damaging agents (Liu, Wang et al. 2005, Varela, Cadinanos et al. 2005), and a permanently activated DNA damage response (DDR) (Figure 3) (Scaffidi and Misteli 2006). Molecular mechanisms contributing to DNA damage in HGPS include: defects in the recruitment of DNA repair factors 53BP1, Rad50 and Rad51 to sites of damage that instead exhibit aberrant accumulation of the nucleotide excision repair protein XPA (Liu, Wang et al. 2005, Liu, Wang et al. 2008); delayed recruitment of phospho-NBS1 and MRE11, components of the MRN complex necessary for sensing DNA lesions (Constantinescu, Csoka et al. 2010); and decreased nuclear levels of components of the DNAPK holoenzyme (DNAPKcs, Ku70 and Ku80) (Liu, Barkho et al. 2011). These deficiencies are consistent with delayed checkpoint response and defects in double-strand break (DSB) repair by non-homologous end joining (NHEJ) and homologous recombination (HR). In mouse models of progeria, defects in ATM-KAP-1 signaling and DNA damage-induced chromatin remodeling were reported (Liu, Wang et al. 2013), and proposed to contribute to deficiencies in the recruitment and retention of DNA repair proteins at heterochromatic lesions.
Interestingly, iPSCs generated from HGPS fibroblasts lack nuclear morphological abnormalities and progerin expression, consistent with the LMNA gene being silenced in stem cells (Liu, Barkho et al. 2011). Differentiation of HGPS-iPSCs to smooth muscle cells (SMCs) results in progerin expression and appearance of progerin-induced phenotypes such as DNA damage, leading to premature senescence. Importantly, these cells exhibit a marked suppression of Poly-(ADP-ribose) polymerase 1 (PARP1), an enzyme that participates in the repair of ssDNA breaks and is critical for the fidelity of DNA replication. Loss of PARP1 in HGPS SMCs leads to an aberrant upregulation of NHEJ during S-phase, contributing to mitotic catastrophe and cell death (Liu, Barkho et al. 2011).
Other factors contributing to genomic instability in HGPS cells include the accumulation of reactive oxygen species (ROS) due to mitochondrial dysfunction. HGPS fibroblasts are hindered in their ability to repair ROS-induced DNA damage, exhibiting higher sensitivity to oxidative stress than normal fibroblasts (Richards, Muter et al. 2011). The accumulation of un-repairable DNA breaks in HGPS cells correlates with proliferation defects, and ROS scavengers reduce DNA damage and improve cellular growth. In addition, SILAC (stable isotope labeling with amino acid) analysis performed in HGPS and normal fibroblasts shows a reduction in the levels of mitochondrial proteins participating in oxidative phosphorylation in HGPS fibroblasts, which is accompanied by mitochondrial dysfunction (Rivera-Torres, Acin-Perez et al. 2013). Similar mitochondrial dysfunction was observed in mouse models of progeria, suggesting that these problems are common in laminopathies.
Impaired NRF2 pathway activity also contributes to increased oxidative stress in HGPS (Kubben, Zhang et al. 2016). Normally, the NRF2 pathway activates antioxidant genes by binding to antioxidant-responsive elements (ARE) motifs. Interestingly, progerin tightly binds NRF2, causing NRF2 subnuclear mislocalization and disrupting formation of transcription factor complexes at ARE motifs. Consequently, NRF2-ARE target genes are repressed in HGPS, and chronic oxidative stress abounds. Excitingly, reactivation of the NRF2 pathway ameliorates classic alterations in HGPS cells, reducing progerin protein levels, lowering oxidative stress, and restoring lamin B1 levels. This finding underscores the importance of oxidative stress to the HGPS phenotype and suggests that NRF2-activiating compounds might be a means for improving HGPS phenotype.
3.5. Telomere shortening
HGPS patient-derived fibroblasts exhibit faster telomere attrition during proliferation in culture than normal fibroblasts, which causes DNA damage and premature entry into senescence (Gonzalo and Kreienkamp 2015). Ectopic expression of telomerase or p53 inactivation reduces DNA damage and suppresses the proliferative defects, suggesting that telomere dysfunction and activation of checkpoints contribute to these defects (Kudlow, Stanfel et al. 2008). Importantly, telomerase expression also reverts progerin-induced changes in gene expression. In particular, many deregulated genes in progerin-expressing cells are linked to senescence, and telomerase can rescue the majority of these changes (Chojnowski, Ong et al. 2015). In addition, this study demonstrated that embryonic stem cells (ESCs), which express high levels of telomerase, are protected from progerin-induced phenotypes. These findings support the notion that progerin causes telomere dysfunction and that telomerase protects HGPS patient cells from the toxic effects of progerin.
Recent studies have also shown that alterations in telomere biology induce accumulation of progerin. For instance, induction of telomere dysfunction by expression of a dominant negative TRF2 protein (TRF2ΔBΔM) results in increased levels of progerin (Cao, Blair et al. 2011). In addition, replicative senescence of normal human fibroblasts is accompanied by an elevated production of progerin, while this effect is not observed during telomere-independent senescence (Cao, Blair et al. 2011). This upregulation of progerin seems to be owed to changes in alternative splicing of the LMNA gene. Interestingly, a whole variety of genes were alternatively spliced in response to telomere damage. Importantly, expression of telomerase in normal fibroblasts reduces the usage of the cryptic splice site that results in progerin production. Although the mechanism whereby telomere dysfunction activates progerin production is not known, it is tempting to speculate that reduced binding of telomeric proteins of the shelterin complex, or activation of the DNA damage response could impact pre-mRNA splicing in general, and alternative splicing of the LMNA gene in particular.
Overall, these studies reveal a reciprocal toxic relationship between telomeres and progerin, with telomere dysfunction inducing progerin production and progerin expression causing telomere dysfunction. This disastrous relationship could contribute to cellular decline during physiological aging. In fact, both telomere shortening and expression of progerin are observed in cells from old individuals. Thus, maintaining telomere function protects from cellular aging not only by preventing chromosomal instability, but also by ensuing proper control of alternative splicing, which in the case of the LMNA gene can have highly detrimental effects.
4. Pathophysiology of HGPS
HGPS patients classically exhibit alopecia (hair loss), bone and joint abnormalities, subcutaneous fat loss, and severe atherosclerosis (Ullrich and Gordon 2015). Patients live for an average of just 14.6 years, with most dying in their early teenage years from myocardial infarction or stroke as a result of rapidly progressive atherosclerosis (Gordon, Massaro et al. 2014) (Figure 4). Fortunately, this disease is extremely rare, with an estimated 350-400 children worldwide. However, despite its paucity, this disease elicits much research attention. Not only is a treatment desperately needed, but better understanding HGPS might elucidate mysteries in the normal aging process. This is because progerin, which is produced at high levels in HGPS, also is produced during aging in the normal aging population (Scaffidi and Misteli 2006).
Figure 4. Pathophysiology of HGPS.

Scheme shows the prominent symptoms and signs of HGPS, as well as potential treatments to test in the future to ameliorate the pathophysiology of this devastating disease.
HGPS disease processes probably begin in utero. Despite this, HGPS patients are seemingly normal at birth and have normal birth weight. Yet, it is likely that some subtle features of disease, like circumoral pallor, are already inconspicuously present at birth. This mild initial phenotype might be surprising given the drastic phenotype that develops later in disease. However, the delay in disease manifestations might result from attenuated expression of lamin A/C and progerin in undifferentiated or embryonic cells, and it may take some time before progerin levels reach a threshold level for provoking disease (Constantinescu, Gray et al. 2006, Zhang, Lian et al. 2011). However, by 9-12 months of age, patients often present with failure to thrive, skin abnormalities, alopecia, circumoral cyanosis, prominent scalp veins, and decreased range of motion (Merideth, Gordon et al. 2008, Ullrich and Gordon 2015). With time, these early problems amplify in magnitude and usher in the characteristic disease phenotype.
HGPS patients develop a unique progeroid appearance. They are distinctively small, most never reaching four feet or 30 kg. A decreased and linear rate of weight gain prevents growth comparable to age matched peers (Gordon, McCarten et al. 2007, Kieran, Gordon et al. 2007). Patients begin to lose cranial hair around 10 months of age. However, with time, patients lose body hair and eyebrows and progress to almost complete alopecia (Rork, Huang et al. 2014). HGPS patients also have distinctive craniofacial characteristics, developing micrognathia, prominent eyes, and a beaked nose (Kieran, Gordon et al. 2007, Domingo, Trujillo et al. 2009). Prominent forehead scalp veins and perioral cyanosis become evident, both likely the result of decreased subcutaneous fat (Rork, Huang et al. 2014). Patients also have multiple dental abnormalities, including both lack of teeth as well as dental crowding, which can manifest as double rows of teeth (Gordon, McCarten et al. 2007, Domingo, Trujillo et al. 2009). Middle ear abnormalities and aberrations in the ear canal also lead to low-frequency hearing loss in many patients (Guardiani, Zalewski et al. 2011).
HGPS is a “segmental aging disease,” since some features of normal aging are present, whereas other features are notably absent. The liver, kidneys, lungs, and gastrointestinal tract are normally proficient in these patients (Kieran, Gordon et al. 2007, Ullrich and Gordon 2015). However, others cell and tissue types, such as those of mesenchymal origin, are particularly susceptible to progerin-induced cellular defects, causing HGPS patients to harness notable fat and bone abnormalities (McClintock, Ratner et al. 2007, Merideth, Gordon et al. 2008, Zhang, Lian et al. 2011). A profound loss of subcutaneous fat is readily apparent in examining these patients. However, loss of subcutaneous fat causes other challenges beyond those readily apparent. Loss of fat in some body areas, such as the feet, can lead to discomfort and often requires supportive therapies (Gordon, Massaro et al. 2014).
Bone and joint abnormalities are a hallmark of HGPS that progress to a skeletal dysplasia (Gordon, Gordon et al. 2011). Bone problems include small clavicles, thin ribs, and acroosteolysis. Patients exhibit reduced bone mineral density with accentuated demineralization at the end of long bones. Avascular necrosis is also present, including at the femoral head, likely resulting from vascular compromise (Cleveland, Gordon et al. 2012). Interestingly, fracture incidence among HGPS patients is not increased compared to the general population, though HGPS patients are more susceptible to skull fractures. This is likely the result of disrupted bone formation in the skull. Patent anterior and posterior fontanels can persist in patients as old as nine years of age, and these patients often also have widened calvarial sutures and a thin calvarium (Ullrich and Gordon 2015).
Skin alterations are often among the first manifestations of HGPS. Though manifestations can present with differing degrees of severity, typical alterations include areas of discoloration, stippled pigmentation, and tightened areas that restrict movement. Sclerodermoid changes, which give the skin a dimpled appearance with varying pigmentation, frequently appear over the abdomen and lower extremities (Rork, Huang et al. 2014).
The most significant problems in HGPS, and which ultimately underlie patient death, are the cardiovascular complications. Patients develop severe and progressive atherosclerosis, eventually leading to myocardial ischemia, infarction, and stroke (Stehbens, Wakefield et al. 1999). Cardiac manifestations include increased afterload and angina (Ullrich and Gordon 2015). Remarkably, it is estimated that 50% of children have radiographically detectable strokes by the age of eight, and infarcts were common on imaging studies of patients between five and 10 years of age (Silvera, Gordon et al. 2013). Most of these strokes are often clinically silent. This suggests that cardiovascular problems are present well before the end of life contributing to both morbidity and mortality.
The atherosclerosis that develops in HGPS has some important differences from the normal aging population, even though calcification, inflammation, and plaque rupture are present in both HGPS and normal aging. Interestingly, HGPS patients do not develop hypercholesterolemia or increased serum high-sensitivity C-reactive protein, two characteristics often seen with cardiovascular disease in the normal population (Stehbens, Wakefield et al. 1999, Olive, Harten et al. 2010). Additionally, vessels have a more complete fibrosis throughout the vessel wall, as arteries and veins show marked adventitial fibrosis with a dense rim of collagen. This complete stiffening of the wall leads to many measurable changes in the vasculature. Patients can become hypertensive, and some patients also have elongated QT intervals by EKG (Merideth, Gordon et al. 2008, Gerhard-Herman, Smoot et al. 2012). Carotid-femoral pulse wave velocity is dramatically elevated, indicating an increase in arterial stiffness. Patients also have abnormally echodense vascular walls by ultrasound, thought to correspond to a dramatically thickened fibrotic matrix. In these patients, as well as mouse models of disease, there is a striking depletion of vascular smooth muscle cells from the media, even in the outermost lamellar units adjacent to the adventitia, that is replaced by proteoglycans and collagen (Varga, Eriksson et al. 2006, Osorio, Navarro et al. 2011, Gerhard-Herman, Smoot et al. 2012, Villa-Bellosta, Rivera-Torres et al. 2013). This is likely due to the extreme sensitivity of vascular smooth muscle cells to progerin expression.
The vascular abnormalities present in HGPS cause neurological disease manifestations as well. Interestingly, HGPS patients have normal cognition and show no evidence of memory or cognitive challenges often associated with the normal aging process (Ullrich and Gordon 2015). This potentially surprising finding might be explained by the observation that lamin A expression is limited in the brain by miR-9, thus preventing significant expression of progerin in neuronal cells and tissues (Jung, Coffinier et al. 2012). Thus, while neuronal cells avoid direct demise from progerin expression, many HGPS patients experience neurological symptoms such as headaches, muscular weakness, or seizures as a result of impaired blood flow and diseased vasculature. Headaches can be single or recurrent and often take on a migraine-type quality. NSAIDs can be used to treat headaches if they occur frequently (Ullrich and Gordon 2015).
With such a multitude of symptoms and organ systems affected, treating disease is challenging. Upon diagnosis, physicians characterize the patient's specific manifestations of disease with a variety of tests (Ullrich and Gordon 2015). Some manifestations can be delayed with adequate therapies. However, the disease has no cure and has limited treatment options for systemically improving disease phenotype.
5. Therapeutic strategies for HGPS
In recent years, a number of therapies have shown promise in preclinical stages for treating disease (Figure 1C). Clinical trials are now being initiated with some compounds to determine their merit in human patients. The first of these compounds to achieve success in HGPS-patient derived cells in vitro and in progeria mouse models in vivo have been FTIs, which work by inhibiting the processing of prelamin A to mature lamin A, or in HGPS, to progerin (Fong, Frost et al. 2006, Yang, Meta et al. 2006, Varela, Pereira et al. 2008, Gordon, Kleinman et al. 2012). Administration of the FTI lonafarnib for two years in a clinical trial of HGPS patients improved secondary outcomes such as pulse-wave velocity, carotid artery wall echodensity, and incidence of stroke, headaches, and seizures (Gordon, Kleinman et al. 2012, Gordon, Massaro et al. 2014). However, the treatment only extended survival by an estimated 1.6 years. As a result, new clinical trials are now being initiated to determine if combination therapies might further improve disease phenotype. An ongoing trial includes two additional compounds to lonafarnib, the statin pravastatin and the bisphosphonate zoledronate, with the goal of inhibiting multiple steps in the farnesyl biosynthetic pathway. This trial is estimated to be completed by July 2017 (https://clinicaltrials.gov/ct2/show/NCT00916747?term=Hutchinson-Gilford+Disease&rank=2).
In addition to prenylation inhibitors, other therapeutic strategies have shown a benefit ameliorating the phenotype of HGPS cells and mouse models of progeria. These include inhibitors of the enzyme responsible for carboxymethylation of the farnesylcysteine of progerin (Ibrahim, Sayin et al. 2013); rapamycin and sulforaphane, compounds that increase progerin clearance by autophagy (Cao, Graziotto et al. 2011, Cenni, Capanni et al. 2011, Gabriel, Roedl et al. 2014); the ROS scavenger N-acetyl cysteine, which reduces the amount of unrepairable DNA damage caused by the increased generation of ROS (Pekovic, Gibbs-Seymour et al. 2011, Richards, Muter et al. 2011, Lattanzi, Marmiroli et al. 2012, Sieprath, Darwiche et al. 2012); methylene blue, a mitochondrial-targeting antioxidant (Xiong, Choi et al., 2015); or resveratrol, an enhancer of SIRT1 deacetylase activity that alleviates progeroid features (Liu, Ghosh et al. 2012). Recently, a phase 1 and 2 clinical trial was initiated to determine the effect of combined administration of everolimus, an inhibitor of the mTOR pathway similar to rapamycin, and lonafarnib (https://clinicaltrials.gov/ct2/show/NCT02579044?term=Hutchinson-Gilford+Disease&rank=1). It is hoped that everolimus might reduce progerin levels by activating its clearance and might synergize with lonafarnib (Cao, Graziotto et al. 2011). However, a comparative study of the effect of three treatments –a FTI, rapamycin, or a combination of zoledronate and pravastatin- on mesodermal stem cells derived from HGPS iPSCs gave variable results (Blondel, Jaskowiak et al. 2014). While all treatments improved nuclear morphology, differences among treatments were observed in the amelioration of other cellular phenotypes. In addition, some combinations had cytotoxic effects. Thus, caution needs to be exercised when designing clinical trials for HGPS patients, since this toxicity will undermine benefits. The best treatment for HGPS patients remains a matter of active discussion and controversy. Understanding the full spectrum of functional effects of the different drugs will allow us to find strategies in the years to come that improve the dramatic phenotypes characteristic of HGPS while lowering toxicity.
Recent studies have identified new compounds that exert a beneficial effect in progeria models. For instance, remodelin, an inhibitor of N-acetyltransferase-10 (NAT10) rescues nuclear morphological abnormalities and proliferation defects. In addition, remodelin increases chromatin compaction and ameliorates the accumulation of DNA damage characteristic of progerin-expressing cells (Larrieu, Britton et al. 2014). Ongoing studies are monitoring the effect of remodelin on gene expression and evaluating its potential as a therapeutic strategy by using mouse models of progeria.
Another set of promising compounds are the retinoids (Swift, Ivanovska et al. 2013). It was recently shown that the LMNA gene promoter contains retinoic acid responsive elements (L-RARE) and that treatment with all trans retinoic acid (ATRA) results in downregulation of LMNA gene expression. In HGPS patient-derived fibroblasts ATRA treatment reduces significantly progerin expression. Interestingly, ATRA synergizes with rapamycin in downregulating progerin levels, which in turn ameliorates a variety of progerin-induced phenotypes (Pellegrini, Columbaro et al. 2015). Retinoids were also identified in a high-throughput, high-content based screening of a library of FDA approved drugs as a class of compounds able to revert cellular HGPS phenotypes (Kubben, Brimacombe et al. 2015). These findings stress the importance of testing in vivo the efficacy of retinoids in ameliorating HGPS defects without inducing toxicity. Furthermore, our recent studies show that activation of vitamin D receptor signaling by ligand (1,25α-dihydroxy-vitamin D3) binding ameliorates a broad repertoire of phenotypes of HGPS patient-derived cells (Kreienkamp, Croke et al. 2016). It is likely that combination therapy is the best strategy to obtain synergy among these compounds, while reducing toxicity owed to lowering the doses of each single compound. Preclinical trials with these new compounds either as single agents or in combination are needed to ascertain their potential therapeutic applicability.
6. Concluding remarks
Despite all that is known about HGPS and the remarkable strides taken in understanding disease, much remains to be learned. Only a few of the paradoxes of this disease have been resolved. For instance, it was surprising that progeroid patients do not develop cancer, given the association between cancer and aging. While some originally hypothesized that the reduced HGPS lifespan masked a heightened propensity for cancer development in progerin-expressing cells, recent research indicates that progerin expression may actually inhibit cell transformation, explaining a normal cancer risk in these patients (Fernandez, Scaffidi et al. 2014). Many other paradoxes and questions remain to be appreciated: What contributes to the variation in the lifespan of these patients? Why do some patients live 24 months and others 20 years? What phenotypes induced by progerin expression are ultimately responsible for progerin-induced cellular compromise? Why are some tissues more susceptible to progerin expression than others? Why are HGPS patients so susceptible to atherosclerosis and cardiovascular disease? What key cellular factors might be targeted to reverse or alleviate progerin production?
Tremendous progress has been made over the last decade in understanding this disease. The next decade represents a kairos for building upon these findings and advancing novel therapeutic strategies for helping these patients. As research proceeds, HGPS continues to reveal unappreciated mysteries of aging. HGPS, with all of its intricacies, offers a unique model for elucidating new roles of lamin A/C and progerin in the cell.
Highlights.
HGPS is caused by LMNA gene mutations and expression of a mutant protein “progerin” that causes cellular and organismal decline.
HGPS cells show disruption of nuclear functions, including epigenetic alterations, misregulated gene expression, DNA repair defects and telomere dysfunction.
HGPS patients exhibit systemic accelerated aging, dying in their teens from myocardial infarction or stroke due to severe atherosclerosis.
Progerin expression increases with age in normal individuals, suggesting its implication in physiological aging.
Current therapies aim to reduce progerin toxicity by downregulating its levels or targeting directly deregulated cellular processes.
Acknowledgments
We wish to thank Victor Carranco for help with figures. We apologize to our colleagues whose work has not been discussed here due to space limitations. Work in the SG lab was supported by NIH RO1 GM094513-01. Work in the PA lab is funded by the Spanish Ministry of Economy and Competitiveness [BFU2013-42709P] and the European Regional Development Fund. RK is supported by a Predoctoral Fellowship from AHA (16PRE27510016).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All the authors declare that there are no conflicts of interest.
References
- Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C. Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review. Gerontology. 2014;60(3):197–203. doi: 10.1159/000357206. [DOI] [PubMed] [Google Scholar]
- Bertrand AT, Chikhaoui K, Ben Yaou R, Bonne G. [Laminopathies: one gene, several diseases]. Biol Aujourdhui. 2011;205(3):147–162. doi: 10.1051/jbio/2011017. [DOI] [PubMed] [Google Scholar]
- Blondel S, Jaskowiak AL, Egesipe AL, Le Corf A, Navarro C, Cordette V, Martinat C, Laabi Y, Djabali K, de Sandre-Giovannoli A, Levy N, Peschanski M, Nissan X. Induced pluripotent stem cells reveal functional differences between drugs currently investigated in patients with hutchinson-gilford progeria syndrome. Stem Cells Transl Med. 2014;3(4):510–519. doi: 10.5966/sctm.2013-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B, Stewart CL. Functional architecture of the cell's nucleus in development, aging, and disease. Curr Top Dev Biol. 2014;109:1–52. doi: 10.1016/B978-0-12-397920-9.00006-8. [DOI] [PubMed] [Google Scholar]
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011;121(7):2833–2844. doi: 10.1172/JCI43578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3(89):89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- Cenni V, Capanni C, Columbaro M, Ortolani M, D'Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, Squarzoni S, Prencipe S, Lattanzi G. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to laminlinked progeria. Eur J Histochem. 2011;55(4):e36. doi: 10.4081/ejh.2011.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarini E, Mozzetta C, Marullo F, Gregoretti F, Gargiulo A, Columbaro M, Cortesi A, Antonelli L, Di Pelino S, Squarzoni S, Palacios D, Zippo A, Bodega B, Oliva G, Lanzuolo C. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J Cell Biol. 2015;211(3):533–551. doi: 10.1083/jcb.201504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnowski A, Ong PF, Wong ES, Lim JS, Mutalif RA, Navasankari R, Dutta B, Yang H, Liow YY, Sze SK, Boudier T, Wright GD, Colman A, Burke B, Stewart CL, Dreesen O. Progerin reduces LAP2alpha-telomere association in Hutchinson-Gilford progeria. Elife. 2015;4 doi: 10.7554/eLife.07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland RH, Gordon LB, Kleinman ME, Miller DT, Gordon CM, Snyder BD, Nazarian A, Giobbie-Hurder A, Neuberg D, Kieran MW. A prospective study of radiographic manifestations in Hutchinson-Gilford progeria syndrome. Pediatr Radiol. 2012;42(9):1089–1098. doi: 10.1007/s00247-012-2423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu D, Csoka AB, Navara CS, Schatten GP. Defective DSB repair correlates with abnormal nuclear morphology and is improved with FTI treatment in Hutchinson-Gilford progeria syndrome fibroblasts. Exp Cell Res. 2010;316(17):2747–2759. doi: 10.1016/j.yexcr.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24(1):177–185. doi: 10.1634/stemcells.2004-0159. [DOI] [PubMed] [Google Scholar]
- Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3(4):235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103(27):10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer TA, Sahni N, Kubben N, Hill DE, Vidal M, Burgess RC, Roukos V, Misteli T. Systematic identification of pathological lamin A interactors. Mol Biol Cell. 2014;25(9):1493–1510. doi: 10.1091/mbc.E14-02-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo DL, Trujillo MI, Council SE, Merideth MA, Gordon LB, Wu T, Introne WJ, Gahl WA, Hart TC. Hutchinson-Gilford progeria syndrome: oral and craniofacial phenotypes. Oral Dis. 2009;15(3):187–195. doi: 10.1111/j.1601-0825.2009.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duband-Goulet I, Woerner S, Gasparini S, Attanda W, Konde E, Tellier-Lebegue C, Craescu CT, Gombault A, Roussel P, Vadrot N, Vicart P, Ostlund C, Worman HJ, Zinn-Justin S, Buendia B. Subcellular localization of SREBP1 depends on its interaction with the C-terminal region of wild-type and disease related A-type lamins. Exp Cell Res. 2011;317(20):2800–2813. doi: 10.1016/j.yexcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisch V, Lu X, Gabriel D, Djabali K. Progerin impairs chromosome maintenance by depleting CENP-F from metaphase kinetochores in Hutchinson-Gilford progeria fibroblasts. Oncotarget. 2016 doi: 10.18632/oncotarget.8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P, Scaffidi P, Markert E, Lee JH, Rane S, Misteli T. Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep. 2014;9(1):248–260. doi: 10.1016/j.celrep.2014.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311(5767):1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116(3):743–752. doi: 10.1172/JCI27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel D, Roedl D, Gordon LB, Djabali K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell. 2014 doi: 10.1111/acel.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, Schwartzman A, Giobbie-Hurder A, Neuberg D, Gordon LB. Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension. 2012;59(1):92–97. doi: 10.1161/HYPERTENSIONAHA.111.180919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesson K, Rescheneder P, Skoruppa MP, von Haeseler A, Dechat T, Foisner R. A-type lamins bind both hetero-and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016;26(4):462–473. doi: 10.1101/gr.196220.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z. Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep. 2015;13(7):1396–1406. doi: 10.1016/j.celrep.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Blasco MA. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle. 2005;4(6):752–755. doi: 10.4161/cc.4.6.1720. [DOI] [PubMed] [Google Scholar]
- Gonzalo S, Eissenberg JC. Tying up loose ends: telomeres, genomic instability and lamins. Curr Opin Genet Dev. 2016;37:109–118. doi: 10.1016/j.gde.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, Blasco MA. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol. 2005;7(4):420–428. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- Gonzalo S, Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr Opin Cell Biol. 2015;34:75–83. doi: 10.1016/j.ceb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW. Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26(7):1670–1679. doi: 10.1002/jbmr.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666–16671. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Massaro J, D'Agostino RB, Sr., Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130(1):27–34. doi: 10.1161/CIRCULATIONAHA.113.008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120(4):824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, Lopez-Otin C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156(3):400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–164. doi: 10.1146/annurev-biochem-060614-034115. [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y, Medalia O. Lamins: the structure and protein complexes. Curr Opin Cell Biol. 2015;32:7–12. doi: 10.1016/j.ceb.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Guardiani E, Zalewski C, Brewer C, Merideth M, Introne W, Smith AC, Gordon L, Gahl W, Kim HJ. Otologic and audiologic manifestations of Hutchinson-Gilford progeria syndrome. Laryngoscope. 2011;121(10):2250–2255. doi: 10.1002/lary.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19(3):413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics. 2013;8(1):28–33. doi: 10.4161/epi.23366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MX, Sayin VI, Akula MK, Liu M, Fong LG, Young SG, Bergo MO. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science. 2013;340(6138):1330–1333. doi: 10.1126/science.1238880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH, 2nd, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 2012;109(7):E423–431. doi: 10.1073/pnas.1111780109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang CS, Cubenas-Potts C, Matunis MJ, Paschal BM. The defective nuclear lamina in Hutchinson-gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol Cell Biol. 2011;31(16):3378–3395. doi: 10.1128/MCB.05087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran MW, Gordon L, Kleinman M. New approaches to progeria. Pediatrics. 2007;120(4):834–841. doi: 10.1542/peds.2007-1356. [DOI] [PubMed] [Google Scholar]
- Kreienkamp R, Croke M, Neumann MA, Bedia-Diaz G, Graziano S, Dusso A, Dorsett D, Carlberg C, Gonzalo S. Vitamin D receptor signaling improves Hutchinson-Gilford progeria syndrome cellular phenotypes. Oncotarget. 2016 doi: 10.18632/oncotarget.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2011;108(30):12325–12330. doi: 10.1073/pnas.1102789108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Brimacombe KR, Donegan M, Li Z, Misteli T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2015 doi: 10.1016/j.ymeth.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH, Misteli T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165(6):1361–1374. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell. 2008;19(12):5238–5248. doi: 10.1091/mbc.E08-05-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344(6183):527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzi G, Marmiroli S, Facchini A, Maraldi NM. Nuclear damages and oxidative stress: new perspectives for laminopathies. Eur J Histochem. 2012;56(4):e45. doi: 10.4081/ejh.2012.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Nobumori C, Tu Y, Choi C, Yang SH, Jung HJ, Vickers TA, Rigo F, Bennett CF, Young SG, Fong LG. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016 doi: 10.1172/JCI85908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K, Zhou Z. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16(6):738–750. doi: 10.1016/j.cmet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11(7):780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang Z, Ghosh S, Zhou Z. Defective ATM-Kap-1-mediated chromatin remodeling impairs DNA repair and accelerates senescence in progeria mouse model. Aging Cell. 2013;12(2):316–318. doi: 10.1111/acel.12035. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang Z, Zhang L, Ghosh S, Zheng H, Zhou Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun. 2013;4:1868. doi: 10.1038/ncomms2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J, 3rd, Izpisua Belmonte JC. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472(7342):221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. Faseb J. 2008;22(2):603–611. doi: 10.1096/fj.07-8598com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi M, Cenni V, Duchi S, Squarzoni S, Lopez-Otin C, Foisner R, Lattanzi G, Capanni C. Barrier-to-Autointegration Factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget. 2015 doi: 10.18632/oncotarget.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marji J, O'Donoghue SI, McClintock D, Satagopam VP, Schneider R, Ratner D, Worman HJ, Gordon LB, Djabali K. Defective lamin A-Rb signaling in Hutchinson-Gilford Progeria Syndrome and reversal by farnesyltransferase inhibition. PLoS One. 2010;5(6):e11132. doi: 10.1371/journal.pone.0011132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The Mutant Form of Lamin A that Causes Hutchinson-Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin. PLoS ONE. 2007;2(12):e1269. doi: 10.1371/journal.pone.0001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013;23(2):260–269. doi: 10.1101/gr.138032.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Lopes-Paciencia S, Huot G, Lessard F, Ferbeyre G. Permanent farnesylation of lamin A mutants linked to progeria impairs its phosphorylation at serine 22 during interphase. Aging (Albany NY) 2016;8(2):366–381. doi: 10.18632/aging.100903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulson CL, Fong LG, Gardner JM, Farber EA, Go G, Passariello A, Grange DK, Young SG, Miner JH. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum Mutat. 2007;28(9):882–889. doi: 10.1002/humu.20536. [DOI] [PubMed] [Google Scholar]
- Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30(11):2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, Depetris D, de Carlos F, Cobo J, Andres V, De Sandre-Giovannoli A, Freije JM, Levy N, Lopez-Otin C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3(106):106ra107. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- Osorio FG, Varela I, Lara E, Puente XS, Espada J, Santoro R, Freije JM, Fraga MF, Lopez-Otin C. Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell. 2010;9(6):947–957. doi: 10.1111/j.1474-9726.2010.00621.x. [DOI] [PubMed] [Google Scholar]
- Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T. Ageingrelated chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11(10):1261–1267. doi: 10.1038/ncb1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekovic V, Gibbs-Seymour I, Markiewicz E, Alzoghaibi F, Benham AM, Edwards R, Wenhert M, von Zglinicki T, Hutchison CJ. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell. 2011;10(6):1067–1079. doi: 10.1111/j.1474-9726.2011.00750.x. [DOI] [PubMed] [Google Scholar]
- Pellegrini C, Columbaro M, Capanni C, D'Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6(30):29914–29928. doi: 10.18632/oncotarget.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013 doi: 10.1111/acel.12070. [DOI] [PubMed] [Google Scholar]
- Reunert J, Wentzell R, Walter M, Jakubiczka S, Zenker M, Brune T, Rust S, Marquardt T. Neonatal progeria: increased ratio of progerin to lamin A leads to progeria of the newborn. Eur J Hum Genet. 2012;20(9):933–937. doi: 10.1038/ejhg.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet. 2011;20(20):3997–4004. doi: 10.1093/hmg/ddr327. [DOI] [PubMed] [Google Scholar]
- Rivera-Torres J, Acin-Perez R, Cabezas-Sanchez P, Osorio FG, Gonzalez-Gomez C, Megias D, Camara C, Lopez-Otin C, Enriquez JA, Luque-Garcia JL, Andres V. Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91:466–477. doi: 10.1016/j.jprot.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Rodriguez S, Eriksson M. Low and high expressing alleles of the LMNA gene: implications for laminopathy disease development. PLoS One. 2011;6(9):e25472. doi: 10.1371/journal.pone.0025472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rork JF, Huang JT, Gordon LB, Kleinman M, Kieran MW, Liang MG. Initial cutaneous manifestations of Hutchinson-Gilford progeria syndrome. Pediatr Dermatol. 2014;31(2):196–202. doi: 10.1111/pde.12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Eguino G, Infante A, Schlangen K, Aransay AM, Fullaondo A, Soriano M, Garcia-Verdugo JM, Martin AG, Rodriguez CI. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl Med. 2012;1(4):309–321. doi: 10.5966/sctm.2011-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312(5776):1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10(4):452–459. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprath T, Darwiche R, De Vos WH. Lamins as mediators of oxidative stress. Biochem Biophys Res Commun. 2012;421(4):635–639. doi: 10.1016/j.bbrc.2012.04.058. [DOI] [PubMed] [Google Scholar]
- Silvera VM, Gordon LB, Orbach DB, Campbell SE, Machan JT, Ullrich NJ. Imaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am J Neuroradiol. 2013;34(5):1091–1097. doi: 10.3174/ajnr.A3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ED, Kudlow BA, Frock RL, Kennedy BK. A-type nuclear lamins, progerias and other degenerative disorders. Mech Ageing Dev. 2005;126(4):447–460. doi: 10.1016/j.mad.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15(3):163–177. doi: 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999;8(1):29–39. doi: 10.1016/s1054-8807(98)00023-4. [DOI] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102(36):12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich NJ, Gordon LB. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol. 2015;132:249–264. doi: 10.1016/B978-0-444-62702-5.00018-4. [DOI] [PubMed] [Google Scholar]
- Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437(7058):564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14(7):767–772. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103(9):3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell. 2008;7(3):383–393. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145(4):401–417. doi: 10.1007/s00418-016-1411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidak S, Kubben N, Dechat T, Foisner R. Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2alpha (LAP2alpha) through expression of extracellular matrix proteins. Genes Dev. 2015;29(19):2022–2036. doi: 10.1101/gad.263939.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enriquez JA, Lopez-Otin C, Andres V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127(24):2442–2451. doi: 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119(7):1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102(29):10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116(8):2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8(1):31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Sarge KD. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J Cell Biol. 2008;182(1):35–39. doi: 10.1083/jcb.200712124. [DOI] [PMC free article] [PubMed] [Google Scholar]