

Abstract
Objective
Abnormal function of high density lipoprotein (HDL) has been implicated as a potential mechanism for the increased cardiovascular disease (CVD) in patients with rheumatoid arthritis. The current work evaluated changes in HDL function and HDL-associated proteins over two years of follow-up in early RA patients receiving either methotrexate (MTX) monotherapy or combination therapies in the Treatment of Early Rheumatoid Arthritis (TEAR) trial.
Methods
The anti-oxidant capacity of HDL, paraoxonase 1 (PON-1) activity, HDL-associated haptoglobin (HDL-Hp), HDL-associated apolipoprotein AI (HDL-apoA-I), and myeloperoxidase (MPO) levels were measured in 550 TEAR participants at 4 time points (0 [pre-treatment], 24, 48, and 102 weeks). Repeated measures analysis was performed using mixed effect linear models with autoregressive covariate structure to model the within-subject covariance over time.
Results
Mixed effect models controlling for traditional CV risk factors, treatment regimen, prednisone use, and statin use demonstrated significant associations of RA disease activity measured by the disease activity score with 28 joint count (DAS28), erythrocyte sedimentation rate (ESR), or C-reactive protein (CRP) with the HDL function profile over time. Specifically, decreases in RA disease activity over time were associated with increases in PON1 activity and HDL-apoA-I levels, and decreases in the HDL inflammatory index (HII), MPO, and HDL-Hp.
Conclusion
Decreases in disease activity in early RA patients treated with MTX, MTX + etanercept, or triple therapy in the TEAR trial were associated with improvements in the HDL function profile. Additional work is warranted to evaluate abnormal HDL function as a potential mechanism and therapeutic target for CV risk in patients with RA.
Keywords: rheumatoid arthritis, etanercept, methotrexate, triple therapy, high density lipoprotein
Patients with rheumatoid arthritis (RA) suffer significantly increased cardiovascular (CV) morbidity and mortality when compared to the general population with a 48% increased risk of incident CV disease (1–3). Levels of systemic inflammation from active RA have been strongly associated with cardiovascular (CV) death as well as subclinical atherosclerosis in RA patients (4;5). Epidemiologic work suggests potential beneficial effects of disease-modifying anti-rheumatic drug therapy (DMARD) on CV mortality in RA patients (6;7), however, mechanisms for CV protective effects are largely unclear. Better understanding of the interaction between systemic inflammation and vascular pathophysiology in RA patients is needed for adequate CV risk assessment and initiation of targeted prevention strategies.
High density lipoproteins (HDL) are complex particles composed of phospholipids, cholesterol, and multiple HDL-associated proteins which actively participate in the particles’ anti-oxidant, anti-atherogenic functions (8;9). Epidemiologic studies in the general population have consistently shown that higher HDL cholesterol (HDL-C) levels are associated with lower CV risk (10;11). However, significant research has suggested that the function of the HDL particle, including its ability to promote cholesterol efflux and inhibit low density lipoprotein (LDL) oxidation, may be more important to CV risk than its cholesterol content, as measured by the HDL-C level (12–14). Recent large clinical trials of cholesterol ester transfer protein (CETP) inhibitors, which increase HDL-associated cholesterol levels but have inconclusive effects on HDL function, did not show therapeutic benefit on CV outcomes (15;16), strengthening the hypothesis that the function rather than the cholesterol content is important to HDL’s anti-atherogenic role.
Our prior work has shown that high levels of inflammation in patients with active RA are associated with abnormal HDL function and alterations in multiple HDL-associated proteins (8;17;18) (Figure 1). In the current study, we evaluated whether treatment of RA can improve the anti-oxidant function of HDL and “reverse” these protein changes. We studied the effect of treatment with either methotrexate (MTX) monotherapy, methotrexate + etanercept (ETA) combination therapy, or triple therapy (TT) [MTX + sulfasalazine (SSZ) + hydroxychloroquine (HCQ)] on HDL function and several HDL-associated proteins in early RA patients participating in the Treatment of Early Rheumatoid Arthritis (TEAR) trial.
Figure 1.
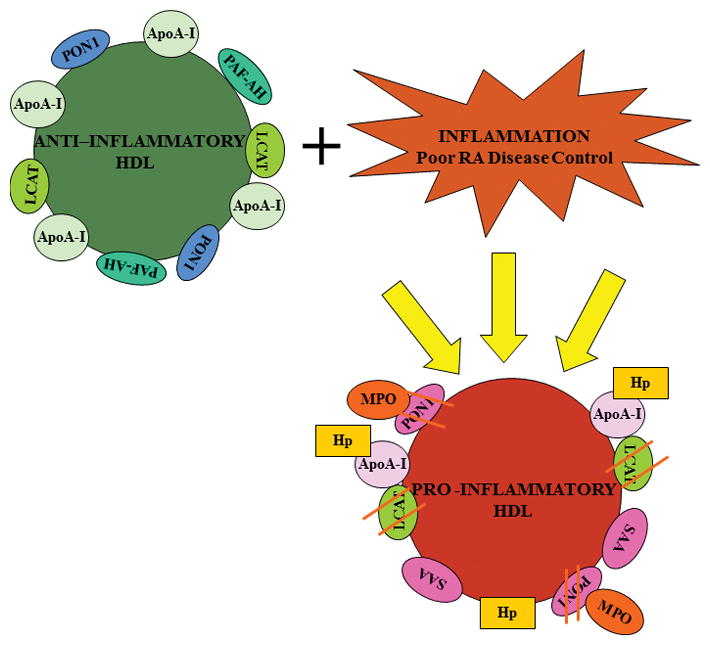
HDL is a particle composed of multiple associated proteins, which perform its anti-inflammatory and anti-atherogenic functions. A normal, anti-inflammatory HDL particle with several of its major associated proteins including paraoxonase 1(PON1), apolipoprotein A-I (apoA-I), lecithin cholesterol acyl transferase (LCAT), and platelet activating factor acetylhydrolase (PAF-AH) is shown. In the setting of active inflammation, HDL may become non-protective, and even pro-inflammatory, by alteration in the level and function of several proteins. Protein changes shown include 1) Displacement of ApoA-I by serum amyloid A (SAA), 2) Increased haptoglobin (Hp) in HDL which binds apoA-I, blocking LCAT activation, and 3) Decreased PON1 activity via oxidative modification of PON1 by the enzyme myeloperoxidase (MPO).
Patients and Methods
Study Design
The TEAR trial was a two-year randomized clinical trial of 755 early RA patients with minimal prior DMARD use who were initially randomized to MTX monotherapy (titrated to 20mg/week), MTX combination therapy with ETA, or TT(19). Patients were allowed prior use of leflunomide, hydroxychloroquine, and sulfasalazine for no more than 2 months and a total dose of ≤ 40mg of methotrexate (19). After 6 months, participants receiving MTX monotherapy who did not achieve low disease activity (disease activity score with 28 joint count (DAS28) [erythrocyte sedimentation rate](ESR)< 3.2) were “stepped up” to either MTX + ETA combination therapy or TT as determined by a baseline randomization algorithm. All treatment arms included matching placebos. No further protocol changes in treatment assignment occurred after 24 weeks of the study.
All patients met the 1987 American College of Rheumatology (ACR) criteria for RA and were serologically positive or had evidence of erosive disease on baseline radiographs of the hands or feet. Low dose prednisone (≤ 10mg/day) was allowed in the study but had to be stable for at least 2 weeks prior to screening and throughout the trial. Full results of the clinical trial have been previously published (19). Patients provided consent for participation in the TEAR trial and separately, for the biorepository substudy. All patients signing consent for the TEAR biorepository study with samples available were included in the current analysis.
Laboratory Testing
Non-fasting serum samples from a total of 550 patients participating in the TEAR biorepository study were available for analysis. Samples were collected over two year follow-up at 0, 24, 48, and 102 weeks and stored at −80°Celsius until analysis. Samples from all time points for individual patients were run together for each of the HDL function assays. Internal controls were used for PON1 and CFA assays and ELISAs were normalized between runs using 4 control samples included with each plate. The Clinical and Epidemiological Research Laboratory at Children’s Hospital in Boston measured C-reactive protein (CRP) levels in mg/L using a high-sensitivity immunoturbidimetric assay on a Hitachi 917 autoanalyzer (Roche Diagnostics, Indianapolis, IN), with the use of reagents and calibrators from Denka Seiken (Tokyo, Japan). ESR, 28 tender and swollen joint counts, and patient/physician global assessments were assessed locally at each site and the (DAS28[ESR]) calculated.
Determination of Paraoxonase 1 (PON1) Activity
PON1 activity was quantified as previously (20)using paraoxon as the substrate and measuring the increase in the absorbance at 405 nm due to the formation of 4-nitrophenol over a period of 12 minutes (at 20 second intervals). Paraoxon was purchased from Sigma (St. Louis, MO) and further purified using chloroform extraction. A unit of PON1 activity was defined as the formation of 1 nmol of 4-ntirophenol per minute per milliliter of sample used.
Evaluation of HDL’s Anti-Oxidant Function
The cell free assay (CFA) was a modification of a previously published method (21) using LDL as the fluorescence-inducing agent. Control LDL was prepared as described previously and HDL-containing supernatants were isolated using dextran bead precipitation (21). To determine the anti-inflammatory properties of HDL, the change in fluorescence intensity as a result of the oxidation of dihydrodichlorofluorescein (DCFH) in incubations with a standard LDL in the absence or presence of the test HDL was assessed and reported as the HDL inflammatory index (HII). In brief, 25μl of LDL-cholesterol (100 μg/ml) was mixed with 125 μl of test HDL (100μg HDL-cholesterol/ml) in black, flat bottom polystyrene microtiter plates and incubated at 37°C with rotation for 30 minutes. 25 μl of DCFH solution (0.2mg/ml) was added to each well, mixed, and incubated at 37°C for one hour with rotation. Fluorescence was determined with a plate reader (Spectra Max, Gemini XS; Molecular Devices) at an excitation wavelength of 485 nm, emission wavelength of 530 nm, and cutoff of 515 nm with photomultiplier sensitivity set at medium. Values for intra- and interassay variability were 0.5 ± 0.37% and 3.0 ± 1.7%, respectively (22).
HDL-Associated Haptoglobin ( Hp) and Apolipoprotein A-I (ApoA-I)
HDL-associated Hp (HDL-Hp) and apoA-I (HDL-ApoA-I) assays were modifications of previously published assays (17) (8) (23). In brief, 96-well microtiter plates were pre-coated with a human HDL antibody at 1:333 dilution overnight at 4°C. Plates were washed and blocked with 1% non-fat milk in 1XPBS for 60 minutes at room temperature. After washing, plates were coated with individual plasma samples diluted at 1:4000 (HDL-Hp)/1:80000 (HDL-ApoA-I) and incubated for 60 minutes at room temperature. Plates were next washed and incubated at room temperature for 60 minutes with a hp horseradish peroxidase (HRP) conjugated antibody at 1:8000 dilution or apoA-I HRP conjugated antibody at 1:4000 dilution. Following incubation with tetramethylbenzidine (TMB) solution for 20 minutes, HRP activity was measured at OD 450 nm. A protein standard for each assay was run by coating a set of standard wells with Hp or ApoA-1 antibody at 1:200 and 1:500 dilution, blocking with 50mM Tris, 0.14 M NaCl, 1% bovine serum albumin (BSA) for 60 minutes at room temperature, and after washing, coating wells with the Protein Calibrator using serial dilutions. The remainder of the assays were performed as described above for the patient samples. All antibodies were purchased from Genway Biotech.
Myeloperoxidase(MPO)
MPO ELISA was done using a kit from Aviscera Bioscience. In brief, 96-well microtiter plates were pre-coated with an MPO capturing antibody at 1:100 dilution overnight at 4°C. Plates were washed and blocked with 50mM Tris, 0.14 M NaCl, 1% BSA overnight at 4°C. After washing, plates were coated with individual plasma samples diluted at 1:10 and incubated for 2 hours at room temperature. Samples were aspirated from plates which were then incubated for 2 hours at room temperature with 1:100 dilution of detection antibody followed by incubation with 1:200 dilution of streptavidin HRP for 60 minutes at room temperature. HRP activity was measured at OD 450 nm after 20 minutes of incubation of plates with TMB solution.
Statistical analysis
Clinical characteristics and biomarkers were compared between treatment groups using one-way ANOVA for continuous variables and Chi-Square test for categorical variables. To determine the relative contribution of RA treatments, RA disease activity/systemic inflammation, and other patient characteristics to changes in HDL function and HDL-associated proteins over time, repeated measures analysis with linear mixed effect models (24) was used to model the within-subject covariance over time. Measures of disease activity/inflammation and HDL function/HDL-associated proteins at four time points were included in the models as fixed effects. Separate models were constructed for each HDL outcome (HII, PON-1 activity, HDL-apoA-I, HDL-Hp, and MPO), and each measure of disease activity/inflammation (ESR, CRP, or DAS28). Other fixed-effect patient covariates included treatment assignment at each time point, age, sex, race, RA disease duration, baseline body mass index (BMI), current smoking status (as measured by baseline cotinine), statin use, prednisone use, diabetes, and presence of cardiovascular disease. Log transformation was performed on all outcome measurements. All statistical testing was two-sided with 0.05 alpha level threshold for declaring significance. Statistical analyses were carried out using SAS version 9.3 (SAS Institute Inc. 2012).
RESULTS
Demographic, Laboratory, and Clinical Characteristics
The baseline clinical characteristics of the TEAR patients participating in the bio-repository study with samples available for analyses are shown in Table 1. The population studied was very similar in demographics to the main TEAR trial population as well as the substudy populations analyzed in previous work (25–27). No significant differences between treatment arms were observed in important demographic and clinical variables including age, sex, race, body mass index (BMI), and smoking status. Over 84% of patients in each group were rheumatoid factor positive with the mean disease duration less than 4.3 months in all groups. Patients had very active arthritis at baseline with mean DAS28 scores in all treatment groups at 5.5 or higher and elevated inflammatory markers (CRP and ESR). The baseline presence of co-morbidities including diabetes and known cardiovascular disease was similar across groups as was the use of prednisone and statins. No differences in baseline HDL function markers or traditional cholesterol levels were observed.
Table 1.
Baseline Clinical Characteristics of TEAR Patients Participating in the Bio-repository Study Analysis by Treatment Group.
| All Subjects N=550 |
MTX Monotherapy N=67 |
MTX + ETA N=198 |
Triple Therapy N=103 |
“Step-up” MTX + ETA N=124 |
“Step up” Triple Therapy N=58 |
p-value | |
|---|---|---|---|---|---|---|---|
| Age (years) | 49.8 ± 12.6 | 47.3 ± 12.0 | 50.8±13.4 | 49.3 ± 12.5 | 49.9± 12.5 | 50.0 ± 11.3 | 0.40 |
| BMI | 30.0± 7.4 | 29.3± 5.6 | 29.6±7.2 | 30.3 ± 8.5 | 30.7± 7.0 | 30.0 ± 8.4 | 0.68 |
| Female | 402(72.8%) | 45(67.2%) | 146(73.7%) | 78(75.7%) | 91(73.4%) | 41(70.7%) | 0.78 |
| RF positive | 490(88.8%) | 60(89.6%) | 172(86.9%) | 95(92.2%) | 112(90.3%) | 49(84.5%) | 0.50 |
| Smoking (baseline) | 177(32.1%) | 23(34.3%) | 68(34.3%) | 29(28.2%) | 38(30.7%) | 19(32.8%) | 0.83 |
| Disease Duration (mos) | 3.6± 6.6 | 3.7 ± 6.9 | 3.5±6.5 | 4.3 ± 7.6 | 3.1± 5.3 | 4.3 ± 7.2 | 0.64 |
| DAS28 (baseline) | 5.8 ± 1.1 | 5.5 ± 1.0 | 5.8±1.0 | 5.7 ± 1.0 | 5.9± 1.1 | 5.8 ± 1.1 | 0.10 |
| CRP (baseline) (mg/l) | 13.9 ± 22.2 | 9.2 ± 11.8 | 16.2±25.6 | 13.2 ± 22.1 | 13.4± 21.3 | 14.4 ± 21.3 | 0.28 |
| ESR (baseline) (mm/h) | 31.9±24.1 | 26.5±18.4 | 32.8 ±25.3 | 31.2±23.7 | 33.8±25.4 | 31.2±22.8 | 0.33 |
| TC (baseline)(mg/dL) | 190.7 ± 47.7 | 188.7 ± 43.8 | 192.1 ± 48.4 | 187.9 ± 46.3 | 189.1 ± 49.1 | 197.9 ± 49.4 | 0.72 |
| HDL-C (baseline) (mg/dL) | 55.6 ± 15.8 | 54.8 ± 13.4 | 56.1 ± 15.7 | 57.8 ± 18.1 | 54.3 ± 14.5 | 56.7 ± 16.9 | 0.52 |
| LDL-C (baseline) (mg/dL) | 105.6 ± 36.5 | 104.3 ± 35.0 | 107.9 ± 36.3 | 98.8 ± 33.7 | 106.0 ± 38.8 | 111.6 ± 38.2 | 0.20 |
| TC/HDL (baseline) (mg/dL) | 3.5 ± 0.9 | 3.6 ± 0.8 | 3.4 ± 0.8 | 3.6 ± 0.9 | 3.6± 0.9 | 3.6 ± 0.9 | 0.26 |
| Race/Ethnicity | 0.25 | ||||||
| African American | 52(9.4%) | 5(7.5%) | 21(10.6%) | 7(6.8%) | 15(12.1%) | 4(6.9%) | |
| Caucasian | 446(80.8%) | 58(86.5%) | 154(77.8%) | 84(81.6%) | 96(77.4%) | 53(91.4%) | |
| Other | 54(9.8%) | 4(6.0%) | 23(11.6%) | 12(11.6%) | 13(10.5%) | 1(1.7%) | |
| Medications | |||||||
| Prednisone Use | 230(41.7%) | 32(47.8%) | 82(41.4%) | 47(45.6%) | 49(39.5%) | 19(32.8%) | 0.43 |
| Statin Use | 78(14.1%) | 8(11.9%) | 32(16.2%) | 14(13.6%) | 15(12.1%) | 7(12.1%) | 0.81 |
| Comorbidities | |||||||
| Diabetes | 53(9.6%) | 4(6.0%) | 19(9.6%) | 10(9.7%) | 13(10.5%) | 6(10.3%) | 0.88 |
| CV Events | 26(4.7%) | 2(3.0%) | 14(7.1%) | 4(3.9%) | 4(3.2%) | 2(3.5%) | 0.43 |
Data shown as mean ± SD, or N (%); p-values are obtained from one-way ANOVA (for continuous variables) or Chi-Square test (for categorical variables). BMI= body mass index, RF= rheumatoid factor, DAS28 = disease activity score using a 28 joint count, CRP = C reactive protein, TC= total cholesterol, HDL-C = high density lipoprotein cholesterol, LDL-C = low density lipoprotein cholesterol. Means and standard deviations for log-transformed variables are presented for the following HDL function variables: HII, PON1 activity, HDL-Hp, HDL-ApoAI and MPO.
HDL function and associated proteins over two-year follow-up
HDL’s anti-oxidant capacity (measured by the HII), HDL-Hp, HDL-ApoA-I, PON-1 activity, and MPO levels are shown at four time points over two year follow-up in each of the five TEAR treatment groups in Table 2. Overall, small mean differences in the HDL function markers over time were noted as shown in the table. No significant differences over long term follow-up between the treatment groups occurred with the exception of HDL-Hp which was lowest in the TT and step-up TT groups at 102 weeks. MPO levels were significantly different between treatment groups at 24 weeks with lowest levels in the TT group, although did not remain significantly different after 102 weeks of treatment.
Table 2.
HDL Function Panel over Two Year Follow-up
| MTX Monotherapy N=67 |
MTX + ETA N=198 |
Triple Therapy N=103 |
“Step-up” MTX + ETA N=124 |
“Step up” Triple Therapy N=58 |
p-value | |
|---|---|---|---|---|---|---|
| Baseline | ||||||
| HII | 1.12±0.63 | 1.06±0.67 | 0.99±0.73 | 1.12±0.66 | 1.15±0.80 | 0.53 |
| PON1 activity | 3.97±0.97 | 3.94±0.97 | 4.05±0.86 | 4.07±0.77 | 3.94±0.74 | 0.70 |
| HDL-Hp | 12.45±0.35 | 12.43±0.41 | 12.44±0.42 | 12.42±0.54 | 12.45±0.34 | 0.99 |
| HDL-ApoAI | 13.63±1.74 | 14.21±2.29 | 14.29±2.48 | 13.64±2.56 | 14.29±1.83 | 0.16 |
| MPO | 6.10±0.60 | 6.12±0.66 | 6.23±0.83 | 6.24±0.66 | 6.09±0.73 | 0.40 |
| 24 weeks | ||||||
| HII | 1.14±0.67 | 1.10±0.67 | 0.92±0.71 | 1.09±0.75 | 1.12±0.76 | 0.33 |
| PON1 activity | 3.79±0.97 | 3.79±0.91 | 3.81±0.86 | 3.92±0.81 | 3.64±0.96 | 0.40 |
| HDL-Hp | 12.27±0.35 | 12.19±0.50 | 12.13±0.73 | 12.26±0.76 | 12.28±0.37 | 0.43 |
| HDL-ApoAI | 13.73±1.14 | 13.96±1.93 | 13.93±1.59 | 13.32±2.11 | 14.05±1.26 | 0.053 |
| MPO | 6.07±0.66 | 5.99±0.64 | 5.96±0.59 | 6.24±0.67 | 6.12±0.77 | 0.017 |
| 48 weeks | ||||||
| HII | 1.00±0.60 | 0.87±0.71 | 0.74±0.68 | 0.94±0.68 | 0.88±0.79 | 0.24 |
| PON1 activity | 3.68±0.91 | 3.81±1.05 | 3.79±0.88 | 3.82±0.99 | 3.56±1.06 | 0.53 |
| HDL-Hp | 12.19±0.57 | 12.18±0.45 | 12.00±1.09 | 12.18±0.50 | 12.08±0.58 | 0.32 |
| HDL-ApoAI | 13.79±1.62 | 13.89±1.95 | 14.18±2.21 | 13.59±2.10 | 14.04±2.22 | 0.51 |
| MPO | 5.80±0.75 | 5.90±0.88 | 5.90±0.79 | 5.90±0.68 | 6.01±0.92 | 0.78 |
| 102 weeks | ||||||
| HII | 0.96±0.59 | 0.91±0.63 | 0.84±0.75 | 0.93±0.75 | 0.71±0.66 | 0.37 |
| PON1 activity | 3.67±1.02 | 3.73±0.96 | 3.63±0.90 | 3.65±1.07 | 3.53±1.06 | 0.82 |
| HDL-Hp | 12.33±0.41 | 12.16±0.60 | 12.02±0.93 | 12.27±0.45 | 11.82±1.02 | 0.0008 |
| HDL-ApoAI | 14.09±1.67 | 14.39±1.69 | 14.24±2.17 | 13.75±1.93 | 13.67±1.81 | 0.12 |
| MPO | 5.80±0.72 | 5.85±0.76 | 5.83±0.79 | 5.81±0.82 | 5.71±0.72 | 0.88 |
Data shown are mean ± SD for log-transformed variable; p-values are obtained from one-way ANOVA. Units: PON1 activity: log(nmoles/min/ml), HDL-Hp: log(ng/ml), HDL-ApoAI: log(ng/ml), MPO: log(ng/ml).
Association of Changes in HDL function with Changes in RA Disease Activity and Systemic Inflammation
Several modest, but significant correlations were noted between changes in CRP and changes in HDL function and associated proteins over the trial follow-up period. Specifically, positive associations of changes in CRP were noted with changes in the HII and HDL-Hp in short and long term follow-up (r = 0.12–0.26, all p values <0.001; correlations of percent changes between baseline and 24/102 weeks). Decreases in CRP over time with treatment were associated with decreases in the HII and HDL-Hp. In contrast, consistently negative associations were noted between changes in CRP and changes in PON-1 activity and HDL-associated apoA-I, although these changes were of more modest magnitude in univariate analysis (maximum r = − 0.15, p = 0.0004; correlation of percent changes between CRP and HDL-apoA-I (24–102 weeks)). This data suggested a potential association between decreases in inflammation and increases in PON-1 activity and HDL-apoA-I.
Determinants of Long Term Changes in HDL Function and Associated Proteins in the TEAR Cohort
In order to determine the relative contribution of RA disease activity, RA treatments, and other patient characteristics to changes in HDL function and associated proteins in the TEAR cohort over time, repeated measures analyses with mixed effect linear models were performed. These models included patient characteristics as well as disease activity and treatment data from four time points over two years of follow-up. In all models tested (Tables 3–5), measures of RA disease activity and inflammation were consistently associated with improvements in the HDL function profile including 1) decreases in the HII, HDL-Hp, and MPO levels and 2) increases in PON1 activity and HDL-apoA-I levels. Specifically, in models controlling for treatment group, age, sex, race, disease duration, BMI, smoking status, statin use, prednisone use, diabetes, and presence of cardiovascular disease, decreases in DAS28, CRP, or ESR over time were associated with decreases in the HII, HDL-Hp, and MPO levels, and increases in PON1 activity and HDL-apoA-I levels. These relationships were all statistically significant in the models with few exceptions (MPO; CRP/ESR models (p=0.07 and 0.09 respectively) and PON-1 activity; DAS28/ESR models (p=0.10 and 0.13 respectively)) (Tables 3–5).
Table 3.
Percentage Change in HDL Function Marker by Covariate in Multivariate Repeated Measures Analysis-CRP models.
| HII | PON1 Activity | HDL-Hp | HDL-ApoAI | MPO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Effects | p | Effects | P | Effects | p | Effects | P | Effects | p | |
| CRP (per 10 unit decrease) | −2.34% | <0.01 | 3.42% | <0.01 | −3.53% | <0.01 | 8.74% | <0.01 | −1.91% | 0.07 |
| Treatment (overall) | 0.05 | 0.63 | 0.07 | 0.30 | 0.49 | |||||
| MTX +ETA vs TT | 9.69% | 0.07 | 2.97% | 0.68 | 10.63% | 0.02 | −18.88% | 0.21 | −3.93% | 0.46 |
| MTX +ETA vs MTX | −3.23% | 0.44 | −4.11% | 0.51 | 3.72% | 0.36 | 5.66% | 0.70 | 3.32% | 0.53 |
| TT vs MTX | −11.78% | 0.02 | −6.87% | 0.35 | −6.24% | 0.18 | 30.25% | 0.13 | 7.55% | 0.23 |
| Race (overall) | 0.05 | <0.01 | 0.03 | 0.21 | <0.01 | |||||
| AA vs Caucasian | −18.53% | 0.03 | 45.64% | <0.01 | −13.82% | 0.05 | 56.88% | 0.13 | 37.64% | <0.01 |
| AA vs Others | −25.27% | 0.02 | 14.90% | 0.39 | −22.74% | 0.01 | 98.60% | 0.09 | 12.84% | 0.30 |
| Others vs Caucasian | 9.03% | 0.36 | 26.76% | 0.05 | 11.55% | 0.14 | −21.01% | 0.43 | 21.98% | 0.02 |
| BMI (per unit increase) | 1.19% | <0.01 | −0.31% | 0.51 | 0.74% | 0.01 | −2.79% | 0.02 | −0.17% | 0.61 |
| Age (per year increase) | −0.03% | 0.89 | 0.24% | 0.41 | −0.08% | 0.64 | 0.85% | 0.25 | −0.22% | 0.31 |
| Female vs Male | −7.91% | 0.18 | 7.17% | 0.37 | −0.52% | 0.91 | 0.99% | 0.96 | −15.09% | <0.01 |
| Disease Duration (per year increase) | 0.59% | 0.15 | −0.03% | 0.95 | 0.06% | 0.86 | 0.23% | 0.86 | −0.94% | 0.01 |
| Baseline Smoking: Yes vs No | 10.34% | 0.09 | 0.43% | 0.95 | 0.28% | 0.95 | −30.94% | 0.05 | 5.41% | 0.32 |
| Baseline Prednisone Use: Yes vs No | 2.34% | 0.67 | 10.93% | 0.13 | 6.96% | 0.11 | 34.47% | 0.08 | 8.00% | 0.12 |
| Baseline Statin Use: Yes vs No | −11.63% | 0.15 | −5.29% | 0.62 | 4.77% | 0.49 | 34.54% | 0.28 | −6.05% | 0.43 |
| Baseline Cardiovascular Disease: Yes vs No | 10.31% | 0.46 | −20.08% | 0.19 | −13.64% | 0.16 | 10.80% | 0.81 | −8.10% | 0.49 |
| Baseline Diabetes Status: Yes vs No | −15.19% | 0.08 | −6.92% | 0.55 | −11.52% | 0.10 | −6.76% | 0.83 | −1.44% | 0.87 |
Percentage change in HDL function variables are shown per categorical variable or per unit change in continuous variables as specified. CRP= C-reactive protein. TT= triple therapy. MTX= methotrexate. ETA= etanercept. AA = African American. BMI= body mass index.
Table 5.
Percentage Change in HDL Function Marker by Covariate in Multivariate Repeated Measures Analysis-DAS28 Models.
| HII | PON1 Activity | HDL-Hp | HDL-ApoAI | MPO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Effects | p | Effects | P | Effects | P | Effects | p | Effects | p | |
| DAS28 (per unit decrease) | −2.98% | <0.01 | 2.60% | 0.10 | −5.36% | <.0001 | 9.20% | 0.01 | −3.72% | 0.01 |
| Treatment (overall) | 0.02 | 0.81 | 0.08 | 0.21 | 0.59 | |||||
| MTX + ETA vs TT | 10.29% | 0.05 | 3.43% | 0.63 | 9.32% | 0.04 | −19.15% | 0.21 | −4.46% | 0.40 |
| MTX + ETA vs MTX | −4.63% | 0.26 | −1.47% | 0.82 | −0.90% | 0.82 | 10.20% | 0.50 | 1.36% | 0.79 |
| TT vs MTX | −13.52% | 0.01 | −4.73% | 0.53 | −9.35% | 0.04 | 36.30% | 0.08 | 6.09% | 0.33 |
| Race (overall) | 0.06 | <0.01 | 0.11 | 0.15 | <0.001 | |||||
| AA vs Caucasian | −17.64% | 0.03 | 44.72% | <0.01 | −11.65% | 0.09 | 64.89% | 0.10 | 39.49% | <0.001 |
| AA vs Others | −24.60% | 0.02 | 17.34% | 0.32 | −18.25% | 0.04 | 116.30% | 0.06 | 18.61% | 0.14 |
| Others vs Caucasian | 9.23% | 0.36 | 23.34% | 0.08 | 8.08% | 0.29 | −23.77% | 0.36 | 17.60% | 0.06 |
| BMI (per unit increase) | 1.16% | 0.00 | −0.40% | 0.40 | 0.67% | 0.02 | −2.81% | 0.02 | −0.29% | 0.41 |
| Age (per year increase) | −0.02% | 0.93 | 0.25% | 0.40 | −0.12% | 0.51 | 1.05% | 0.16 | −0.26% | 0.23 |
| Female vs Male | −8.97% | 0.13 | 9.89% | 0.23 | −3.93% | 0.40 | −13.35% | 0.48 | −16.33% | <0.01 |
| Disease Duration (per year increase) | 0.50% | 0.22 | 0.06% | 0.91 | −0.13% | 0.68 | 0.13% | 0.93 | −1.07% | 0.01 |
| Baseline Smoking: Yes vs No | 11.34% | 0.06 | 1.17% | 0.87 | −0.75% | 0.87 | −31.72% | 0.04 | 4.82% | 0.38 |
| Baseline Prednisone Use: Yes vs No | −2.65% | 0.62 | 10.72% | 0.14 | 7.41% | 0.09 | 36.38% | 0.07 | 7.70% | 0.14 |
| Baseline Statin Use: Yes vs No | −11.63% | 0.15 | −4.60% | 0.67 | 6.02% | 0.39 | 38.18% | 0.25 | −7.46% | 0.33 |
| Baseline Cardiovascular Disease: Yes vs No | 11.05% | 0.43 | −20.13% | 0.18 | −14.89% | 0.12 | 8.79% | 0.85 | −7.63% | 0.52 |
| Baseline Diabetes Status: Yes vs No | −14.36% | 0.10 | −2.54% | 0.83 | −11.99% | 0.09 | −6.96% | 0.83 | 0.26% | 0.98 |
Percentage change in HDL function variables are shown per categorical variable or per unit change in continuous variables as specified. CRP= C-reactive protein. TT= triple therapy. MTX= methotrexate. ETA= etanercept. AA = African American. BMI= body mass index.
Treatment assignment was not consistently associated with changes in all of the HDL function profile markers. However, TT was significantly associated with a decrease in the HII, consistent with improvement in HDL’s overall anti-oxidant function, when compared to MTX monotherapy in all multivariate models using DAS28, ESR, or CRP as the measure of RA activity (p values =0.01–0.02). TT was also associated with a modest improvement in the HII compared to combination therapy with MTX and ETA(p values = 0.05–0.07). Finally, TT was associated with a significant decrease in HDL-associated Hp compared to combination therapy with MTX and ETA therapy (p values = 0.02–0.04 all models) as well as compared to MTX monotherapy in the DAS28 model (p=0.04) (Tables 3–5).
Table 3 shows results from the mixed effect linear models for the HII, PON1 activity, HDL-Hp, HDL-apoA-I and MPO levels using CRP as the disease activity covariate. Decreases in CRP over time were associated with significant decreases in HII and HDL-Hp, and significant increases in PON-1 activity and HDL-apoA-I. Specifically, a 10 unit decrease in CRP was associated with a 2.34% decrease in the HII, a 3.53% decrease in HDL-Hp, a 3.42% increase in PON-1 activity, and a 8.74% increase in HDL-associated apoA-I. A strong trend was also observed for a decrease in MPO levels by 1.91% per 10 unit decrease in CRP (p=0.07). Use of TT over time was associated with a lower HII by 11.78% compared to MTX monotherapy (p=0.02) and by 9.69% compared to combination therapy with MTX and ETA therapy (p= 0.07). TT was also associated with significantly lower HDL-Hp levels over time by 10.63% compared to MTX + ETA therapy (p = 0.02). These data suggest a potential beneficial effect of TT on the HDL particle. Similar effects of TT compared to MTX and MTX + ETA therapies were observed in the ESR and DAS28 models (Tables 4–5).
Table 4.
Percentage Change in HDL Function Marker by Covariate in Multivariate Repeated Measures Analysis-ESR Models.
| HII | PON1 Activity | HDL-Hp | HDL-ApoAI | MPO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Effects | p | Effects | P | Effects | p | Effects | p | Effects | P | |
| ESR (per 10 unit decrease) | −2.88% | <0.001 | 1.72% | 0.13 | −4.14% | <.0001 | 9.08% | <0.001 | −1.61% | 0.09 |
| Treatment (overall) | 0.02 | 0.74 | 0.11 | 0.24 | 0.45 | |||||
| MTX + ETA vs TT | 10.61% | 0.05 | 3.31% | 0.65 | 9.54% | 0.04 | −20.15% | 0.18 | −4.23% | 0.42 |
| MTX + ETA vs MTX | −3.97% | 0.33 | −2.57% | 0.68 | 1.31% | 0.74 | 6.54% | 0.65 | 3.36% | 0.52 |
| TT vs MTX | −13.18% | 0.01 | −5.69% | 0.44 | −7.51% | 0.10 | 33.42% | 0.10 | 7.93% | 0.21 |
| Race (overall) | 0.03 | <0.01 | 0.04 | 0.08 | <0.001 | |||||
| AA vs Caucasian | −20.26% | 0.02 | 47.05% | <0.01 | −14.96% | 0.03 | 85.02% | 0.04 | 38.35% | <0.001 |
| AA vs Others | −26.43% | 0.02 | 19.03% | 0.28 | −21.05% | 0.02 | 132.84% | 0.04 | 17.18% | 0.17 |
| Others vs Caucasian | 8.39% | 0.39 | 23.54% | 0.08 | 7.73% | 0.31 | −20.54% | 0.44 | 18.07% | 0.06 |
| BMI (per unit increase) | 1.16% | 0.00 | −0.42% | 0.37 | 0.70% | 0.02 | −2.78% | 0.02 | −0.24% | 0.50 |
| Age (per year increase) | −0.05% | 0.82 | 0.26% | 0.38 | −0.14% | 0.43 | 1.10% | 0.14 | −0.25% | 0.24 |
| Female vs Male | −9.00% | 0.12 | 9.72% | 0.24 | −4.13% | 0.38 | 15.18% | 0.48 | −16.01% | <0.01 |
| Disease Duration (per year increase) | 0.52% | 0.21 | 0.04% | 0.94 | 0.70% | 0.75 | 0.19% | 0.89 | −1.03% | <0.01 |
| Baseline Smoking: Yes vs No | 11.47% | 0.06 | 1.18% | 0.87 | 0.14% | 0.94 | −32.04% | 0.04 | 4.72% | 0.39 |
| Baseline Prednisone Use: Yes vs No | −2.68% | 0.61 | 10.88% | 0.13 | 6.85% | 0.11 | 37.53% | 0.07 | 7.32% | 0.16 |
| Baseline Statin Use: Yes vs No | −13.62% | 0.09 | −4.38% | 0.68 | 5.60% | 0.42 | 39.74% | 0.23 | −7.77% | 0.31 |
| Baseline Cardiovascular Disease: Yes vs No | 10.73% | 0.44 | −19.92% | 0.19 | −15.38% | 0.11 | 9.48% | 0.83 | −7.97% | 0.50 |
| Baseline Diabetes Status: Yes vs No | −14.29% | 0.10 | −2.68% | 0.82 | −11.90% | 0.09 | −6.62% | 0.83 | 0.52% | 0.95 |
Percentage change in HDL function variables are shown per categorical variable or per unit change in continuous variables as specified. CRP= C-reactive protein. TT= triple therapy. MTX= methotrexate. ETA= etanercept. AA = African American. BMI= body mass index.
Several patient characteristics were associated with changes in the HDL function profile over time in the mixed effect models (Table 3–5). Female sex was associated with lower MPO levels by as much as 16% compared to male sex (all p values <0.01). Higher BMI was associated with a worse HDL function profile including a higher HII (worse anti-oxidant capacity), higher HDL-Hp, and lower HDL-apoA-I levels (p ≤ 0.02 all models). Smoking was also associated with worse HDL function including a higher HII and lower HDL-apoA-I levels (p = 0.04–0.09). African American (AA) race was associated with higher PON1 activity (44.72–47.05%; p values <0.01) and higher MPO levels (>37%; p values <0.01) compared to Caucasians, and a lower HII (p values = 0.02–0.03) compared to both Caucasian and other races. Strong trends were also observed for lower HDL-Hp levels and higher HDL-apoA-I levels in AA compared to Caucasians and other races in all models (p values 0.02 – 0.13). Finally, baseline statin use was modestly associated with lower HIIs consistent with better HDL anti-oxidant function (p values = 0.09–0.15).
Discussion
Investigators have previously suggested that HDL may have evolved as part of the innate immune response which uses rapid induction of an oxidative state as a means of combating bacteria and viruses (28). In the absence of acute or chronic inflammation, HDL is an anti-inflammatory, protective particle in mice, rabbits, and humans. However, with onset of a systemic inflammatory state, HDL becomes pro-inflammatory and non-protective, in part due to alteration in the level and function of several of its associated proteins (9;28;29).
Under normal conditions, HDL inhibits oxidation of LDL to an inflammatory form which promotes the development of atherosclerosis, and HDL also promotes cholesterol efflux from peripheral tissues including the artery wall (12;30). During systemic inflammation, these functions of HDL are impaired by oxidative modification of the HDL particle including its associated protein, PON1(31). This process occurs in part via pro-oxidant enzymes such as MPO and promotes the development of a dysfunctional, even “pro-oxidant” particle (32)(Figure 1). Additional changes in HDL’s protein cargo which occur include increases in acute phase proteins such as serum amyloid A (SAA), fibrinogen, and Hp, as well as decreases in the major HDL-associated protein, ApoA-I (Figure 1)(8;29).
Active RA has been associated with abnormal HDL function and structure and a markedly increased CV risk (3;4;8;17). In particular, data suggests that both the cholesterol efflux function and the anti-oxidant function of HDL are impaired in patients with high RA disease activity and are correlated (17;18). Impaired cholesterol efflux by HDL and lower anti-oxidant capacity have also been associated with CV events and death in the general population (12–14). Therefore, impairment in HDL function has been proposed as a mechanism by which active RA increases CV risk. In the current study, we evaluated whether improvement in RA disease control in the TEAR trial could “reverse” the deleterious effects of inflammation on the HDL particle. We examined HDL’s overall anti-oxidant function as well as a panel of four HDL-associated proteins.
A major strength of this study was the robust repeated measures analyses which incorporated data from 4 different time points over 2 years of follow-up to evaluate potential associations between changes in RA disease activity/inflammation and changes in the HDL function profile, while controlling for RA treatments as well as other patient characteristics. Separate models were done for each measure of RA disease activity including DAS28, ESR, and CRP and showed very similar results. Improvement in RA disease activity was associated with improvement in the HDL function profile including: 1) decreases in the HII (improved anti-oxidant capacity), HDL-Hp, and MPO levels and 2) increases in PON1 activity and HDL-apoA-I levels.
Studies of other DMARDs in RA patients have also shown improvement in select markers of HDL function. RA patients with an inadequate response to MTX who were treated with tocilizumab (n= 69) had significant increases in PON1 activity and decreases in HDL-associated SAA compared to placebo (n= 63)(33). Tocilizumab also decreased levels of inflammation as measured by CRP; however, specific relationships between changes in RA disease activity and PON1 activity/HDL-SAA were not reported (33). Tofacitinib treatment of active RA patients similarly decreased HDL-SAA levels (p=0.06) in a small study (n=36)(34). Popa et al. reported that infliximab improved both PON1 activity (n= 45) and HDL’s anti-oxidative capacity (n=15), describing modest but significant correlations of PON1 activity with ESR or DAS28 at baseline and after 2 weeks of therapy(35). Finally, Raterman et al. studied HDL’s protein cargo using mass spectrometry in 6 RA patients with an excellent response to rituximab as compared to 6 non-responders (36). RA patients with excellent responses to rituximab had significant decreases in HDL-associated SAA which were not seen in the non-responders (36).
The current work is the first large, randomized controlled clinical trial including both biologic and non-biologic therapies with TT to examine changes in a panel of HDL function markers over long term, two year follow-up. Improvement in RA disease activity was consistently associated with improvement in the HDL function panel in all multivariate models. Associations were also noted between select markers of HDL function and several patient characteristics including treatment. Unexpectedly, TT was associated with improved anti-oxidant function of HDL over time as measured by a lower HII compared to both MTX monotherapy and compared to combination therapy with MTX + ETA therapy. TT was also associated with lower HDL-associated Hp over time compared MTX + ETA therapy in all models with a trend for lower HDL-Hp compared to MTX monotherapy. The clinical significance of lower HDL-Hp over time in RA patients warrants further study.
Previous observational work has suggested that hydroxychloroquine is associated with a favorable lipid profile in RA patients (37). Our recent study confirmed this work, reporting that TT was associated with lower LDL-C levels over long term follow-up in the TEAR trial (25). Interestingly, a small study by Breton et al. in non-RA patients (n= 50) reported that lower LDL-C levels were predictors of better anti-oxidant function of HDL measured by a similar assay as used in the current work (38). While these data suggest that TT, and perhaps hydroxychloroquine, may lead to better anti-oxidant function of HDL by lowering LDL-C levels, further mechanistic work to understand this relationship is needed. In addition, the association of TT with lower HDL-Hp warrants confirmation in additional studies.
Several other patient characteristics were significantly associated with HDL function over long term follow-up in the TEAR trial. Many of these patient characteristics such as smoking, race and BMI showed similar effects on HDL function in RA patients as previously reported in the general population. For example, smoking use was associated with lower HDL-associated apoA-I levels by approximately 30% as well as a higher HII in all models. These results are consistent with early work by Berg et al. which demonstrated significantly lower apoA-I levels in smokers compared to non-smokers in non-RA patients (39). Other work has shown that cigarette smoke oxidatively modifies the HDL particle and is associated with decreased HDL function (40).
AA race in the TEAR cohort was associated with a significantly higher PON1 activity, lower HII, and trends for higher HDL-apoA-I levels and lower HDL-Hp levels compared to Caucasian race in all models. A study of non-RA southern AAs previously reported that AAs have higher PON1 activity compared to Caucasians (41) and work by Enknmao et al. also reported significantly higher apoA-I levels in non-RA AAs compared to non-RA Caucasians (42). MPO levels were consistently higher in AAs compared to Caucasians in the TEAR trial, similar to results of the Dallas Heart Study in non-RA patients (43).
Obesity has previously been linked to impaired HDL function in non-RA patients (44). In the current work, a higher BMI was consistently associated with a worse HDL function profile including higher HII and HDL-Hp, and lower HDL-apoA-I. Previous work has also suggested beneficial effects of statins on HDL’s anti-oxidant function (13;22), and a strong trend was noted in the current work for statin users to have better HDL anti-oxidant function compared to non-statin users. In summary, the above data reinforces the importance of both RA specific and non-specific patient factors to HDL function over long term follow-up.
Few cardiovascular events occurred during the two year TEAR trial with 3 deaths due to cardiac disorders (general [unattended death], coronary heart failure, and ventricular septal defect) (19). A CV adjudication process was not used during the study. While associations between the HDL function markers and CV events could not be made for these reasons, data in the general population suggests strong links between HDL function and CV risk (12–14). In particular, impaired cholesterol efflux by HDL has been associated with CV events and Ansell et al. reported worse anti-oxidant function of HDL in patients with coronary heart disease compared to matched controls, even in patients with high HDL-C levels (13). Bhattacharya et al. reported significantly lower PON1 activity in over 1100 patients with CVD compared to non-CVD controls (14). Lower plasma PON1 activity has also been significantly associated with CV risk as assessed by carotid plaque in a 168 RA patient cohort study (20). Finally, multiple studies have shown strong associations of MPO levels with risk of CVD in non-RA patients (45) (46;47).
There are some limitations to the current work. The study describes a very early RA patient population with high disease activity at baseline, primarily seropositive, and naïve to prior DMARDs; these results may not be generalized to RA patients with more established disease or with lower levels of disease activity. Additional studies are warranted. In addition, data on prednisone and statin use were only available at the baseline visit for the TEAR patients. It is possible that changes in these medications over two-year follow-up could have affected the HDL function assays in the study. However, it is unlikely that these effects would be of significant magnitude to alter the strong and consistent relationships between RA disease activity and the multiple assays of HDL function described in the several models tested. In addition, the use of statins at baseline was associated with the expected trend for better HDL anti-oxidant function(13;22). Finally, additional medications including non-statin cholesterol-lowering medications and supplements, which could have affected HDL function were not available for the analyses.
In summary, the current work demonstrated that improvement in RA disease activity, whether measured by DAS28, ESR, or CRP, in early RA patients treated in the TEAR trial was associated with an improvement in the HDL function profile including: 1) decreases in the HII (improved anti-oxidant capacity), HDL-Hp, and MPO, and 2) increases in PON1 activity and HDL-apoA-I. Growing epidemiologic work suggests that aggressive treatment of active RA decreases CV morbidity and mortality despite variable increases in traditional cholesterol levels (7;48–50). The current data provide a potential mechanism for the beneficial effects of improved disease control on CV risk by improvement in HDL function. The identification of specific mechanisms linking RA disease activity to CV risk is particularly important to patients with active RA despite currently available treatments. Better understanding of these mechanisms may lead to development of alternative, targeted therapeutics for prevention of CVD in these high risk patients.
Acknowledgments
Funding: Amgen provided funding for the TEAR trial but was not responsible for data collection, analysis, or interpretation of results; UCLA, UAB, and co-authors, are solely responsible for all data collection, management, and statistical analysis. The TEAR bio-repository was funded by the NIH (R01 AR052658). Dr. Charles-Schoeman received support from the NHLBI (5K23HL094834, R01HL123064) and NIAMS (R21AR057913). Dr. Wang received support from NCRR (1UL1RR033176). Dr. Curtis received support from the NIH (AR053351) and the Agency for Healthcare Research and Quality (R01HS018517). This ancillary study was funded by NIAMS (R21 AR 057913-01A1) to CCS.
Reference List
- 1.Agca R, Heslinga SC, van Halm VP, Nurmohamed MT. Atherosclerotic cardiovascular disease in patients with chronic inflammatory joint disorders. Heart. 2016;102(10):790–5. doi: 10.1136/heartjnl-2015-307838. [DOI] [PubMed] [Google Scholar]
- 2.Roman MJ, Moeller E, Davis A, Paget SA, Crow MK, Lockshin MD, et al. Preclinical carotid atherosclerosis in patients with rheumatoid arthritis. Ann Intern Med. 2006;144(4):249–56. doi: 10.7326/0003-4819-144-4-200602210-00006. [DOI] [PubMed] [Google Scholar]
- 3.Nurmohamed MT, Heslinga M, Kitas GD. Cardiovascular comorbidity in rheumatic diseases. Nat Rev Rheumatol. 2015;11(12):693–704. doi: 10.1038/nrrheum.2015.112. [DOI] [PubMed] [Google Scholar]
- 4.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005;52(3):722–32. doi: 10.1002/art.20878. [DOI] [PubMed] [Google Scholar]
- 5.Del Rincon I, Williams K, Stern MP, Freeman GL, O’Leary DH, Escalante A. Association between carotid atherosclerosis and markers of inflammation in rheumatoid arthritis patients and healthy subjects. Arthritis Rheum. 2003;48(7):1833–40. doi: 10.1002/art.11078. [DOI] [PubMed] [Google Scholar]
- 6.Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(3):480–9. doi: 10.1136/annrheumdis-2014-206624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi HK, Hernan MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002;359(9313):1173–7. doi: 10.1016/S0140-6736(02)08213-2. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe J, Charles-Schoeman C, Miao Y, Elashoff D, Lee YY, Katselis G, et al. Proteomic profiling following immunoaffinity capture of HDL: Association of acute phase proteins and complement factors with pro-inflammatory HDL in rheumatoid arthritis. Arthritis Rheum. 2012 doi: 10.1002/art.34363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM. High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation. 2001;103(18):2283–8. doi: 10.1161/01.cir.103.18.2283. [DOI] [PubMed] [Google Scholar]
- 10.Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370(9602):1829–39. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 12.Khera AV, Cuchel M, de lL-M, Rodrigues A, Burke MF, Jafri K, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–35. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108(22):2751–6. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya T, Nicholls SJ, Topol EJ, Zhang R, Yang X, Schmitt D, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008;299(11):1265–76. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 17.Charles-Schoeman C, Watanabe J, Lee YY, Furst DE, Amjadi S, Elashoff D, et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009;60(10):2870–9. doi: 10.1002/art.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charles-Schoeman C, Lee YY, Grijalva V, Amjadi S, Fitzgerald J, Ranganath VK, et al. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis. 2012;71(7):1157–62. doi: 10.1136/annrheumdis-2011-200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreland LW, O’Dell JR, Paulus HE, Curtis JR, Bathon JM, St Clair EW, et al. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012;64(9):2824–35. doi: 10.1002/art.34498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charles-Schoeman C, Lee YY, Shahbazian A, Gorn AH, Fitzgerald J, Ranganath VK, et al. Association of paraoxonase 1 gene polymorphism and enzyme activity with carotid plaque in rheumatoid arthritis. Arthritis Rheum. 2013;65(11):2765–72. doi: 10.1002/art.38118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42(8):1308–17. [PubMed] [Google Scholar]
- 22.Charles-Schoeman C, Khanna D, Furst DE, McMahon M, Reddy ST, Fogelman AM, et al. Effects of High-dose Atorvastatin on Antiinflammatory Properties of High Density Lipoprotein in Patients with Rheumatoid Arthritis: A Pilot Study. J Rheumatol. 2007;34(7):1459–64. [PubMed] [Google Scholar]
- 23.Watanabe J, Grijalva V, Hama S, Barbour K, Berger FG, Navab M, et al. Hemoglobin and its scavenger protein haptoglobin associate with apoA-1-containing particles and influence the inflammatory properties and function of high density lipoprotein. J Biol Chem. 2009;284(27):18292–301. doi: 10.1074/jbc.M109.017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–74. [PubMed] [Google Scholar]
- 25.Charles-Schoeman C, Wang X, Lee YY, Shahbazian A, Navarro-Millan I, Yang S, et al. Association of Triple Therapy with Improvement in Cholesterol Profiles over Two Year Follow-up in the TEAR Trial. Arthritis Rheumatol. 2015 doi: 10.1002/art.39502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro-Millan I, Charles-Schoeman C, Yang S, Bathon JM, Bridges SL, Jr, Chen L, et al. Changes in lipoproteins associated with methotrexate or combination therapy in early rheumatoid arthritis: results from the treatment of early rheumatoid arthritis trial. Arthritis Rheum. 2013;65(6):1430–8. doi: 10.1002/art.37916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreland LW, O’Dell JR, Paulus HE, Curtis JR, Bathon JM, St Clair EW, et al. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012;64(9):2824–35. doi: 10.1002/art.34498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15(4):158–61. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96(6):2758–67. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fogelman AM. Mechanisms of disease: proatherogenic HDL--an evolving field. Nat Clin Pract Endocrinol Metab. 2006;2(9):504–11. doi: 10.1038/ncpendmet0245. [DOI] [PubMed] [Google Scholar]
- 31.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114(4):529–41. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest. 2013;123(9):3815–28. doi: 10.1172/JCI67478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis. 2015;74(4):694–702. doi: 10.1136/annrheumdis-2013-204345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charles-Schoeman C, Fleischmann R, Davignon J, Schwartz H, Turner SM, Beysen C, et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015;67(3):616–25. doi: 10.1002/art.38974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popa C, van Tits LJ, Barrera P, Lemmers HL, van den Hoogen FH, van Riel PL, et al. Anti-inflammatory therapy with TNF{alpha} inhibitors improves HDL-cholesterol anti-oxidative capacity in rheumatoid arthritis patients. Ann Rheum Dis. 2008 doi: 10.1136/ard.2008.092171. [DOI] [PubMed] [Google Scholar]
- 36.Raterman HG, Levels H, Voskuyl AE, Lems WF, Dijkmans BA, Nurmohamed MT. HDL protein composition alters from proatherogenic into less atherogenic and proinflammatory in rheumatoid arthritis patients responding to rituximab. Ann Rheum Dis. 2013;72(4):560–5. doi: 10.1136/annrheumdis-2011-201228. [DOI] [PubMed] [Google Scholar]
- 37.Morris SJ, Wasko MC, Antohe JL, Sartorius JA, Kirchner HL, Dancea S, et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res (Hoboken ) 2011;63(4):530–4. doi: 10.1002/acr.20393. [DOI] [PubMed] [Google Scholar]
- 38.Breton CV, Yin F, Wang X, Avol E, Gilliland FD, Araujo JA. HDL anti-oxidant function associates with LDL level in young adults. Atherosclerosis. 2014;232(1):165–70. doi: 10.1016/j.atherosclerosis.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berg K, Borresen AL, Dahlen G. Effect of smoking on serum levels of HDL apoproteins. Atherosclerosis. 1979;34(3):339–43. doi: 10.1016/s0021-9150(79)80011-8. [DOI] [PubMed] [Google Scholar]
- 40.Ueyama K, Yokode M, Arai H, Nagano Y, Li ZX, Cho M, et al. Cholesterol efflux effect of high density lipoprotein is impaired by whole cigarette smoke extracts through lipid peroxidation. Free Radic Biol Med. 1998;24(1):182–90. doi: 10.1016/s0891-5849(97)00214-1. [DOI] [PubMed] [Google Scholar]
- 41.Davis KA, Crow JA, Chambers HW, Meek EC, Chambers JE. Racial differences in paraoxonase-1 (PON1): a factor in the health of southerners? Environ Health Perspect. 2009;117(8):1226–31. doi: 10.1289/ehp.0900569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyroler HA, Heiss G, Schonfeld G, Cooper G, Heyden S, Hames CG. Apolipoprotein A-I, A-II and C-II in black and white residents of Evans County. Circulation. 1980;62(2):249–54. doi: 10.1161/01.cir.62.2.249. [DOI] [PubMed] [Google Scholar]
- 43.Chen LQ, Rohatgi A, Ayers CR, Das SR, Khera A, Berry JD, et al. Race-specific associations of myeloperoxidase with atherosclerosis in a population-based sample: the Dallas Heart Study. Atherosclerosis. 2011;219(2):833–8. doi: 10.1016/j.atherosclerosis.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuo Y, Oberbach A, Till H, Inge TH, Wabitsch M, Moss A, et al. Impaired HDL function in obese adolescents: impact of lifestyle intervention and bariatric surgery. Obesity (Silver Spring) 2013;21(12):E687–E695. doi: 10.1002/oby.20538. [DOI] [PubMed] [Google Scholar]
- 45.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286(17):2136–42. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 46.Kataoka Y, Shao M, Wolski K, Uno K, Puri R, Murat TE, et al. Myeloperoxidase levels predict accelerated progression of coronary atherosclerosis in diabetic patients: insights from intravascular ultrasound. Atherosclerosis. 2014;232(2):377–83. doi: 10.1016/j.atherosclerosis.2013.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem. 2011;57(1):33–9. doi: 10.1373/clinchem.2010.152827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solomon DH, Reed GW, Kremer JM, Curtis JR, Farkouh ME, Harrold LR, et al. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015;67(6):1449–55. doi: 10.1002/art.39098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao VU, Pavlov A, Klearman M, Musselman D, Giles JT, Bathon JM, et al. An evaluation of risk factors for major adverse cardiovascular events during tocilizumab therapy. Arthritis Rheumatol. 2015;67(2):372–80. doi: 10.1002/art.38920. [DOI] [PubMed] [Google Scholar]
- 50.Greenberg JD, Kremer JM, Curtis JR, Hochberg MC, Reed G, Tsao P, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(4):576–82. doi: 10.1136/ard.2010.129916. [DOI] [PubMed] [Google Scholar]