

Abstract
Macrocyclic lactones (ML) are important anthelmintics used in animals and humans against parasite nematodes, but their therapeutic success is compromised by the spread of ML resistance. Some ABC transporters, such as p-glycoproteins (Pgps), are selected and overexpressed in ML-resistant nematodes, supporting a role for some drug efflux proteins in ML resistance. However, the role of such proteins in ML transport remains to be clarified at the molecular level. Recently, Caenorhabditis elegans Pgp-1 (Cel-Pgp-1) has been crystallized, and its drug-modulated ATPase function characterized in vitro revealed Cel-Pgp-1 as a multidrug transporter. Using this crystal structure, we have developed an in silico drug docking model in order to study the binding of ML and other anthelmintic drugs to Cel-Pgp-1. All tested ML bound with high affinity in a unique site, within the inner chamber of the protein, supporting that ML may be transported by Cel-Pgp-1. Interestingly, interacting residues delineate a ML specific fingerprint involving H-bonds, including T1028. In particular, benzofurane and spiroketal moieties bound to specific sub-sites. When compared with the aglycone ML, such as moxidectin and ivermectin aglycone, avermectin anthelmintics have significant higher affinity for Cel-Pgp-1, likely due to the sugar substituent(s) that bind to a specific area involving H-bonds at Y771. Triclabendazole, closantel and emodepside bound with good affinities to different sub-sites in the inner chamber, partially overlapping with the ML binding site, suggesting that they could compete for Cel-Pgp-1-mediated ML transport. In conclusion, this work provides novel information on the role of nematode Pgps in transporting anthelmintics, and a valuable tool to predict drug-drug interactions and to rationally design new competitive inhibitors of clinically-relevant nematode Pgps, to improve anthelmintic therapeutics.
Keywords: Anthelmintics, Macrocyclic lactones, Nematodes, Caenorhabditis elegans, ABC transporters, Molecular docking
Abbreviations: ABC, ATP-binding cassette; ABA, abamectin; AH, anthelmintic; Cel, Caenorhabditis elegans; Ceg, Cylicocylus elongatus; Cgr, Cricetulus griseus; CID, Chemspider; CLO, closantel; DB, Drugbank; DOR, doramectin; EMD, emodepside; EPR, eprinomectin; Hco, Haemonchus contortus; Hsa, Homo sapiens; IVM, ivermectin; IVA, ivermectin-aglycone; LEV, levamisole; M, Merck Index; MDR, multidrug resistance; ML, macrocyclic lactone(s); Mmu, Mus musculus; MNP, monepantel; MOX, moxidectin; NBD, nucleotide binding domain; Oar, Ovis aries; PDB, Protein Data Bank; Pgp, p-glycoprotein; RMSD, root mean square deviation; SEL, selamectin; TBZ, thiabendazole; TCZ, triclabendazole; TMD, transmembrane domain
Graphical abstract
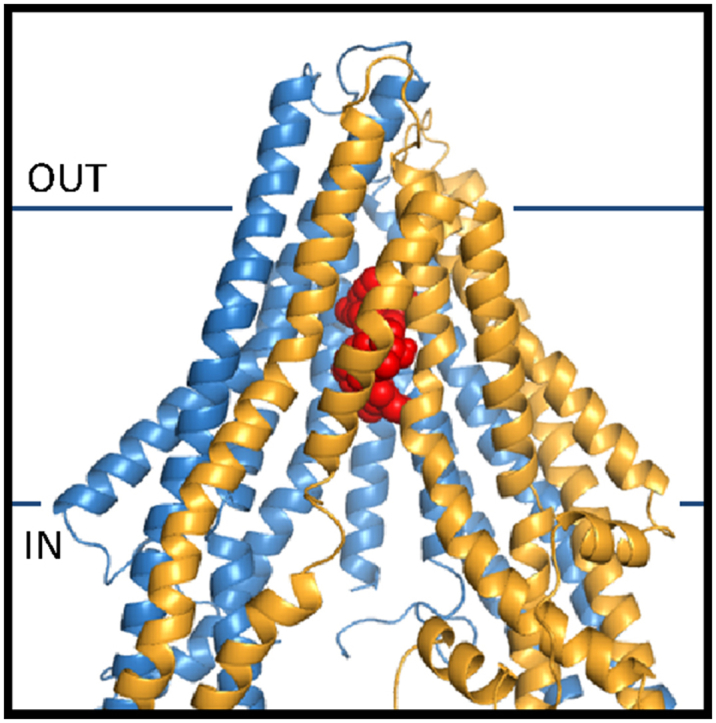
Ivermectin (red spheres) binding to Cel-Pgp-1 (blue ribbon: N-term, yellow ribbon: C-term).
Highlights
-
•
Cel-Pgp-1 cristal provides a valuable tool to study anthelmintic interactions.
-
•
Cel-Pgp-1 recognizes a broad panel of anthelmintics, including macrocyclic lactones.
-
•
Cel-Pgp-1 is predicted to transport MLs and contribute to anthelmintic resistance.
-
•
Sugars in avermectins increase the affinity to Cel-Pgp-1 compared with moxidectin.
-
•
These data provide a basis for modelling ABC homologs in parasitic nematodes.
1. Introduction
Macrocyclic lactones (ML) are the most important anthelmintic (AH) drugs used today for the control of parasitic nematodes and ectoparasites in animals and humans. Since the discovery of ivermectin (IVM) in 1980, several structurally-related ML drugs, belonging to the avermectin or milbemycin families, have been marketed. They all exert their anthelmintic effect by binding to glutamate-gated chloride channels expressed in the nervous system, causing paralysis of somatic, pharyngeal or excretory cell muscles that leads to paralysis, starvation, loss of tolerance to host immunity and death of the nematode (Cully et al., 1994, Forrester et al., 2003, Moreno et al., 2010). Overall, besides their exceptional safety margin and broad-spectrum activity, ML were previously effective against parasite strains resistant to older deworming medications. However, the long-term use of ML has led to the development of drug resistance in animal parasites (Jabbar et al., 2006, Kaplan, 2004), and this phenomenon has also emerged in human parasites (Osei-Atweneboana et al., 2007, Osei-Atweneboana et al., 2011). Because there are few therapeutic alternatives and new drug discovery is a very long and expensive process, a challenge for sustainable control of nematode parasites is to retain the efficiency of the existing AH drugs, including ML, by overcoming the process of AH resistance. This indicates the urgency to decipher the molecular mechanisms of anthelmintic resistance in nematodes.
Some active membrane transporters from the ATP-binding cassette (ABC) family are associated with ML resistance, and possibly resistance against other AH drugs, in parasite nematodes (Lespine et al., 2012). Many ABC transporters are present in various kingdoms, including different infectious agents (Koenderink et al., 2010, Lage, 2003), where they lead to various multidrug resistance phenotypes (Jones and George, 2005). Interestingly, all these transporters have two nucleotide binding domains (NBDs), which are highly conserved sequences giving the hallmark for belonging to the ABC family (Higgins, 1992). These NBDs bind ATP for hydrolysis, providing energy for substrate translocation during the transport cycle. In addition, ABC efflux transporters are all similarly organized in two transmembrane domains (TMDs) of 6 α-helices each. These TMDs delineate an “inner chamber” within the membrane where substrates can bind. Although TMDs harbor less conserved sequences when compared with NBDs, they generally present a fairly conserved conformational folding between sub-families and species. These typical structural characteristics support the notion that these proteins share similar transport mechanisms among species (Seeger and van Veen, 2009). In mammals, multidrug ABC transporters such as p-glycoprotein (MDR1/ABCB1/Pgp) can efflux various, structurally unrelated drugs, and Pgp is responsible for the multidrug resistance (MDR) in mammalian tumor cells (Eckford and Sharom, 2009, Leonard et al., 2003). Pgp is also physiologically expressed in the tissue barriers and is involved in intestinal absorption, cerebral distribution and biliary and urinary excretions. Thereby, it contributes in a significant manner to the pharmacokinetics of a broad range of molecules of pharmacological interest (Schinkel, 1997). Noticeably, amongst many drugs, IVM and other ML are transported by mammalian Pgp (Lespine et al., 2007, Roulet et al., 2003, Schinkel et al., 1994), which contributes to modulating their AH efficacy in the host.
The model free-living nematode, Caenorhabditis elegans (Cel) genome contains 14 homologs of ABCB/Pgps gene products and 10 are found in the parasitic nematode Haemonchus contortus (Hco) (Laing et al., 2011, Laing et al., 2013). There is little information on their functions, except that the loss of each of the 14 Pgps increases susceptibility of C. elegans to IVM to various degrees (Ardelli and Prichard, 2013, Janssen et al., 2013). Also, induction of expression of some Pgp genes after selection under IVM pressure is associated with IVM resistance in C. elegans and in several parasitic nematodes, which can be partly reversed by using mammalian Pgp inhibitors (James and Davey, 2009, Lespine et al., 2012, Menez et al., 2016). In addition, ML were shown to inhibit parasitic nematode Pgp-mediated drug transport in heterologous recombinant systems overexpressing Hco-Pgp-2, Hco-Pgp-9.1, Hco-Pgp-16, Cylicocylus elongatus (Ceg)-Pgp-9 or Dirofilaria immitis (Dim)-Pgp-11 (Godoy et al., 2015a, Godoy et al., 2015b, Godoy et al., 2016, Kaschny et al., 2015, Mani et al., 2016). Nevertheless, all these data give only indirect evidence that ML can be substrates of nematode Pgps. Considerable progress was made when Cel-Pgp-1 was purified, allowing functional characterization. Indeed, its stimulation of ATPase activity by mammalian Pgp-substrate drugs provides the most compelling evidence of a multidrug transport function for a nematode Pgp, but AH drugs have not been tested on this relevant experimental system. Therefore, direct information about the molecular relationships between AH drugs and ABC transporters in nematodes is still crucially lacking.
In this context, the determination of the crystal structure of Cel-Pgp-1 at a good atomic resolution (3.4 Å, Protein Data Bank PDB structure 4F4C) realizes a clear breakthrough as it is a unique and accurate molecular description of a nematode Pgp (Jin et al., 2012). Plus, it presents an open inward-facing conformation, defining a large cavity (the “inner chamber”), which is expected to be competent for substrate uptake as the first step of its transport enzymatic cycle. Thus, it provides a reliable structural basis to undertake in silico investigations of drug binding by molecular docking approaches. In this work, we studied the capacity of ML and other AHs of therapeutic interest to interact with Cel-Pgp-1 using in silico docking techniques. In order to validate and strengthen the computational modelling approach, we first performed docking calculations with the drugs identified as substrates of Cel-Pgp-1 on the basis of their ability to stimulate its ATPase activity, as previously demonstrated in vitro (Jin et al., 2012). Briefly, using AutoDock 4 scoring function, we found for all 6 ATPase activators, valinomycin, vinblastine, actinomycin D, dipyridamole, progesterone and paclitaxel, a good to very good binding energy (in the range from −7.0 to −17.0 kcal mol−1) for the best docking poses, which were all located within the inner chamber of the protein (David et al., work in process). Furthermore, docking calculations for positively charged rhodamine 123, taken as a negative control since it did not stimulate Cel-Pgp-1 ATPase activity, gave two poses that were either clearly outside the inner chamber or presented a very weak binding energy (−3.8 kcal mol−1). This good qualitative and quantitative agreement for the correlation between in vitro enzymological data and in silico calculations provided confidence for using our modelling strategy to investigate putative interactions with Cel-Pgp-1 of compounds belonging to several AH classes. For the first time, we determined the parameters of nematode Pgp-AH interaction, including binding energy and location of the binding sites in the protein, with a model nematode Pgp. In particular, we delineated their predictive binding sites by identifying amino acid residues that interact with different drug substituents of importance in the specific binding of each ML. We thus proposed a molecular view of the binding of several ML, which showed a unified handling by the transporter since they all share a common binding domain in the inner chamber. Using this model, we compared ML binding with the binding modes of other AH classes on Cel-Pgp-1, which all presented different binding sites within the inner chamber. Our findings thus provide a significant breakthrough in understanding how AHs bind to nematode Pgps, and provide strong evidence to indicate that ML can be transported by parasite nematode Pgp-1 homologs.
2. Computational methods
2.1. Structure of Cel-Pgp-1
The Cel-Pgp-1 X-ray structure, determined at a resolution of 3.4 Å (PDB code 4F4C) (Jin et al., 2012), was used in all docking calculations. The whole chain A was taken into account, with the exception of two detergent molecules (undecyl 4-O-alpha-D-glucopyranosyl-1-thio-beta-D-glucopyranoside, PDB entry name 0SA), bound in the inner chamber, and that have been removed for grid maps calculations. The 4F4C structure includes the full glycosylated Cel-Pgp-1 sequence (1321 amino acids), but N-terminal (M1-R3) and C-terminal (G1307-K1321) segments are missing in the structure, as well as a short segment (A52-E54) located in an extended loop of the first TMD domain, and a 49-residues segment (K666-E715) belonging to the linker region. Interestingly, an additional helix-turn-helix motif (Q9-V32) is present in the N-terminal domain, a structural feature that has not been observed in other Pgp structures released in the PDB.
Orientations of proteins in membranes database (http://opm.phar.umich.edu/) allowed the determination of the position of the lipid bilayer relative to Cel-Pgp-1, which defined the grid box position. The atomic coordinates PDB file was then converted into a PDBQT file by AutoDock Tools 4 (Morris et al., 2009) for docking calculations. PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC) was used as visualization tools for various tasks (3D alignments, ligands and hotspots location, grid box positioning for AutoGrid 4) and for structure rendering in figures.
2.2. Preparation and conformational analysis of ligands
The molecular structures of ligands were extracted from Chemspider (CID), Drugbank (DB) or Merck Index (M) (Suppl. Fig. S1 and S3), depending on the availability of structures. Each molecular structure was carefully scrutinized for chirality, and sometimes corrected when inconsistencies were found in the literature. For abamectin (ABA), the dry compound is a mixture of B1a (substituent isobutyl on C25) and B1b (substituent isopropyl on C25) forms. As B1a is largely predominant in natural mixtures, the docking calculations were performed only with B1a compound. Several AHs, i.e. levamisole (LEV), closantel (CLO) and monepantel (MNP) are used in vivo as racemic mixtures for treatment, which led us to investigate the docking of each enantiomer.
In the semi-flexible mode, the ligand is handled as flexible around all the rotatable bonds. However, the conformational space of the ligand can be poorly explored when it contains ring structures as in avermectins, since AutoDock does not consider single bonds in non-aromatic cycles as rotatable bonds. To overcome this limitation, in order to better sample the initial conformational space accessible to the ligand, we generated for each ring-containing compound 10 different low energy conformations. For this, we used Marvin Sketch and the minimization under the MMFF94 force field provided in Marvin Suite (https://www.chemaxon.com/products/marvin/marvinsketch/). The diversity of the 10 lowest energy conformers was evaluated by their pairwise root mean square deviations (RMSD) for atomic positions after superimposition under PyMOL. For each obtained cluster of close conformers, a representative one, defined as the center of the group according to the calculated RMSDs, was selected. One to five different conformers were thus selected as starting points for further docking procedures. In all cases, the selected conformers presented rather close energies, corresponding to rapidly interconverting forms of the molecule, and the docking results were generally comparable between each conformer. Thus, the most representative conformer for docking results was chosen for data presentation.
2.3. Docking calculations
Molecular docking experiments were performed using AutoDock 4 (release 4.2.6) in the semi-flexible mode with the Cel-Pgp-1 4F4C PDB structure kept rigid, and prepared with AutoDock Tools (Morris et al., 2009). AutoDock, which is the most cited docking software (Sousa et al., 2006, Sousa et al., 2013), has a free-energy scoring function based on AMBER force field and a large set of diverse protein-ligand complexes with known inhibition constants. Few residues of the protein could have been declared as flexible in the PDBQT file, but the program restrains the total number of torsional degrees of freedom to 32, shared between the ligand and the receptor. This is a drawback in the case of the Cel-Pgp-1 structure, since the inner chamber is large, and in the absence of consistent indications about the exact location of the binding sites of the various drugs, different cavities have to be taken into account in the calculation. Thus, we privileged an approach based on better coverage of ligand flexibility and a grid box extended to the whole membrane part of the receptor protein. Indeed, for all ligands tested, the docking box, in which grid maps were computed using program AutoGrid 4, encompassed all the TM helices and the whole internal cavity, including lateral access channels and protein surface, to allow a large sampling of potential poses. The grid built by AutoGrid included 100, 124, and 126 points in x, y, and z directions, with a grid spacing of 0.375 Å to allow a good compromise between resolution of the explored volume and the size of the binding area (box dimensions 37.5 × 46.5 × 47.3 Å, centered in the inner cavity of Cel-Pgp-1, at the point x = 22.2 Å; y = 77.6 Å; z = −1.4 Å). For each ligand conformer, 100 independent calculations were performed using the Lamarckian genetic algorithm. All the other parameters were set at the default value.
The 100 generated poses were assigned a score calculated by AutoDock that can be considered as an estimated free energy of ligand binding (indicative of binding affinity). They were then clustered as a function of the closeness of their positions and conformations with RMSD set at 2.0 Å, and finally ranked by their binding score (for the best pose in the cluster). The results are displayed in an energy scores histogram, which reproducibility could be assessed by comparing docking calculations performed on close ligand conformers, or on a truly duplicated calculation. As a result, binding energies (positions of the best pose in each cluster of the histogram) were found to fall within a range of 0.25 kcal/mol, and the number of poses in a cluster within 10%. This gives an indication of the accuracy of the histogram parameters in our series of runs, i.e., the binding energies and the overall distribution of clusters.
2.4. Data analysis
Different parameters and observables can be used in the interpretation of docking data issued by AutoDock: binding energies (i.e. docking scores), histogram bars energy range, profile of the histogram, and location of calculated positions in the protein structure. The position of clusters in figures and tables corresponds to the binding energy value of the lowest energy pose in the cluster. The spreading of clusters in the energy scores histogram was considered as a general indication for ligand docking calculation reliability: the less scattered the energies, the higher specificity of binding can be expected. Conversely, a pseudo-Gaussian profile for a group of histogram bars may suggest a non-specific docking. However, in some cases, very negative binding energies (i.e. very good docking scores) were found in poorly populated clusters whereas the general aspect of the histogram was scattered. We considered these poses as relevant, despite the lack of sampling, in view of the gap energy with the next clusters, revealing high binding site specificity.
The question arose as to whether the lowest energy cluster or the most populated cluster (highest histogram bar) had to be considered. The AutoDock docking score is based on an empirical free-energy force field which has been parameterized using a large number of protein-inhibitor complexes for which both structure and inhibition constants were known, and thus should reliably reflect the affinity of the ligand for the receptor, and the stability of the ligand-protein complex. In contrast, the number of poses found in a cluster reflects the number of times that conformers are found in very close (within RMSD) binding sites, without any anticipation of their stability. Thus considering the highest cluster in the energy histogram may be not relevant per se for identifying the most probable docking site of the considered ligand. Practically, for each ligand, when the difference in the energies between the two best-scored clusters was more than 2 kcal/mol, the lowest energy one was considered as the most representative, since the other ones correspond to ligand-protein complexes associated to negligible lifetime. Alternatively, when the energy gap was narrower, we manually analyzed all the clusters within 2 kcal/mol below the lowest one for the localization of the included poses in the protein 3D structure. Finally, one or two main clusters were selected as representative, and they corresponded most often to the first and second minimum energy clusters, except when clusters corresponding to non-relevant positions in the protein were found interleaved in the ranking. In some cases, this protocol of validation led us to consider a double binding site on the protein, leading to a stoichiometry of one or two depending on whether these two docking positions were partially overlapping or not. Such a possibility simply reflects the large size of the multispecific binding domain, which likely encompasses the whole “inner chamber”.
Lastly, criteria for discriminating relevant from non-relevant docking poses had to be defined. This delineation was made necessary by the fact that we enlarged the zone of docking search to the whole membrane part of the protein. The large size of the grid box ensured an exhaustive conformational exploration not biased by preliminary beliefs, although it contributed to the scattering of the docking results. Poses that were outside of the expected ligand binding pocket were not considered for further analysis. These were essentially poses located either “outside” of the Cel-Pgp-1 structure, i.e., at the protein/lipid interface, or lowermost in the transmembrane domain, i.e., at the level of interface with the cytosolic medium (“cytosolic antechamber”), where the phospholipid polar headgroups are likely to be invaginated into the cavity between the transmembrane helices in the inward-facing Cel-Pgp-1 conformation (Haubertin et al., 2006). These poses can have a functional relevance, for example as allosteric modulator sites, or not, but likely not as binding sites for transport. Finally, a search of the possible access channels allowing ligands to reach the inner pocket was performed using Mole 2.0. (http://mole.upol.cz/). In addition to the wide opening of the protein towards the cytosolic interface, the result showed two lateral tunnels located between TM10 and TM12, communicating with the cytosolic leaflet of the membrane (data not shown).
For each lowest energy pose of selected clusters, the number and nature of interacting residues were analyzed within the protein. Among these, particular interest was given to residues belonging to the “hotspots for drug binding”, described hereafter.
2.5. Determination of the residues constituting the “hotspots for drug binding”
A number of experimental works have been conducted in order to determine the key residues responsible for multidrug recognition by mammalian Pgp (human and rodent isoforms). They initially included various directed-mutagenesis analyses that allowed identification of residues whose mutation led to alterations of the MDR profile, in contrast to numerous mutations that led to unspecific global decrease of Pgp function. The significance of these mutations was shown by cytotoxicity assays, which were a good indication of their involvement in the multi-specific drug recognition and binding. Furthermore, some additional data were collected with a chemical cross-linking approach, using a few drug derivatives bearing a moiety that could be activated, and ensuring specificity by testing protection by the native drug. All these data have been compiled in a review by Shilling et al. (2006), and were completed by including some references by Loo & Clarke (Bessadok et al., 2011, Loo et al., 2006a, Loo et al., 2006b, Loo and Clarke, 2001, Loo and Clarke, 2002). Finally, the only crystal structures released so far for mammalian Pgps are of murine Pgp. They were co-crystallized with hydrophobic cyclic peptide inhibitors than can be considered as functionally relevant ligands, and which pointed to a set of contact residues. However, two versions of the interpretation of the experimental X-ray diffraction data have been published (Aller et al., 2009, Li et al., 2014) that differed in the orientation of some transmembrane helices and side chains. Therefore, we consider the lists of contact residues from the two versions equally. All these identified residues form a collection of 62 residues listed in Suppl. Table S1 with their corresponding numbering in the 4 mammalian proteins sequences (Hsa-ABCB1, Mmu-ABCB1a and B1b, Cgr-ABCB1). Moreover, 14 of these 62 residues are common between at least two different techniques. They are all situated in the transmembrane part of the protein. Altogether, they provide a frame in the inner chamber that offers a set of anchoring points for multi-specific recognition and binding, and eventual translocation, of various transport ligands.
Multiple protein sequence alignments have been performed on Cel-Pgp-1, human ABCB1, murine ABCB1a and B1b, and Chinese hamster ABCB1, using Muscle software (Edgar, 2004). We checked that the transmembrane segments were satisfactorily aligned. Among the 62 human ABCB1 hotspot residues, 16 (26%) were found identical and 12 homologous with the corresponding residues in Cel-Pgp-1, representing a global conservation ratio of 45%. As a comparison, human ABCB1 and Cel-Pgp-1 display a global similarity of 63% (BLASTP positive matches), and still 58% when considering the TMDs only. This indicates that hotspots are subjected to more genetic variation than the overall sequence. More precisely, this set of residues is mainly hydrophobic, but less markedly for Cel-Pgp-1: the hydrophobic (F-Y-A-L-I-V-M)/hydrophilic (S-T-N-Q-H) ratio is 44/14 for Hsa-Pgp and 37/20 for Cel-Pgp-1, respectively.
3. Results
3.1. Binding mode of macrocyclic lactones on Cel-Pgp-1
We have first docked seven ML of pharmacological interest, whose structures are presented in Suppl. Fig. S1. A model for the binding site of ivermectin (IVM), and four other avermectins, abamectin (ABA), eprinomectin (EPR), doramectin (DOR) and selamectin (SEL), the ivermectin-aglycone derivative (IVA), and the milbemycin, moxidectin (MOX) on Cel-Pgp-1 was hence proposed.
The energy clustering histograms obtained from the docking calculations allowed us to identify two major possible positions for each ML (except one for DOR), based on the lowest (i.e. most negative) binding energies calculated (Fig. 1A and Suppl. Fig. S2) that were considered relevant for predicting ML binding site. The binding energies for all the considered clusters (13 for the 7 ML) ranged from −13.0 to −9.4 kcal/mol, which are indicative for high affinity binding to Cel-Pgp-1. All positions were found in the Cel-Pgp-1 deepest part of the inner transmembrane cavity, at the top of the inner chamber, and the identification of drug interacting residues, some forming H-bonds, allowed us to delineate a specific ML binding site, which involved several amino acids identified as hotspot residues on mammalian Pgp (detailed in Table 2 and Suppl. Table S2).
Fig. 1.

Ivermectin and moxidectin binding to Cel-Pgp-1. A and B. Histograms of energy scores, clusterized at RMSD = 2 Å. The first (1) and second (2) lowest energy clusters are framed with red and green dotted lines, respectively. C and D. Binding sites of the lowest energy clusters of IVM (C) and MOX (D). E and F. Binding sites of the 2nd lowest energy clusters of IVM (E) and MOX (F). IVM is represented in red spheres, MOX is represented in green spheres, Cel-Pgp-1 in light blue (N-term) and yellow (C-term) ribbon. Images were generated using PyMol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
Table 2.
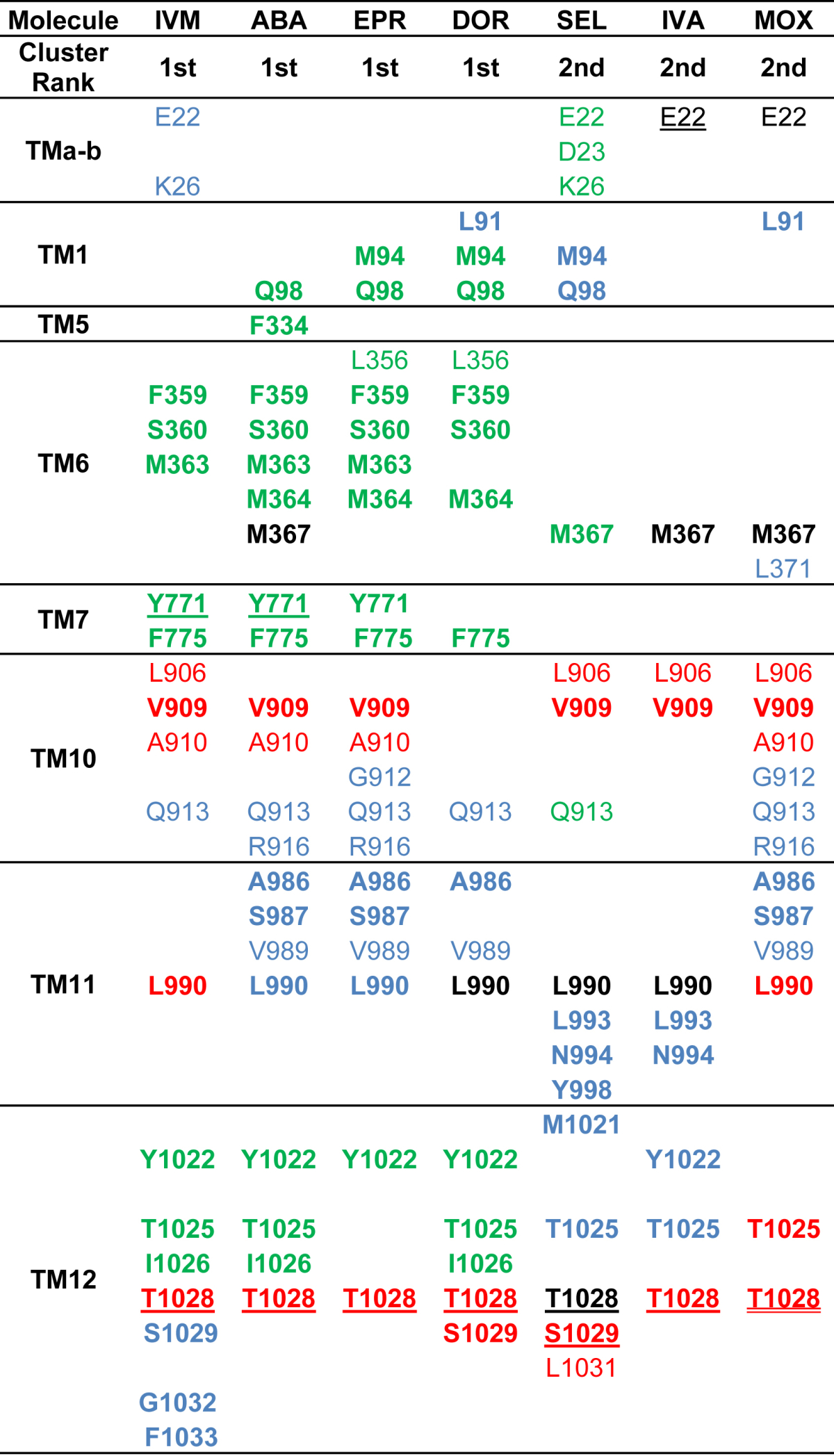
List of interacting residues of each transmembrane helix of Cel-Pgp-1 with the first or second lowest energy clusters of ivermectin (IVM1), abamectin (ABA1), eprinomectin (EPR1), doramectin (DOR1), selamectin (SEL2), ivermectin-aglycne (IVA2) and moxidectin (MOX2). Bold: hotspot residues. Underscored: residues establishing a H-bond. Black: residues interacting with macrocycle. Red: residues interacting with benzofurane. Blue: residues interacting with spiroketal unit. Green: residues interacting with the mono/di-saccharide moiety.
3.1.1. Docking of avermectins and the milbemycin, moxidectin
For IVM, the positions of the two lowest energy clusters on Cel-Pgp-1 were very close, with IVM1 (−12.2 kcal/mol, 20 poses) being slightly deeper in the inner chamber than IVM2 (−11.1 kcal/mol, 6 poses) (Table 1 and Fig. 1). Overall, they interacted with 19 (including 14 hotspot residues, 2 H-bonds) and 13 residues (6 hotspot residues, 3 H-bonds), respectively, and they shared 6 common interacting residues, including 4 hotspot residues and 1 H-bond (Table 2 and Suppl. Table S2).
Table 1.
Physico-chemical properties and docking characterization of macrocyclic lactones to Cel-Pgp-1: ivermectin (IVM), abamectin (ABA), eprinomectin (EPR), doramectin (DOR), selamectin (SEL), ivermectin-aglycone (IVA) and moxidectin (MOX).
| Molecule | IVM | ABA | EPR | DOR | SEL | IVA | MOX | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW (Da) | 875 | 873 | 914 | 899 | 770 | 587 | 640 | ||||||
| logP | 4.3 | 4.4 | 4.2 | 4.7 | 5.2 | 3.4 | 5.4 | ||||||
| Cluster rank | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 1 | 2 | 1 | 2 | 1 | 2 |
| Binding Energy (kcal/mol) | −12.2 | −11.1 | −12.3 | −11.1 | −12.5 | −12.0 | −13.0 | −12.9 | −12.4 | −9.6 | −9.4 | −10.5 | −10.1 |
| Nb of poses | 20 | 6 | 8 | 37 | 7 | 6 | 19 | 52 | 23 | 33 | 49 | 6 | 68 |
| Nb of inter-acting residues | 19 | 13 | 21 | 15 | 20 | 17 | 17 | 14 | 18 | 10 | 10 | 19 | 16 |
| Nb of hotspot residues | 14 | 6 | 17 | 10 | 14 | 12 | 14 | 13 | 12 | 7 | 8 | 14 | 8 |
| Nb of H-bonds | 2 | 3 | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 3 | 2 | 3 | 2 |
For ABA, the two lowest-energy positions identified (−12.3 and −11.1 kcal/mol, 8 and 37 poses, respectively) interacted with 21 and 15 residues, including 17 and 10 hotspot residues, respectively, and forming 2 H-bonds (Table 1). They shared 8 common interacting residues (including 6 hotspot residues and 0 H-bond) that closely overlapped with IVM positions (Table 2 and Suppl. Table S2).
For EPR, the clustering was more scattered than that of ABA or IVM (Suppl. Fig. S2), but nevertheless two lowest energy clusters were found at −12.5 kcal/mol (EPR1, 7 poses) and −12.0 kcal/mol (EPR2, 6 poses) (Table 1). They both bound in the deep pocket in the inner chamber, with EPR2 being deeper than EPR1. These two clusters interacted with 14 and 12 hotspot residues (6 were shared by both positions), respectively, among 20 and 17 interacting residues (8 shared), and formed 1 and 2 H-bonds, respectively (Table 2 and Suppl. Table S2).
For DOR, only one energy cluster was considered (−13.0 kcal/mol, 19 poses) (Table 1), as it was significantly separated from all the other clusters (Suppl. Fig. S2). DOR1 interacted with 17 residues in Cel-Pgp-1 transmembrane inner chamber, including 14 hotspot residues, with 1 H-bond formed (Table 2 and Suppl. Table S2).
For SEL, the two lowest energy clusters were found at −12.9 kcal/mol (SEL1, 52 poses), and −12.4 kcal/mol (SEL2, 23 poses) (Table 1). These two clusters were positioned close to each other, with 14 and 18 interacting residues, 13 and 12 being hotspot residues, respectively. They each formed 2 H-bonds with different hotspot residues, and they shared 8 common interacting residues, including 7 hotspot residues (Table 2 and Suppl. Table S2).
For IVA, the two lowest energy clusters together comprising most of the poses (82%), indicating a good specificity of binding to this site, were close in energy, at −9.6 and −9.4 kcal/mol, and both bound very similarly to a unique binding site (Table 1). IVA1 and IVA2 shared 6 common interacting residues out of 10 for each, with 4 hotspot residues out of 7 and 8, respectively, and formed 3 and 2 H-bonds, respectively (Table 2 and Suppl. Table S2).
For MOX, the two lowest energy clusters were found at −10.5 kcal/mol (MOX1, 6 poses) and −10.1 kcal/mol (MOX2, 68 poses) (Table 1 and Fig. 1). They interacted with 19 and 16 residues, 8 being common to the 2 clusters, including 14 and 8 hotspot residues, respectively; 5 of which being common and forming 3 and 2 H-bonds, respectively (Table 2 and Suppl. Table S2).
In summary, the very low binding energies calculated in the docking modes (from −13.0 to −11.1 kcal/mol), strongly suggest that avermectins can bind with high affinity to Cel-Pgp-1 (Table 1). IVA and MOX positions showed significantly less negative binding energies (from −10.5 to - 9.4 kcal/mol) than the avermectins, reflecting a lower affinity, but they were still consistent with specific binding to Cel-Pgp-1. Interestingly, all ML comparably bound to a unique site located deep in the transmembrane domain of the nematode transporter, and subtle differences were observed in relation with the specific substituents of each ML (Table 2).
3.1.2. Relation between ML structure and their binding characteristics
We showed above that all ML bound to a similar and specific binding site (Fig. 2A) on the transmembrane domain of Cel-Pgp-1, with very negative energies reflecting high affinity binding (Table 1). In addition, ML binding involved some hotspot residues, which have been previously identified to be important for drug binding to mammalian Pgp (Table 2 and Suppl. Table S2). Nevertheless, it was possible to point out noticeable specificities between each ML, due to their different substituents that appeared important in the respective orientation of each ML in their binding site. Thus, we could propose several common features and some specific aspects for each ML in terms of binding characteristics, based on the analysis of their two lowest energy clusters.
Fig. 2.

Binding domain characteristics of macrocyclic lactones. A. Front view of Cel-Pgp-1 represented in transparent light blue (N-term) and yellow (C-term) ribbon, with the binding sites of the first and second lowest energy clusters of abamectin (ABA1 and ABA2, black), ivermectin (IVM1 and IVM2, red), eprinomectin (EPR1 and EPR2, sand), doramectin (DOR1, blue), selamectin (SEL1 and SEL2, purple), ivermectin-aglycone (IVA1 and IVA2, orange), and moxidectin (MOX1 and MOX2, green), all represented in sticks and transparent surfaces. TM helices numbers are indicated as their number only, on each helix. B. Zoom, without Cel-Pgp-1, on the front and lateral views of the overlap between the binding sites of the first lowest energy clusters of abamectin (ABA1, black), ivermectin (IVM1, red), eprinomectin (EPR1, sand), doramectin (DOR1, blue). C. Zoom, without Cel-Pgp-1, on the front and lateral views of the overlap between the binding sites of second lowest energy clusters of selamectin (SEL2, purple), ivermectin-aglycone (IVA2, orange), moxidectin (MOX2, green), with the lowest energy cluster of ivermectin (IVM1, red) as a marker of the binding position of other ML. All molecules are represented in sticks. Images were generated with PyMol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
3.1.2.1. Influence of the macrocyclic ring
The docking of each ML on Cel-Pgp-1 revealed that a total of 42 residues interacted with ML (Table 2). Among them, 14 residues, including 10 hotspot residues, were found to interact at least 5 times when considering the 10 most clearly overlapping cluster positions, which delineated a specific binding pocket, and gave a specific “fingerprint” for ML binding (Table 2 and Suppl. Table S2). Remarkably, the following ML cluster positions: IVM1, ABA1, EPR1, DOR, SEL2, IVA2 and MOX2 tightly superimposed (Fig. 2B and C), and they aligned quite perfectly at the macrocyclic ring level. Therefore, the binding characteristics of these relevant clusters were further analyzed. In particular, the residues E22, M367 and L990 were respectively interacting with the C12–13, C14–15 and C17-18 of the macrocyclic ring: interactions of E22 with IVA and MOX; M367 with ABA, IVA and MOX; and L990 with DOR, SEL and IVA (black colored in Table 2).
3.1.2.2. Influence of the benzofurane moiety
The benzofurane moiety is also a common structural feature that superimposed for all ML on their binding site in Cel-Pgp-1, although it was found in an inverted orientation for SEL and IVA when compared to the other ML. Indeed, in most of the tested ML, the benzofurane moiety interacted with 6 residues, i.e. L906, V909, A910, L990, T1028 and S1029 (red colored in Table 2). Interestingly, all ML interacted with T1028 on TM12, and in all the considered cluster positions, the C5-hydroxyl group formed an H-bond with T1028 (Table 2 and Fig. 3). T1028 has also been identified as a hotspot residue, and therefore we considered it as an important factor for the binding of all ML.
Fig. 3.

Key residues of Cel-Pgp-1 for interaction with macrocyclic lactones. A and B. Cel-Pgp-1 key residues surrounding the binding site or forming H-bonds with ivermectin (IVM1, red lines) (A) and moxidectin (MOX2, green sticks) (B). Cel-Pgp-1 is represented in light blue (N-term) and yellow (C-term) transparent ribbons for TM helices and sticks for specifically interacting residues. Atoms of Cel-Pgp-1 residues and substrates are colored in blue for N, red for O and grey for H. H-bonds are represented in yellow dotted lines with red shadow. C and D. Ivermectin (C) and moxidectin (D) plane structures with H-bonds to residues of Cel-Pgp-1 represented as dotted lines. Images were generated with PyMol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
3.1.2.3. Influence of the spiroketal moiety
The spiroketal moiety, commencing at C17, differs between ML in various ways, and it introduces stereochemical diversity that impacts the binding to Cel-Pgp-1. ABA (an avermectin B1) is the natural compound from which most of avermectins are derived, and it has a C22—C23 double bond instead a saturated bond for IVM (C22—C23 dihydro-avermectin B1). Consequently, the spiroketal moiety of ABA has higher structural rigidity, and it bound to Cel-Pgp-1 in a slightly different orientation than that of IVM (Fig. 2B); both sharing only one common interacting residue (blue colored in Table 2). This reveals that the saturation of the C22-23 bond played a role in the conformation of the ML, and impacted the binding characteristics of the IVM spiroketal moiety. EPR (4′-epiacetylamino-4′-deoxy-avermectin B1) differs from ABA at the spiroketal moiety level by having at C25 a terminal oleandrose group. Nevertheless, the spiroketal moieties of EPR and ABA matched strictly on their binding site (Fig. 2B), interacting with residues mainly present on TM10 (Q913, R916) and TM11 (A986, S987, V989, L990) (Table 2). MOX has a substituted olefinic side chain at the 25-position and a methoxime group at the 23-position, which are two characteristics specific to this drug, not found in other commercial milbemycins or avermectins. Remarkably, although the spiroketal moieties of MOX, DOR, ABA and EPR differ considerably (Suppl. Fig. 1), the substituents bound to the same binding site with similar orientation for the four drugs (Fig. 2B and C), interacting with TM10 and TM11, and sharing Q913, A986 and V989 as common interacting residues (Table 2). Conversely, although DOR and SEL have similar spiroketal moieties characterized by a C25-cyclohexyl substituent, they did not overlap on the Cel-Pgp-1 binding site (Fig. 2 B and C). SEL and IVA have also similar spiroketal moieties that are both partly aligned with the disaccharide group of the four di-glycosylated avermectins (Fig. 2 B and C), interacting partly with TM11 and partly with TM12 (Table 2). This clearly revealed that common structural features, other than the spiroketal moiety, determine the orientation of ML on the binding site. This could be the presence of one or two sugars, or the saturated bond on C22-23 in SEL and IVM.
3.1.2.4. Influence of the disaccharide moiety
The four avermectins IVM, ABA, EPR and DOR have in common a disaccharide moiety linked to the C13 of the macrolide. This substituent mostly superimposed for these four ML (Fig. 3B), and interacted with TM1, TM6, TM7 and TM12 (green colored in Table 2). They all shared a number of interacting hotspot residues, i.e. F359, S360, F775 and Y1022, plus Q98, M363, M364, Y771, T1025 and I1026 when considering 3 ML out of the 4, that clearly characterized the binding fingerprint specific of di-glycosylated avermectins. Interestingly, ABA and IVM disaccharide moieties both formed an H-bond with the hotspot residue Y771, showing the importance of this residue in the orientation of ML that contain a disaccharide (Table 2 and Fig. 3A and C). Apart from Y1022 and T1025, which interacted with spiroketal or benzofurane, none of the residues listed above interacted with IVA and MOX. Given the absence of glucide substituent on these two ML, and the monosaccharide of SEL, these residues are interacting specifically with the distal sugar unit of the disaccharidic avermectins. Indeed, the orientation of the monosaccharide of SEL and the hydroxyl on the C13 of IVA superimposed in a different orientation than that of the corresponding structure of other ML, which overlapped with the spiroketal moiety of IVM, and shared with it the residues E22 and K26 in TMa-b, and Q913 in TM10 as common interacting residues (Fig. 2 and Table 2). Because their only structural difference relies on the presence or absence of disaccharide, the comparison of IVM and IVA binding provided interesting information. Remarkably, 7 of the 10 residues interacting with IVA1 were all found to interact with IVM1, with a common H-bond on T1028, showing that the binding site of these two closely related molecules are very similar (Suppl. Table S2). It should be mentioned that the three ML lacking the disaccharide (SEL, IVA and MOX) exhibited an energetic clustering clearly less scattered than the four di-glycosylated avermectins (see Fig. 1A and Suppl. Fig. S2), suggesting that the absence of the two sugar units significantly increased the selectivity of the calculated interactions with the recognition domain. In addition, the two unglycosylated ML (IVA and MOX) have significant higher (i.e. less negative) binding energies when compared with other ML, which supports the important role of sugar in anchoring IVM onto the binding site.
Globally, these data reveal that all ML bound with good energy in the same pocket buried within the Cel-Pgp-1 inner chamber, and were clearly superimposed. Consideration of the residues involved in the binding of each chemical moiety of the ML, presented in different colors in Table 2, clearly supports a typical and unique binding pocket for all ML (Fig. 2, Fig. 3). Furthermore, as the four ML that contain a disaccharide moiety were aligned for their lowest energy clusters, while this happens only for the second clusters of SEL, IVA and MOX, we propose that the main determinant of the orientation of ML, in their binding pocket, is the disaccharide moiety.
3.2. Binding mode of other anthelmintic drugs with Cel-Pgp-1
3.2.1. Docking results
The structures of all AHs tested are shown in Suppl. Fig. S3. For predicting AH binding site, a similar approach was used to choose the optimal cluster energy from the histogram obtained from the docking calculations (Suppl. Fig. S4).
For triclabendazole (TCZ), the lowest energy cluster (TCZ1, 97 poses) appeared at −8.0 kcal/mol, whereas the only other cluster (TCZ2, 3 poses) was found at −7.4 kcal/mol (Table 3). On the one hand, TCZ1 was found deep in the inner chamber of Cel-Pgp-1 (Fig. 4), interacting with 8 residues, (all hotspot residues, 1 H-bond) (Table 4). On the other hand, the binding site of the second cluster was in the middle of the inner chamber, at the level of the middle of the transmembrane bilayer (Fig. 4). For this position, 9 interacting residues were found (6 hotspot residues, 1 H-bond) (Table 4). These two positions were totally separated (Table 4 and Fig. 4), hence consistent with a binding stoichiometry of 2.
Table 3.
Physico-chemical properties and docking characterization of anthelmintics to Cel-Pgp-1: triclabendazole (TCZ), thiabendazole (TBZ), levamisole-R (LEV-R), closantel-R (CLO-R), monepantel (MNP-S) and emodepside (EMD). IVM data from Table 1 are given in italic for the sake of comparison. (*) cluster positioned outside the inner chamber.
| Molecule | TCZ | TBZ | LEV-R | CLO-R | MNP-S | EMD | IVM | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW (Da) | 360 | 201 | 204 | 663 | 473 | 1119 | 875 | |||||
| logP | 5.8 | 2.2 | 2.9 | 7.6 | 5.6 | 7.3 | 4.3 | |||||
| Cluster rank | 1 | 2 | 1* | 2 | 1* | 2 | 1 | 1 | 4 | 1 | 1 | 2 |
| Binding Energy (kcal/mol) | −8.0 | −7.4 | −7.2 | −5.7 | −6.8 | −6.1 | −10.3 | −8.1 | −7.8 | −9.5 | −12.2 | −11.1 |
| Nb of poses | 97 | 3 | 94 | 3 | 31 | 65 | 80 | 3 | 1 | 5 | 20 | 6 |
| Nb of inter-acting residues | 8 | 9 | 12 | 7 | 12 | 8 | 10 | 14 | 14 | 17 | 19 | 13 |
| Nb of hotspot residues | 8 | 6 | 3 | 6 | 5 | 7 | 6 | 6 | 12 | 6 | 14 | 6 |
| Nb of H-bonds | 1 | 1 | 0 | 2 | 1 | 2 | 2 | 1 | 0 | 3 | 2 | 3 |
Fig. 4.

Binding domains of anthelmintic drugs. A. Front view of Cel-Pgp-1 represented in transparent light blue (N-term) and yellow (C-term) ribbon, with the binding sites of the first and second lowest energy clusters of triclabendazole (TCZ1 and TCZ2, light pink), thiabendazole (TBZ1 and TBZ2, purple), levamisole-R (LEV1 and LEV2, dark green), closantel-R (CLO1, yellow), monepantel-S (MNP1 and MNP4, light green), emodepside (EMD1, dark blue), all represented in sticks and transparent surfaces. TM helices numbers are indicated as their number only, on each helix. B. Zoom, without Cel-Pgp-1, on the front view of all anthelmintics docking positions in the binding sub-domains, represented in transparent surfaces. C. Zoom, without Cel-Pgp-1, on the front and lateral views of the overlaps between the binding sites of triclabendazole (TCZ1 and TCZ2, light pink), thiabendazole (TBZ2, purple), levamisole (LEV2, dark green), closantel (CLO1, yellow), monepantel (MNP1 and MNP4, light green) and emodepside (EMD1, dark blue). All molecules are represented in sticks. Images were generated with PyMol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
Table 4.
List of interacting residues of each transmembrane helix of Cel-Pgp-1 with the selected lowest energy clusters of triclabendazole (TCZ), thiabendazole (TBZ), levamisole-R (LEV-R), closantel-R (CLO-R), monepantel-S (MNP-S) and emodepside (EMD). The interacting residues with the reference ML, ivermectin (Table 1), are also presented for comparison. Bold: hotspot residues. Underscored: residues establishing a H-bond. Green: common interacting residues to TCZ1. Blue: common interacting residues to TCZ2. Orange: common interacting residues to TBZ1. Dark green: common interacting residues to TBZ2. Purple: common interacting residues to LEV2. Brown: common interacting residues to CLO1. Pink: common interacting residues to MNP1. Black: interacting residues never shared with any other AH cluster.

For thiabendazole (TBZ), the lowest energy cluster (TBZ1, 94 poses) was at −7.2 kcal/mol (Table 3), but it was found at a very lateral position in the TM helices of Cel-Pgp-1 (Fig. 4). TBZ1 interacted with 12 residues (3 hotspot residues, no H-bond) (Table 4). Alternatively, the second cluster (TBZ2, 3 poses), found at −5.7 kcal/mol, was found deep in the inner chamber, totally apart from TBZ1 (Fig. 4). It showed 7 interacting residues, (6 hotspot residues, 2 H-bonds) (Table 4) and shared 5 common residues, including 4 hotspots, with TCZ1 (Table 4).
For levamisole (LEV), the lowest energy cluster, (LEV1, 31 poses) of the two enantiomers R and S, which gave similar results for all clusters, was found at −6.8 kcal/mol (Table 3). It bound to Cel-Pgp-1 on a very lateral position in the TM helices bundle, close to TBZ1 position, and sharing 3 common interacting residues with TBZ1 (Table 4 and Fig. 4). LEV1 interacted with 12 binding residues (5 hotspot residues, 1 H-bond) (Table 4). The second lowest energy cluster, LEV2 (65 poses), formed a cluster at −6.1 kcal/mol (Table 3). It interacted deep in the inner chamber, very differently from LEV1, and overlapping with TCZ1 and TBZ2 (Fig. 4). It bound to 8 residues, (7 hotspot residues, 2 H-bonds), and showed 7 common interacting residues, including 6 hotspot residues, with TBZ2, and 5 common interacting residues with TCZ1, all hotspot residues (Table 4) .
For closantel (CLO), similar lowest energy clusters CLO1 were found whatever the enantiomer considered, S or R. For the latter, it contained the maximum number of poses (80) at −10.3 kcal/mol (Table 3 and Fig. 5A). The binding site of this cluster was in the middle of the inner chamber (Fig. 5B), similar to the three other clusters found between 9.5 and 9.9 kcal/mol, and showed 10 interacting residues (6 hotspot residues, 2 H-bonds) (Table 4). It shared 3 common interacting hotspot residues with TCZ1, 2 with TBZ2 and LEV2, as well as 2 common interacting residues with TCZ2 (Table 4 and Fig. 4).
Fig. 5.

Closantel-R binding to Cel-Pgp-1. A. Histogram of energy scores, clusterized at RMSD = 2 Å. The lowest (1) energy cluster is framed with red dotted lines. B. Binding site of the lowest energy cluster, generated using PyMol. Closantel-R is represented in light yellow spheres, Cel-Pgp-1 in light blue (N-term) and yellow (C-term) ribbon. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article).
For monepantel (MNP), two main docking positions were found for both enantiomers S and R. For MNP-S, on one hand, the lowest energy cluster (MNP1, 3 poses) was found at −8.1 kcal/mol (Table 3), and bound “horizontally” at the level of the middle of the bilayer (Fig. 4). The 2 following clusters docked at very similar positions. On the other hand, the 4th cluster (MNP4, 1 pose) showed an energy of −7.8 kcal/mol, and was very close to most of the clusters LEV2, TBZ2 and TCZ2, at the deepest part of the inner chamber (Fig. 4). They both interacted with 14 residues, including 6 hotspot residues and 1H-bond for MNP1, and 12 hotspot residues without forming any H-bond for MNP4 (Table 3). These two binding locations were totally separated (Table 4), consistent with a stoichiometry of 2. In addition, MNP1 shared 9 common interacting residues with TCZ2 and 2 with CLO1. MNP4 shared 7 common interacting residues with TCZ1 and TBZ2, 8 with LEV2, and 4 with CLO1 (Table 4 and Fig. 4).
For emodepside (EMD), the lowest energy cluster (EMD1, 5 poses) was found at −9.5 kcal/mol (Table 3). Its docking position was found near the cytosolic aperture of the inner chamber (Fig. 4). EMD1 bound to 17 residues (6 hotspot residues, 3 H-bonds) (Table 4), and shared 4 interacting residues with CLO1, 1 with TCZ2, and 2 with MNP1. All the other clusters bound very closely to the same site.
3.2.2. Analysis of the binding sites of AHs
The binding domain in Cel-Pgp-1 for AHs belonging to different classes could be analyzed by comparing their respective interacting sites and residues (Table 4 and Fig. 4). First, the two benzimidazoles, TCZ and TBZ, as well as the imidazothiazole LEV, displayed docking positions (TCZ1, TBZ2 and LEV2) that were remarkably highly overlapping (Table 4 and Fig. 4): they shared 5 common interacting residues (Q98, N994, M1021, Y1022, T1025), all hotspot residues. Their related chemical structures and small sizes made possible their interaction with the deepest part of the inner chamber of Cel-Pgp-1.
Second, this inner sub-domain is not specific to the structurally related AHs, TCZ, TBZ and LEV, since CLO1 and MNP also bound on it. Nevertheless, with its two binding positions, MNP, along with CLO1 and TCZ2, can describe a bigger surface of the inner chamber by filling various connected sub-sites (Fig. 4). Therefore, in the core of the chamber, the three AHs, TCZ, CLO and MNP share a high number of common interacting residues with IVM (taken as the representative molecule for all ML, gathering IVM1 and IVM2): 8 (including 7 hotspot residues) for TCZ1 & TCZ2, 8 (including 5 hotspot residues) for CLO1, and 9 (including 6 hotspot residues) for MNP1 and MNP4 (Table 4).
Finally, the large molecule EMD was well accommodated by the binding sub-domain close to the cytoplasmic entrance of the inner chamber of Cel-Pgp-1. Indeed, it also shared some common interacting residues with MNP1, TCZ2 and CLO1 (Fig. 4). Moreover, among its 17 interacting residues found in the docking, 9 (including 5 hotspot residues) were common to IVM1 and IVM2 (Table 4).
4. Discussion
Resistance against anthelmintic drugs in parasite nematodes, including against ML, compromises the therapeutic benefit of AH-based treatments (Lespine et al., 2012). Taking account of the serious concern for animal and human health caused by nematode infections, it is urgent to gain an understanding of the molecular factors involved in limiting drug efficacy in nematodes. It is now well established that some ABC transporters are central in limiting ML efficacy. Mammalian Pgp transports ML out of the host organism thereby limiting the active concentration reaching the nematode target (Kiki-Mvouaka et al., 2010). In addition, some nematode Pgps also interact with ML, and they are involved in the process of development of AH resistance (Lespine et al., 2012). Nevertheless, tools are needed to understand in detail the role of these proteins in ML transport, and to consider them as relevant targets for reversion of AH resistance. The release of the 3D crystal structure of Cel-Pgp-1, with good resolution (Jin et al., 2012) and QMEAN Z-score (Domicevica and Biggin, 2015) provides an opportunity to launch a sound in silico structural study of drug binding to this protein. This is particularly relevant in the view of multispecific recognition, and hence expected transport capacity, of this model protein. Indeed, the open inward-facing conformational state, in which Cel-Pgp-1 has been crystalized as revealed by the study of Jin et al. (2012), is by far the most frequent found conformation among the ABC proteins structurally determined today (Ward et al., 2007), and particularly the unique one found for mammalian Pgps (Aller et al., 2009, Li et al., 2014, Szewczyk et al., 2015). This conformation seems to be stable and ubiquitous, and is now commonly admitted to be the competent one for substrate recognition and uptake. This is corroborated by the fact that the only Pgp structures co-crystalized with a drug involve Mmu-Pgp with QZ59 derivatives, all bound in the same location within the inner chamber. From all the structures today available, either from bacterial or eukaryotic organisms, the general scheme admitted for drug translocation by multidrug transporters implies that the initial drug chelation step, obviously necessary for drug transport, occurs before and independently from the following trans-conformational steps constituting the catalytic cycle of the active ATP-dependent transmembrane drug translocation.
However, a critical point is that our in silico approach needs to be validated by an in silico/in vitro correlation. Such a validating correlation could actually be achieved by considering the enzymatic data reported in the same publication (Jin et al., 2012) and a series of in silico docking calculations performed for a set of 6 molecules that have been shown to stimulate Cel-Pgp-1 ATPase activity, compared to a negative control without effect on this activity. This previous analysis allowed us to draw the conclusion that the inner chamber of Cel-Pgp-1 should be considered as the binding domain competent for recognizing transport substrates, thereby initiating the active transport cycle leading to their transmembrane translocation. We have thus defined a threshold value for binding energies between “ligands” and “weak- or non-ligands”, at about −4 to −5 kcal/mol (David et al., work in process).
Finally, since the inner chamber is in direct connection with the lipid phase of the surrounding membrane, it can be expected to be filled with phospholipid molecules when considering the limits of the hydrophobic thickness of the protein and its positioning in the bilayer calculated by the OPM server (http://opm.phar.umich.edu). In a first approximation, the lipid phase can be considered as fluid, so that, similarly to solvent molecules, lipid molecules are not considered as ligands bound to the protein, and thus not included in the grid maps calculated by Autodock. Lipids are rather expected to influence the kinetics of interaction but not the drug binding affinity for the receptor at equilibrium. However, in a multispecific context, the specific binding of lipids cannot be excluded. If such specific lipid binding site exists, with an affinity comparable to the considered drug, possible competitive interactions can occur between drug and lipid, depending on the location of the corresponding binding sites within the multispecific inner chamber. The scope of this study is nevertheless to model and analyze possible AH drugs interactions with Cel-Pgp-1 in a pharmacological perspective.
4.1. Structural characterization of the ML binding site in Cel-Pgp-1
For the first time, our docking data provides strong evidence that Cel-Pgp-1 is involved in ML high affinity recognition and eventual active transport. All the tested ML displayed very low binding energies (between −13.0 and −9.4 kcal/mol), in the range of binding energies found for the set of validation ligands, as discussed elsewhere (David et al., work in process). The whole docking data collected on seven ML revealed a striking homogeneity in the predominant bound conformations found, in favor of a very similar binding mode for all of them within the inner chamber of Cel-Pgp-1. This convergence is indicative of the robustness of the docking results and contributes to give them high confidence.
Remarkably, all ML were bound in the most buried part of the transmembrane inner chamber. More specifically, the macrocycle and the benzofurane moieties, which together constitute the common core of the ML chemical structures, always clearly superimposed in the same sub-sites. In particular, the hotspot residue T1028 was found to establish various H-bonds with benzofurane groups of all tested ML. At variance, the spiroketal moiety, also present in all ML, proved to be able to accommodate two distinct sub-sites in the same area, suggesting possible alternate binding modes for this substituent. More noticeably, all the conformations found for the 7 tested ML involved a high ratio of hotspot residues: indeed, over the 13 considered docking positions, a total of 209 interacting residues were detected by AutoDock, of which 149 (about 70%) were hotspot residues. This emphasizes a possible underlying structural homology with mammalian Pgp for binding large amphiphilic/hydrophobic ligands.
Regarding the disaccharide moiety harbored by four of the tested avermectins (ABA, IVM, EPR, DOR), the comparison with the data obtained with SEL (bearing one monosaccharide), IVA and MOX (no sugar unit), highlighted the effects of a sugar moiety on binding affinity for Cel-Pgp-1. First, SEL displayed binding energies (−12.9/-12.4 kcal/mol) typically within the range of values calculated for the other tested avermectins (−13.0 to −11.1 kcal/mol), whereas IVA and MOX clearly displayed less favorable binding energies (−10.5 to −9.4 kcal/mol). This indicates that the proximal sugar unit plays a major role in establishing energetic interactions with the receptor binding domain, while the distal sugar is less influential in stabilizing the interaction. This is reinforced by the significant number of hotspot residues (13) on Cel-Pgp-1 interacting with the sugar moiety. Second, SEL, IVA and MOX were all found in two overlapping but distinct docking positions, both relevant since they correspond to their two lowest energy clusters. One position overlaid well with that of the four disaccharide-branched avermectins, whereas the other one, although still in the same sub-site, was oriented differently. Although highlighting the remarkable common binding mode of all the tested ML, these data also illustrate the role of the disaccharide moiety in anchoring ML in the summit part of the inner chamber in Cel-Pgp-1, in which it is well fitted due to favorable steric interactions.
Our data are consistent with reports showing interactions between ML and C. elegans Pgps, with increased Cel-Pgp-1 expression in IVM-resistant C. elegans strains (James and Davey, 2009, Yan et al., 2012), and increased C. elegans sensitivity to IVM (Ardelli and Prichard, 2013, Janssen et al., 2013) and MOX (Bygarski et al., 2014) after disruption of the Cel-Pgp-1 gene. In addition, major differences have been reported on interaction of IVM and MOX with mammalian Pgp. Indeed, the apparent affinity of IVM for Cgr-Pgp calculated in vitro was higher than that of MOX (Lespine et al., 2007, Prichard et al., 2012), and disaccharide moities were then suggested to modify the binding property of ML. Similarly, both drugs interact differently with parasitic nematode Pgps (Godoy et al., 2015a, Godoy et al., 2015b, Godoy et al., 2016, Kaschny et al., 2015, Kerboeuf and Guegnard, 2011, Mani et al., 2016). Thus, our data strengthen the hypothesis that the presence of sugar substituents could partly determine the affinity binding of ML to various mammalian and nematode Pgps.
4.2. Binding of other anthelmintics on Cel-Pgp-1
In silico docking calculations for other AH drugs belonging to various chemical classes brought additional information about the molecular mechanisms of multispecific recognition properties exhibited by Cel-Pgp-1. The chemical diversity of the tested AHs revealed the wide range of ligand-protein interactions accessible in this putative multidrug transporter. In particular, a large majority of docking positions were found within the inner chamber, indicating its pivotal role in the initial step of recognition and binding of substrate drugs. The very negative binding energies of the four hydrophobic AHs, TCZ, CLO, MNP and EMD, indicate that Cel-Pgp-1 might efficiently transport them, favoring the possible contribution of this protein to AH drug resistance. However, two noticeable exceptions were observed for TBZ and LEV, whose best binding energy clusters included poses located at the membrane limit of the transmembrane helix bundle. Such a binding position makes the translocation of these two substrates unlikely. The hypothesis that these drug binding sites are not relevant for active transmembrane drug translocation is thus favored. We cannot, however, exclude that these binding sites can play some functional, allosteric regulatory role, which would be desirable to test in vitro. In contrast, the conformations of the second lowest energy clusters for TBZ and LEV appeared to be localized within the inner chamber, but with less favorable binding energy (−5.7 and −6.1 kcal/mol, respectively). It remains to be evaluated, in vitro, whether these “weak ligands” actually interact with Cel-Pgp-1 at biochemically/pharmacologically relevant concentrations.
Remarkably, among the total 87 interacting residues encompassed by the docking positions of the 6 tested AHs found in the inner chamber, 57 (i.e. 66%) are hotspot residues. Once again, this high ratio provides an indication of a common structural template for multispecific recognition shared by Cel-Pgp-1 and mammalian Pgp, although their respective drug handling profiles do not fully overlap, possibly due to differences in the balance of hydrophobic/polar residues lining the inner chamber. Interestingly, TCZ, CLO and EMD have been reported to interact with mammalian Pgp (Dupuy et al., 2010, Elmshauser et al., 2014), whereas TBZ and LEV were shown not to interact with mammalian Pgp (Dupuy et al., 2010, Efferth and Volm, 1993, Hayeshi et al., 2006).
4.3. Usefulness and applicability of this structural model of multispecific drug binding
Globally, this in silico analysis of ML binding on Cel-Pgp-1 provides a new, detailed vision of the fine molecular mechanisms of multispecific recognition capacities of a putative multidrug transporter. Altogether, our data based on a large selection of potential ligands are convergent to reveal that the inner chamber forms a large multi-specific binding domain composed of some continuously connected sub-sites. This representation can help in understanding or predicting mutual relationships of recognized ligands, depending on the sub-site(s) they potentially bind and the overlaps between sub-sites. In particular, the smallest size ligands, such as TCZ and MNP, can be predicted to bind with a stoichiometry of 2, while some others, such as MOX, display “dual” binding site with two different, partially overlapping positions. These different configurations illustrate the palette of possible interactions offered by all the contact residues lining this chamber, and the large size of the multidrug binding domain. This is clearly at variance with the standard depiction of ligand-receptor interaction typically given by the “lock-and-key” mechanism. Overall, Cel-Pgp-1 and mammalian Pgp appear to share common structural features in their respective inner chambers based on the high proportion of hotspots residues in Cel-Pgp-1, aligned (but often not identical) with residues involved in multidrug recognition in mammalian Pgp.
More relevant pharmacologically is the observation of overlapping docking positions for ML and the AH drugs, TCZ, CLO, EMD and MNP, which permits the prediction that simultaneous presence of a ML and one of these compounds would lead to competitive binding on, and hence transport by, Cel-Pgp-1. This needs of course to be confirmed experimentally, but if so, it could be inferred as advantageous to administer a Cel-Pgp-1-mediated resistant worm a combination of a ML with one of these compounds.
This model can be the basis for establishing similar drug binding models on ABC transporters of parasitic nematodes showing sufficient sequence similarity with Cel-Pgp-1, in order to allow reliable structure rebuilding by homology modelling. This strategy could be powerful to test in silico the transporters capacities to exhibit either selective or multiple AH drug binding. In the C. elegans genome, 14 Pgp-related ABC genes show differential expression levels during the worm life cycle and various tissue localizations (Zhao et al., 2004) (Wormbase: http://www.wormbase.org/). The closest homologs of Cel-Pgp-1, expressed in the intestine, are the ABC full transporters of the sub-family B and its orthologs in other nematodes. For example, in H. contortus, if we restrict the computation of sequence identity to TMD1-TMD2 regions, we observe that Hco-Pgp-1 displays 63% sequence identity with Cel-Pgp-1, while the latter shares only between 22% (Cel-Pgp-11) and 48% (Cel-Pgp-9) of sequence identity with all other Cel-Pgp sequences of the same genome. Thus, Hco-Pgp-1 would be the most direct candidate in this parasite to benefit from a similar in silico approach for AH drugs docking on its structural model.
Therefore, based on the Cel-Pgp-1 structural model of multidrug binding, describing the set of interacting residues lining the inner chamber where ML bind specifically on a given nematode Pgp, will be pivotal for the screening or rational design of optimized ligands with high affinity and selectivity. Such molecules would competitively inhibit the transport of AH drugs in nematodes and could be used to increase AH efficacy. For this, it will be important to check for the specificity of the designed inhibitor for nematode Pgps, to avoid toxic effects in hosts. This is especially important in some mammals, such as some collie dogs with defective Pgp transporters in the blood-brain barrier which are highly sensitive to ivermectin, due to excess of drug accumulation into the brain and subsequent neurotoxicity (Roulet et al., 2003, Schinkel et al., 1994). Such data would point to in vitro and in vivo experiments relevant for investigating the involvement of the ABC transporters in nematode drug transport and resistance, and they could provide a clue for finding specific efficient AH resistance reversing agents.
In conclusion, in this in silico work based on a recently released crystal structure of the nematode C. elegans ABC transporter Pgp-1, we showed for the first time that most of the AH drugs currently used in veterinary and human medicine to treat helminth infections can be substrates of Cel-Pgp-1. We clearly identified several hotspots for drug binding, as well as common motifs of different ML interacting with specific sub-sites of the binding pocket of Cel-Pgp-1, which can be targeted to design molecules that would inhibit ML binding to this transporter in the perspective of necessary high host/nematode selectivity. Such information on the key residues, in mammalian Pgp and in nematode Pgp, which interact with the different compounds, should also provide insights for the design of novel anthelmintic analogs which are non-ligands, thus unlikely to be subject to efflux-based mechanisms of drug resistance development in nematodes. The model that we have developed could hence generally be used to assess the possible interactions of new chemical entities with nematode Pgp. This model is a critical insight into the possible role of Pgp homologs in expelling AH drugs out of parasite nematodes, making them resistant to treatment. Furthermore, this is the necessary step to identify in combination with in silico-guided in vitro experiments, the ABC transporters (at least from the B sub-family) specifically responsible for AH resistance, in order to rationally design or select specific inhibitors that could be novel therapeutic weapons against AH resistant parasites.
Acknowledgements
We deeply acknowledge Chantal Lebrun and Nicolas Loiseau for valuable help with data analysis, presentation and comments on the manuscript. We also thank Fabien Jourdan and Clément Frainay for extensive technical support, and the GenoToul Bioinformatics hardware infrastructure that was used for computing. This work was supported by the Natural Sciences and Engineering Research Council of Canada, the FRQNT Centre for Host-Parasite Interactions, Quebec, and EMIDA ERA-NET project CARES n 11-EMID-003-02.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ijpddr.2016.09.001.
Contributor Information
François André, Email: francois.andre@cea.fr.
Anne Lespine, Email: anne.lespine@toulouse.inra.fr.
Appendix A. Supplementary data
The following is/are the supplementary data related to this article:
References
- Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P.M., Trinh Y.T., Zhang Q., Urbatsch I.L., Chang G. Structure of p-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardelli B.F., Prichard R.K. Inhibition of P-glycoprotein enhances sensitivity of Caenorhabditis elegans to ivermectin. Vet. Parasitol. 2013;191:264–275. doi: 10.1016/j.vetpar.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Bessadok A., Garcia E., Jacquet H., Martin S., Garrigues A., Loiseau N., Andre F., Orlowski S., Vivaudou M. Recognition of sulfonylurea receptor (ABCC8/9) ligands by the multidrug resistance transporter P-glycoprotein (ABCB1): functional similarities based on common structural features between two multispecific ABC proteins. J. Biol. Chem. 2011;286:3552–3569. doi: 10.1074/jbc.M110.155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bygarski E.E., Prichard R.K., Ardelli B.F. Resistance to the macrocyclic lactone moxidectin is mediated in part by membrane transporter P-glycoproteins: implications for control of drug resistant parasitic nematodes. Int. J. Parasitol. Drugs Drug Resist. 2014;4:143–151. doi: 10.1016/j.ijpddr.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully D.F., Vassilatis D.K., Liu K.K., Paress P.S., Van der Ploeg L.H., Schaeffer J.M., Arena J.P. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature. 1994;371:707–711. doi: 10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
- Domicevica L., Biggin P.C. Homology modelling of human P-glycoprotein. Biochem. Soc. Trans. 2015;43:952–958. doi: 10.1042/BST20150125. [DOI] [PubMed] [Google Scholar]
- Dupuy J., Alvinerie M., Menez C., Lespine A. Interaction of anthelmintic drugs with P-glycoprotein in recombinant LLC-PK1-mdr1a cells. Chem. Biol. Interact. 2010;186:280–286. doi: 10.1016/j.cbi.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Eckford P.D., Sharom F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T., Volm M. Reversal of doxorubicin-resistance in sarcoma 180 tumor cells by inhibition of different resistance mechanisms. Cancer Lett. 1993;70:197–202. doi: 10.1016/0304-3835(93)90231-w. [DOI] [PubMed] [Google Scholar]
- Elmshauser S., Straehle L.C., Kranz J., Krebber R., Geyer J. Brain penetration of emodepside is increased in P-glycoprotein-deficient mice and leads to neurotoxicosis. J. Vet. Pharmacol. Ther. 2014;38:74–79. doi: 10.1111/jvp.12149. [DOI] [PubMed] [Google Scholar]
- Forrester S.G., Prichard R.K., Dent J.A., Beech R.N. Haemonchus contortus: HcGluCla expressed in Xenopus oocytes forms a glutamate-gated ion channel that is activated by ibotenate and the antiparasitic drug ivermectin. Mol. Biochem. Parasitol. 2003;129:115–121. doi: 10.1016/s0166-6851(03)00102-6. [DOI] [PubMed] [Google Scholar]
- Godoy P., Che H., Beech R.N., Prichard R.K. Characterization of Haemonchus contortus P-glycoprotein-16 and its interaction with the macrocyclic lactone anthelmintics. Mol. Biochem. Parasitol. 2015;204:11–15. doi: 10.1016/j.molbiopara.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Godoy P., Che H., Beech R.N., Prichard R.K. Characterisation of P-glycoprotein-9.1 in Haemonchus contortus. Parasit. Vectors. 2016;9:52. doi: 10.1186/s13071-016-1317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy P., Lian J., Beech R.N., Prichard R.K. Haemonchus contortus P-glycoprotein-2: in situ localisation and characterisation of macrocyclic lactone transport. Int. J. Parasitol. 2015;45:85–93. doi: 10.1016/j.ijpara.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Haubertin D.Y., Madaoui H., Sanson A., Guerois R., Orlowski S. Molecular dynamics simulations of E. coli MsbA transmembrane domain: formation of a semipore structure. Biophys. J. 2006;91:2517–2531. doi: 10.1529/biophysj.106.084020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayeshi R., Masimirembwa C., Mukanganyama S., Ungell A.L. The potential inhibitory effect of antiparasitic drugs and natural products on P-glycoprotein mediated efflux. Eur. J. Pharm. Sci. 2006;29:70–81. doi: 10.1016/j.ejps.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Higgins C.F. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Jabbar A., Iqbal Z., Kerboeuf D., Muhammad G., Khan M.N., Afaq M. Anthelmintic resistance: the state of play revisited. Life Sci. 2006;79:2413–2431. doi: 10.1016/j.lfs.2006.08.010. [DOI] [PubMed] [Google Scholar]
- James C.E., Davey M.W. Increased expression of ABC transport proteins is associated with ivermectin resistance in the model nematode Caenorhabditis elegans. Int. J. Parasitol. 2009;39:213–220. doi: 10.1016/j.ijpara.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Janssen I.J., Krucken J., Demeler J., von Samson-Himmelstjerna G. Caenorhabditis elegans: modest increase of susceptibility to ivermectin in individual P-glycoprotein loss-of-function strains. Exp. Parasitol. 2013;134:171–177. doi: 10.1016/j.exppara.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Jin M.S., Oldham M.L., Zhang Q., Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.M., George A.M. Multidrug resistance in parasites: ABC transporters, P-glycoproteins and molecular modelling. Int. J. Parasitol. 2005;35:555–566. doi: 10.1016/j.ijpara.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Kaplan R.M. Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol. 2004;20:477–481. doi: 10.1016/j.pt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Kaschny M., Demeler J., Janssen I.J., Kuzmina T.A., Besognet B., Kanellos T., Kerboeuf D., von Samson-Himmelstjerna G., Krucken J. Macrocyclic lactones differ in interaction with recombinant P-glycoprotein 9 of the parasitic nematode Cylicocylus elongatus and ketoconazole in a yeast growth assay. PLoS Pathog. 2015;11:e1004781. doi: 10.1371/journal.ppat.1004781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerboeuf D., Guegnard F. Anthelmintics are substrates and activators of nematode P glycoprotein. Antimicrob. Agents Chemother. 2011;55:2224–2232. doi: 10.1128/AAC.01477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiki-Mvouaka S., Menez C., Borin C., Lyazrhi F., Foucaud-Vignault M., Dupuy J., Collet X., Alvinerie M., Lespine A. Role of P-glycoprotein in the disposition of macrocyclic lactones: a comparison between ivermectin, eprinomectin, and moxidectin in mice. Drug Metab. Dispos. 2010;38:573–580. doi: 10.1124/dmd.109.030700. [DOI] [PubMed] [Google Scholar]
- Koenderink J.B., Kavishe R.A., Rijpma S.R., Russel F.G. The ABCs of multidrug resistance in malaria. Trends Parasitol. 2010;26:440–446. doi: 10.1016/j.pt.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Lage H. ABC-transporters: implications on drug resistance from microorganisms to human cancers. Int. J. Antimicrob. Agents. 2003;22:188–199. doi: 10.1016/s0924-8579(03)00203-6. [DOI] [PubMed] [Google Scholar]
- Laing R., Hunt M., Protasio A.V., Saunders G., Mungall K., Laing S., Jackson F., Quail M., Beech R., Berriman M., Gilleard J.S. Annotation of two large contiguous regions from the Haemonchus contortus genome using RNA-seq and comparative analysis with Caenorhabditis elegans. PLoS One. 2011;6:e23216. doi: 10.1371/journal.pone.0023216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing R., Kikuchi T., Martinelli A., Tsai I.J., Beech R.N., Redman E., Holroyd N., Bartley D.J., Beasley H., Britton C., Curran D., Devaney E., Gilabert A., Hunt M., Jackson F., Johnston S.L., Kryukov I., Li K., Morrison A.A., Reid A.J., Sargison N., Saunders G.I., Wasmuth J.D., Wolstenholme A., Berriman M., Gilleard J.S., Cotton J.A. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 2013;14:R88. doi: 10.1186/gb-2013-14-8-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard G.D., Fojo T., Bates S.E. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- Lespine A., Martin S., Dupuy J., Roulet A., Pineau T., Orlowski S., Alvinerie M. Interaction of macrocyclic lactones with P-glycoprotein: structure-affinity relationship. Eur. J. Pharm. Sci. 2007;30:84–94. doi: 10.1016/j.ejps.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Lespine A., Menez C., Bourguinat C., Prichard R.K. P-glycoproteins and other multidrug resistance transporters in the pharmacology of anthelmintics: prospects for reversing transport-dependent anthelmintic resistance. Int. J. Parasitol. Drugs Drug Resist. 2012;2:58–75. doi: 10.1016/j.ijpddr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Jaimes K.F., Aller S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T.W., Bartlett M.C., Clarke D.M. Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem. J. 2006;396:537–545. doi: 10.1042/BJ20060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T.W., Bartlett M.C., Clarke D.M. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T.W., Clarke D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 2001;276:14972–14979. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- Loo T.W., Clarke D.M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- Mani T., Bourguinat C., Keller K., Ashraf S., Blagburn B., Prichard R.K. Interaction of macrocyclic lactones with a Dirofilaria immitis P-glycoprotein. Int. J. Parasitol. 2016;46:631–640. doi: 10.1016/j.ijpara.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Menez C., Alberich M., Kansoh D., Blanchard A., Lespine A. Acquired tolerance to ivermectin and moxidectin after drug selection pressure in the nematode Caenorhabditis elegans. Antimicrob. Agents Chemother. 2016;60:4809–4819. doi: 10.1128/AAC.00713-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Y., Nabhan J.F., Solomon J., Mackenzie C.D., Geary T.G. Ivermectin disrupts the function of the excretory-secretory apparatus in microfilariae of Brugia malayi. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20120–20125. doi: 10.1073/pnas.1011983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Atweneboana M.Y., Awadzi K., Attah S.K., Boakye D.A., Gyapong J.O., Prichard R.K. Phenotypic evidence of emerging ivermectin resistance in Onchocerca volvulus. PLoS Negl. Trop. Dis. 2011;5:e998. doi: 10.1371/journal.pntd.0000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Atweneboana M.Y., Eng J.K., Boakye D.A., Gyapong J.O., Prichard R.K. Prevalence and intensity of Onchocerca volvulus infection and efficacy of ivermectin in endemic communities in Ghana: a two-phase epidemiological study. Lancet. 2007;369:2021–2029. doi: 10.1016/S0140-6736(07)60942-8. [DOI] [PubMed] [Google Scholar]
- Prichard R., Menez C., Lespine A. Moxidectin and the avermectins: consanguinity but not identity. Int. J. Parasitol. Drugs Drug Resist. 2012;2:134–153. doi: 10.1016/j.ijpddr.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulet A., Puel O., Gesta S., Lepage J.F., Drag M., Soll M., Alvinerie M., Pineau T. MDR1-deficient genotype in Collie dogs hypersensitive to the P-glycoprotein substrate ivermectin. Eur. J. Pharmacol. 2003;460:85–91. doi: 10.1016/s0014-2999(02)02955-2. [DOI] [PubMed] [Google Scholar]
- Schinkel A.H. The physiological function of drug-transporting P-glycoproteins. Semin. Cancer Biol. 1997;8:161–170. doi: 10.1006/scbi.1997.0068. [DOI] [PubMed] [Google Scholar]
- Schinkel A.H., Smit J.J., van Tellingen O., Beijnen J.H., Wagenaar E., van Deemter L., Mol C.A., van der Valk M.A., Robanus-Maandag E.C., te Riele H.P. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- Seeger M.A., van Veen H.W. Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta. 2009;1794:725–737. doi: 10.1016/j.bbapap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Shilling R.A., Venter H., Velamakanni S., Bapna A., Woebking B., Shahi S., van Veen H.W. New light on multidrug binding by an ATP-binding-cassette transporter. Trends Pharmacol. Sci. 2006;27:195–203. doi: 10.1016/j.tips.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Sousa S.F., Fernandes P.A., Ramos M.J. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- Sousa S.F., Ribeiro A.J., Coimbra J.T., Neves R.P., Martins S.A., Moorthy N.S., Fernandes P.A., Ramos M.J. Protein-ligand docking in the new millennium–a retrospective of 10 years in the field. Curr. Med. Chem. 2013;20:2296–2314. doi: 10.2174/0929867311320180002. [DOI] [PubMed] [Google Scholar]
- Szewczyk P., Tao H., McGrath A.P., Villaluz M., Rees S.D., Lee S.C., Doshi R., Urbatsch I.L., Zhang Q., Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta crystallogr. Sect. D Biol. Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A., Reyes C.L., Yu J., Roth C.B., Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Urdaneta-Marquez L., Keller K., James C.E., Davey M.W., Prichard R.K. The role of several ABC transporter genes in ivermectin resistance in Caenorhabditis elegans. Vet. Parasitol. 2012;190:519–529. doi: 10.1016/j.vetpar.2012.06.038. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Sheps J.A., Ling V., Fang L.L., Baillie D.L. Expression analysis of ABC transporters reveals differential functions of tandemly duplicated genes in Caenorhabditis elegans. J. Mol. Biol. 2004;344:409–417. doi: 10.1016/j.jmb.2004.09.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.