

Abstract
The first enantioselective total synthesis of (−)-cycloclavine has been accomplished in 8 steps and 7.1% overall yield. Key features include the first catalytic asymmetric cyclopropanation of allene, mediated by dirhodium catalyst Rh2(S-TBPTTL)4, and the enone 1,2-addition of a new TEMPO-carbamate methyl carbanion. An intramolecular strain-promoted Diels-Alder methylenecyclopropane (IMDAMC) reaction provided a pivotal tricyclic enone intermediate in >99% ee after crystallization. The synthesis of (−)-1 was completed by a late stage intramolecular Diels-Alder furan (IMDAF) cycloaddition to install the indole.
Keywords: Clavine alkaloids, allene, enantioselective, cyclopropanation, sequential cycloadditions
Graphical Abstract

Cycloclavine (1) was originally isolated from the seeds of the African morning glory shrub Ipomoea hildebrandtii and later from Aspergillus japonicus, a species of filamentous fungus.[1,2] In preliminary biological evaluations, it demonstrated promising insecticidal and anti-parasitic activity.[3] Cycloclavine belongs to the clavine subset of ergot alkaloids, a prominent group of natural products that share a fused indole core (Figure 1). Members of this family have long served as an inspiration for the design of therapeutic agents and agrochemicals due to their potent and diverse biological activities.[4] In recent years, major advances in elucidating the biogenetic origin of ergot alkaloids have been made.[4] A unique structural feature of cycloclavine is a pyrrolidine-fused cyclopropane in place of the piperidine that is typical for other clavine and lysergic acid alkaloids.
Figure 1.

Structures of natural ergot alkaloids of clavine and lysergic acid subclasses and cycloclavine in its unnatural configuration, (−)-1.
While its biological properties remain largely unexplored, cycloclavine’s unusual pentacyclic scaffold has attracted the interest of synthetic chemists. Particularly, the assembly of three consecutive stereogenic carbons at C(5), C(8) and C(10), two of them all-carbon quaternary stereocenters, requires robust synthetic methodologies and high stereochemical control.
To date, three total syntheses and three formal syntheses of racemic cycloclavine have been reported, including a prior total synthesis accomplished by our group.[5,6,7,8–9] However, no enantioselective total synthesis of cycloclavine has been accomplished. In order to meet this objective and establish a concise route to either enantiomer as well as analogs of the natural product, we fundamentally revised our previous 14-steps/1.2% yield approach to racemic product and developed a streamlined asymmetric total synthesis of the unnatural enantiomer (−)-cycloclavine, (−)-(1) (Figure 2). Our main goals were: (a) to introduce the enone moiety in 2 directly from the ketone product of the methylenecyclopropane (MCP)-Diels-Alder reaction; (b) to develop an asymmetric cyclopropanation of unsubstituted allene 5 to install the quaternary stereocenter that directs the formation of the two neighboring stereogenic carbons in (−)-1; and (c) to generate the amide linkage in 3 by direct aminolysis of a diazopropanoate 6.
Figure 2.

Retrosynthetic strategy to enantiomerically enriched cycloclavine.
Diazopropanoates are difficult substrates for transition metal mediated cyclopropanations due to their propensity to undergo β-hydride elimination.[10] However, we hoped that dirhodium complexes with hindered carboxylate ligands would be amenable to our strategy and also enable the unprecedented use of volatile allene in this reaction.[11, 12] The coupling of vinylogous amide 4 to the activated ester eliminates several linear steps. A conventional approach would have required ester conversion into a more reactive acid chloride or anhydride for N-acylation of the vinylogous amide. The successful implementation of this two-step approach to 3 would represent a major improvement compared to our previous[6] synthesis, which required seven steps to access an analogous intermediate.
We first explored an experimental validation of the key cyclopropanation. Allene (5) represents an ideal, atom-economical precursor to MCPs. However, little is known about the fundamental behavior of unsubstituted allene in transition metal catalyzed cyclopropanations.[13a,b] In addition, allene is well known to oligomerize in the presence of certain metals and therefore its successful use in an enantioselective metal carbenoid cyclopropanation remained speculative.[14, 15] We were thus pleased to see that the reaction proceeded smoothly in the presence of 1 mol% of dirhodium catalyst 8 (Table 1, Entry 1). After screening several chiral variants of 8, we identified complex 9[12] as the most promising catalyst for enantioselective conversions. The degree of enantioinduction depended on the nature of the ester, with the sterically hindered t-butyl diazopropanoate providing the best er at low temperatures (entries 2 and 3). However, MCPs bearing an activated ester facilitated direct aminolysis and were therefore strategically more appealing (entries 4–6). While the succinimide and pentachlorophenyl diazopropanoates provided only moderate enantioinduction (entries 4 and 5), the pentafluorophenyl diazopropanoate provided the desired cyclopropane in high yield with a good er of 87:13 that could be further upgraded to an excellent er by crystallization (vide infra).
Table 1.
Optimization of Catalytic Asymmetric Allene Cyclopropanation.
 | |||||
|---|---|---|---|---|---|
| Entry | R | Catalyst | T (°C) | Yield (%) |
er |
| 1 | Et | 8 | −78 | 88 | 50:50[a] |
| 2 | t-Bu | 9 | −78 | 51 | 91:9[a] |
| 3 | t-Bu | 9 | −98 to −80 | 47 | 93:7[a] |
| 4 | Su[b] | 9 | −78 to rt | 27 | 77:23[a] |
| 5 | Pcp[c] | 9 | −78 | 74[f](84)[g] | 77:23[a,e] |
| 6 | Pfp[d] | 9 | −78 | 86 | 87:13 |
Determined by 1H NMR analysis of diastereomeric Mosher ester derivatives (see SI);
succinimide;
pentachlorophenyl;
pentafluorophenyl;
crystallization provided enrichment of the mother liquor up to 90:10 er;
reaction on 0.2 mmol scale;
reaction on 9 mmol scale.
After confirming the feasibility of the first key reaction, our focus shifted to the elaboration of enone 2 (Scheme 1). The pivotal cyclopropanation with 10 proceeded well on gram-scale and afforded the enantiomerically enriched 11 in 86% yield. The vinylogous amide 4 was deprotonated with n-BuLi at −78 °C in THF, then acylated with activated ester 11 to afford amide 3. The E/Z ratio of 4 proved to be inconsequential as both starting isomers provided the E-alkene product 3, probably due to rapid isomerization of the enamine upon deprotonation and preferential trapping of the E-enamide. Chiral SFC analysis of amide 3 revealed an er of 87:13, reflecting the enantiomeric enrichment obtained in the prior cyclopropanation step. Treatment of amide 3 with NaHMDS triggered enolization to the corresponding enolate that was trapped in situ with TBSCl to deliver the highly reactive amino silyloxy diene 12. This diene underwent an anti-selective intramolecular [4+2] cycloaddition with the pendant methylenecyclopropane upon microwave irradiation at 95 °C. The crude enol ether products were directly subjected to TBAF cleavage to afford separable diastereomers of ketone 14 in 4.8:1 dr at C(5).
Scheme 1.

Eight-step total synthesis of (−)-cycloclavine
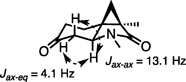
The relative configuration at the new ring-junction in the major diastereomer was assigned as trans through a combination of NMR and X-ray analyses.[16] An ab initio calculation at the MP6-31+G* level of the two transition states leading to the trans- and cis-ring fusion products in the IMDAMC[17] reaction revealed that the trans-stereochemistry in 13‡ is only slightly favored (<0.5 kcal/mol), rationalizing the relatively modest diastereoselectivity (4.8:1 dr) observed for this process.
Next, a Pd(DMSO)2(TFA)2 catalyzed direct dehydrogenation of ketone 14 was achieved using a modification of the protocol originally reported by Diao and Stahl for the aerobic dehydrogenation of ketones.[18a,b] Under optimized conditions, the reaction proceeded with excellent regioselectivity, forming the less-substituted enone 2 exclusively. This one-step protocol is particularly appealing because it uses molecular O2 and catalytic PdII. In prior work, this transformation required two steps and the use of stoichiometric transition metal.[6] Enone 2 was obtained as a crystalline solid and provided 2 in >99% ee after recrystallization.
The final challenge of the synthesis was the introduction of the indole core through an IMDAF reaction.[19, 22] A Boc-carbamate stabilized furylamine methyllithium suffered stereo- and chemoselectivity problems in the 1,2-addition on enone 2; i.e. lactam addition was competitive with ketone addition. Therefore, we developed a new TEMPO carbamate that led to a more thermodynamically stable and chemoselective furyl lithium species 15. This reagent underwent 1,2-addition to enone 2 at low temperatures in Et2O to afford the tertiary allylic alcohol 16 as a separable 1.5:1 mixture of diastereomers at C(3). The relative configuration of the major diastereomer was assigned based on a combination of 1-D NOE/ 2-D ROESY correlations and X-ray analysis. Alcohol 16 was subjected to the IMDAF reaction in toluene at 135 °C. This cyclization/ aromatization sequence was accompanied by the concurrent thermolysis[23a,b] of the TEMPO-carbamate protecting group to deliver the desired, protective group free indole. Reduction of the lactam with LiAlH4 yielded (−)-cycloclavine, (−)-(1), in 48% yield.
In conclusion, we report the first enantioselective synthesis of (−)-cycloclavine. In terms of efficiency, this 8-step/ 7.1% overall yield route from allene is a significant improvement on previous syntheses. Key features include the first use of unsubstituted allene in a transition metal mediated enantioselective cyclopropanation to deliver a methylenecyclopropane adorned with an active ester handle for immediate further derivatization. This transformation represents a direct and atom-economical method to access MCPs and could be further expanded for the synthesis of diverse ‘spring-loaded’ reagents in organic synthesis.[24] Additional noteworthy features include the application of a regioselective aerobic dehydrogenation to form bicyclic enone 2 and the development of a TEMPO-carbamate as a new carbanion-stabilizing nitrogen-protecting group that can be cleaved in situ by thermolysis.
Experimental Section
Supplementary Material
Acknowledgments
The authors thank Dr. Steven Geib (University of Pittsburgh) for X-ray structure analyses and gratefully acknowledge funding from the NIH (GM067082) and NSF (CHE-0910560). SRM also acknowledges support from the Mary E. Warga and the University of Pittsburgh School of Arts and Sciences Predoctoral Fellowships.
Footnotes
Supporting Information
References
- 1.Stauffacher D, Niklaus P, Tscherter H, Weber HP, Hofmann A. Tetrahedron. 1969;25:5879–5887. doi: 10.1016/s0040-4020(01)83095-7. [DOI] [PubMed] [Google Scholar]
- 2.Furuta T, Koike M, Abe M. Agric. Biol. Chem. 1982;46:1921–1922. [Google Scholar]
- 3.Körber K, Song D, Rheinheimer J, Kaiser F, Dickhaut J, Narine A, Culbertson DL, Thompson S, Rieder J. WO2014096238 A1. 2014
- 4.McCabe SR, Wipf P. Org. Biomol. Chem. 2016;14:5894–5913. doi: 10.1039/c6ob00878j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Incze M, Dörnyei G, Moldvai I, Temesvári-Major E, Egyed O, Szántay C. Tetrahedron. 2008;64:2924–2929. [Google Scholar]
- 6.Petronijevic FR, Wipf P. J. Am. Chem. Soc. 2011;133:7704–7707. doi: 10.1021/ja2026882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jabre ND, Watanabe T, Brewer M. Tetrahedron Lett. 2014;55:197–199. doi: 10.1016/j.tetlet.2013.10.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang W, Lu J-T, Zhang H-L, Shi Z-F, Wen J, Cao X-P. J. Org. Chem. 2014;79:122–127. doi: 10.1021/jo4023588. [DOI] [PubMed] [Google Scholar]
- 9.Netz N, Opatz T. J. Org. Chem. 2016;81:1723–1730. doi: 10.1021/acs.joc.5b02815. [DOI] [PubMed] [Google Scholar]
- 10.DeAngelis A, Dmitrenko O, Fox JM. J. Am. Chem. Soc. 2012;134:11035–11043. doi: 10.1021/ja3046712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeAngelis A, Dmitrenko O, Yap GPA, Fox JM. J. Am. Chem. Soc. 2009;131:7230–7231. doi: 10.1021/ja9026852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goto T, Takeda K, Anada M, Ando K, Hashimoto S. Tetrahedron Lett. 2011;52:4200–4203. [Google Scholar]
-
13.
Davies HML, Antoulinakis EG. Org. React. 2001;57:73.
Applequist DE, Johnston MR, Fisher F. J. Am. Chem. Soc. 1970;92:4614–4617.
There are only two prior literature reports of metal mediated cyclopropanations of allene. However, both required elevated temperatures and proceeded in low yield to give racemic product:
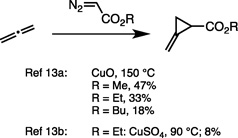
- 14.Griesbaum K. Angew. Chem. Int. Ed. 1966;5:933–946. [Google Scholar]; Angew. Chem. 1966;78:953–966. [Google Scholar]
- 15.Jones FN, Lindsey RV. J. Org. Chem. 1968;33:3838–3841. [Google Scholar]
-
16.The configuration of 14 was assigned by analysis of vicinal coupling constants. A single crystal X-ray structure of ±-(14) confirmed this assignment (see SI).
- 17.Heiner T, Kozhushkov SI, Noltemeyer M, Haumann T, Boese R, de Meijere A. Tetrahedron. 1996;52:12185–12196. [Google Scholar]
- 18.a) Diao T, Stahl SS. J. Am. Chem. Soc. 2011;133:14566–14569. doi: 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stahl SS, Diao T. Comp. Org. Synth. 2014;7:178–212. [Google Scholar]
- 19.Petronijevic F, Timmons C, Cuzzupe A, Wipf P. Chem. Commun. 2009:104–106. doi: 10.1039/b816989f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padwa A, Flick AC. Adv. Heterocycl. Chem. 2013;110:1–41. [Google Scholar]
- 21.Boonsompat J, Padwa A. J. Org. Chem. 2011;76:2753–2761. doi: 10.1021/jo200125c. [DOI] [PubMed] [Google Scholar]
- 22.LaPorte M, Hong KB, Xu J, Wipf P. J. Org. Chem. 2013;78:167–174. doi: 10.1021/jo3022605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Henry-Riyad H, Kobayashi S, Tidwell TT. ARKIVOC. 2005;vi:266–276. [Google Scholar]; b) Henry-Riyad H, Tidwell TT. ARKIVOC. 2008;x:113–126. [Google Scholar]
- 24.Milligan JA, Wipf P. Nat. Chem. 2016;8:296–297. doi: 10.1038/nchem.2485. [DOI] [PubMed] [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.