

Abstract
Glial fibrillary acidic protein (GFAP), microtubule-associated protein tau, and amyloid β peptide (Aβ42) have been proposed as diagnostic and prognostic biomarkers in traumatic brain injury (TBI). Single molecule array (Simoa) is a novel technology that employs highly sensitive immunoassays for accurate measurements of candidate biomarkers found at low concentration in biological fluids. Our objective was to trace the trajectory of tau, GFAP, and Aβ42 levels in plasma from the acute through subacute stages after TBI, compared with controls. Samples from 34 TBI subjects enrolled in the Citicoline Brain Injury Treatment Trial (COBRIT) were studied. Injury severity was assessed by Glasgow Coma Scale (GCS) and admission CT. Glasgow Outcome Scale Extended (GOSE) was assessed 6 months after injury. Plasma was collected within 24 h (Day 0), and 30 and 90 days after the TBI. Plasma collected from 69 healthy volunteers was used for comparison. At every time point, increases were noted in plasma GFAP (p < 0.0001 for all comparisons), tau (p < 0.0001, p < 0.0001, and p = 0.0044, at Days 0, 30, and 90, respectively), and Aβ42 (p < 0.001, p < 0.0001, and p = 0.0203, respectively) in TBI cases compared with controls. The levels were maximal at Day 0 for GFAP and tau and at Day 30 for Aβ42. Area under curve (AUC) analyses for Day 0 GFAP and tau were excellent for discrimination of complicated mild TBI (cmTBI) from controls (0.936 and 0.901, correspondingly). Discriminant component analysis (DCA) for all three biomarkers at Days 0 and 30 differentiated controls from cmTBI (91.1% and 89.7% correctly classified, at each time point). Duration of post-traumatic amnesia (PTA) correlated weakly with tau levels at 30 days (Spearman's r = 0.40; 95% CI 0.0003–0.60, p = 0.044). The Marshall CT Grade on admission correlated weakly with Day 30 tau levels (Spearman's r = 0.41; 95% CI 0.04–0.68, p = 0.027). Day 30 Aβ42 correlated with GOSE (standardized β −0.486, p = 0.042). GFAP, tau and Aβ42 were increased up to 90 days after TBI compared with controls. Total tau levels correlated with clinical and radiological variables of TBI severity. Plasma Aβ42 correlated with clinical outcome. Combination of all three biomarkers at Days 0 and 30 can be used to differentiate controls from cmTBI populations, and may be useful as biomarkers of TBI in both acute and subacute phases.
Keywords: : diagnostic and prognostic biomarkers, GFAP, highly sensitive immunoassays, TBI, total tau
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability in people <45 years of age in industrialized countries.1 Biomarkers have historically been critical to advances in a broad range of clinical conditions.2 Diagnostic and therapeutic breakthroughs in fields as diverse as cardiology and oncology have relied on quantitative biomarkers that are reliable indicators of the underlying pathology as well as predictors of outcome.3,4 The absence of validated biomarkers in the TBI field is a major factor limiting our understanding of the natural history and the long-term effects of TBI, as well as a barrier to TBI drug development.5 Additionally, most of the work performed to date has focused on the first few days after injury,6–8 and much less is known about biomarkers during the subacute (8–90 days) and chronic (> 90 days) periods after TBI,9 further hindering diagnostic and therapeutic development in the later stages of the disease. Because TBI exposure has been associated with long-term neurodegeneration, the late effects of TBI are an important and insufficiently studied issue.
Glial fibrillary acid protein (GFAP),10,11 microtubule-associated protein tau,12–14 and amyloid β peptide (particularly the 1–42 fragment, Aβ42)12 have been proposed as promising diagnostic and prognostic biomarkers in TBI. GFAP is a structural protein expressed almost exclusively in astrocytes and released upon disintegration of the cytoskeleton.15 GFAP has been widely studied in TBI, and elevated levels in plasma show promise as a diagnostic and prognostic biomarker.10,11 In moderate and severe TBI, GFAP levels are elevated in cerebrospinal fluid (CSF) and serum, particularly in patients who experienced an unfavorable outcome.16 In addition, GFAP had excellent sensitivity to discriminate TBI patients from controls (with area under curve [AUC] 0.87) in the Transforming Research and Clinical Knowledge in TBI (TRACK-TBI) study in which 85% of the patient cohort had mild TBI.11 It is of note that superior sensitivity and specificity for diagnosing TBI were obtained when GFAP was combined with ubiquitin terminal C - hydrolase 1 (UCH-L1) (AUC 0.94), thus supporting that combination of biomarkers may be supportive for diagnosis compared with each alone for diagnosis and prognosis of TBI.11 However, current assays for GFAP in blood are suboptimal,17 as up to one quarter of TBI patients had GFAP levels below the lower limit of detection (LLOD).11
Tau is a microtubule-associated protein that acts as a structural element in the axonal cytoskeleton and is linked to axonal damage.13,18 Most widely used assays for tau are adequate for CSF, but not sensitive enough to reliably detect low concentrations found in blood.12 Increases of tau were observed in CSF after severe TBI, peaking 1 week after the brain trauma.12 Acute increases of tau in CSF of severe TBI correlated with clinical outcome.19 Studies using the highly sensitive single molecule array (Simoa) assay indicate that sports-related TBIs are also associated with increases in tau in plasma.8,20
Increased Aβ42 is associated with long-term neurodegenerative conditions, such as Alzheimer's disease (AD).21 Even a single TBI may be associated with aβ42 deposits in the postmortem brains of short-term (6 h to 7 days) or long-term (1–47 years) TBI survivors.22,23 Further, positron emission tomography studies using carbon 11–labeled Pittsburgh Compound B ([11C]PiB) showed increased amyloid deposition following acute TBI compared to controls.24
Simoa is a novel technology that employs highly sensitive immunoassays and allows accurate measurements of candidate biomarkers found at low concentrations in blood.25 Simoa detected increases of serum tau and Aβ42 after hypoxic brain injury.26,27 Plasma tau was increased in hockey players after concussion,8 and in military personnel who sustained a TBI.28 There was a correlation between tau and severity of post-concussive symptoms (r = 0.37; p = 0.003).28 However, to date, the validity and reliability of a combination of plasma levels of GFAP, tau and Aβ42 in TBI subjects using the ultrasensitive Simoa technology have never been assessed, particularly during the subacute period.
The Citicoline Brain Injury Treatment Trial (COBRIT), a phase 3, double-blind randomized clinical trial conducted between July 20, 2007, and February 4, 2011, enrolled 1213 patients at eight United States level 1 trauma centers, to compare the effects of citicoline versus placebo in patients with complicated mild, moderate, or severe TBI. Extensive clinical and imaging data were collected while participants were hospitalized, and functional and cognitive status was assessed at 90 and 180 days after injury. There was no benefit from citicoline treatment noted in the COBRIT trial.29 A subset of the COBRIT TBI participants enrolled at a single site donated blood for future biomarker research, and these well-characterized samples were tested in this study.
We hypothesized that:
1. Plasma levels of GFAP, tau, and Aβ42 are elevated after TBI compared with controls.
2. Plasma levels of tau and Aβ42 remain elevated for several weeks after TBI, whereas GFAP levels decline.
3. Elevations of GFAP, tau, and Aβ42 are associated with injury severity.
4. Elevations of GFAP, tau, and Aβ42 are associated with clinical outcome 6 months after injury.
Methods
Study population
A total of 34 TBI subjects enrolled in the COBRIT at the University of Texas Southwestern Medical Center were included in the study. All participants or their legally authorized representatives (LAR) gave informed consent for the use of the participant's blood samples and clinical data in TBI research. If an LAR provided consent originally, the participant directly provided consent for continued involvement upon recovery of decision-making capacity. Baseline CT scans, vital signs, medical history, demographics, and injury information were obtained and recorded as described elsewhere.29
Non-fasting blood samples were collected from COBRIT participants within 24 h of injury and again at 30 and 90 days after injury. Blood was collected into plastic dipotassium ethylenediaminetetraacetic acid (EDTA) tubes, immediately placed on ice, centrifuged (15 min, 2000g, room temperature) and frozen in aliquots within 1 h after collection. Samples were stored at −80°C until tested.
Blood samples of controls without TBI were processed and stored identically. One set of control samples (n = 19) was collected at the National Institutes of Health (NIH) Clinical Center under a clinical protocol designed to recruit healthy individuals. All research participants signed informed consent before participating in the study. TBI was excluded by questionnaire, as well as severe psychiatric conditions, congestive heart failure, active cancer, sleep disorder, pregnancy or lactation in women, uncontrolled diabetes, drug and/or addiction abuse or dependence, and medication with diuretics, beta blockers, or narcotics. A second set of control samples (n = 50) was purchased from a commercially available source (Bioreclamation) when controls were screened by a questionnaire and were determined to be free from hepatitis B and C and from HIV infections. There was no difference in age, race, or gender distribution between the two sets of controls. There were no significant differences in biomarker levels between the two sources of control samples. There was no gender difference in biomarkers levels in the TBI or the control group.
Immunoassays
GFAP, tau, and Aβ42 concentrations in plasma samples were measured with a digital array technology (Quanterix Corporation, Lexington, MA), which uses a single-molecule enzyme-linked immunoarray method previously described.30 Simoa25 is a novel technology that employs highly sensitive immunoassays with LLOD ≤100 fg/mL. The lowest limit of quantification (LLOQ), LLOD, intra-assay coefficient of variation (%CV) and inter assay %CV for GFAP, tau, and Aβ42 measured by Simoa are shown in Table 1.
Table 1.
The Lowest Limit of Quantification (LLOQ) and the Lowest Limit of Determination (LLOD), Intra-assay Coefficient of Variation (%CV), and Inter-assay %CV for Glial Fibrillary Acidic Protein (GFAP), Tau and Aβ42 Measured by Simoa
| Assays | LLODa, pg/mL | LLOQa, pg/mL | Intra-assay %CV | Inter-assay %CV |
|---|---|---|---|---|
| GFAP | 0.8 | 1.6 | 14% | 12% |
| Tau | 0.068 | 0.104 | 6% | 5% |
| Aβ42 | 0.176 | 0.548 | 14% | 2% |
Values are corrected for pre-dilutions (4 × ).
Each plate of samples tested included an eight point calibration curve (run in triplicate), two controls (run in duplicate), and plasma samples (run in duplicate) with the tau, GFAP, and Aβ42 assays. Samples were diluted offline manually using appropriate sample diluents and preferred dilution factors according to the product inserts for each assay following the recommended procedures. The laboratory scientists who undertook the analyses were blinded to the participant groups, and there was no difference in the distribution of cases and controls on the Simoa plates. Data analysis showed that 14% of GFAP of TBI and 70% of control samples were below LLOD. However, only 1 out of 34 of TBI samples was below LLOD on Day 0. Importantly, all data for tau and Aβ42 in controls and in TBI subjects were above the LLODs.
Evaluation of CT scans
All patients underwent CT imaging of the brain at the time of initial presentation to the Emergency Department (ED). Each patient's head CT was characterized using the recommendation of the TBI – Common Data Element (CDE) Neuroimaging working group including Marshall Grade.31 Each CT was de-identified, and reviewed by a blinded reader, who had been qualified to read CT CDEs through use of a training and evaluation set. Imaging features were extracted in the database for the analysis.
Outcomes
The primary outcome measure of COBRIT was the 180 day Glasgow Outcome Scale Extended score (GOSE).32 The GOSE provides eight categories of outcome (1–8): Dead, Vegetative State, Lower Severe Disability, Upper Severe Disability, Lower Moderate Disability, Upper Moderate Disability, Lower Good Recovery, and Upper Good Recovery. Ratings are based on patient consciousness, social relationships, and other sequela of TBI. An Upper Good Recovery score (8) indicates return to pre-injury baseline with no residual effects from the TBI.
Statistical analysis
Descriptive statistics with means and proportions were used to describe categorical variables. Biomarker levels were treated as continuous data. The Shapiro–Wilk test was used for testing normality of the distribution. The Wilcoxon rank test or the Mann–Whitney test was used for continuous variables. Intracranial lesions shown on initial CT scans were scored and analyzed as categorical variables. We assessed the ability of each biomarker level to separate patients with complicated mild TBI (cmTBI) from controls. This was quantified by receiver operating characteristic (ROC) and AUC. In line with current statistical consensus, AUC of 0.8–0.9 is considered very good, AUC of 0.7–0.8 is considered adequate, and AUC <0.07 is considered poor. Spearman correlation and linear regression were used to test the association of variables. The Friedman test was used in a subgroup of patents for whom data at all three time points were available, to compare paired biomarker levels. Discriminant component analysis (DCA) was used to discriminate cmTBI from controls. Results with two tailed p < 0.05 were considered significant. P values were not corrected for multiple comparisons. Statistical analysis was conducted using GraphPad Prism (v. 6.02) (Graph Pad Software, San Diego, CA) or with Statistical Package for the Social Sciences SPSS (version 22, IBM Corporation).
Results
Demographic and clinical characteristics
The majority (67%) of the COBRIT trial participants were diagnosed with cmTBI (GCS in the ED 13–15, with evidence of traumatic intracranial injury on CT scan) with the remainder diagnosed with moderate to severe TBI (msTBI) (GCS in the ED 3–12). Median Marshall Grade of TBI severity on admission CT scan was 3 and it ranged from 2 to 5. Table 2 summarizes the demographic information on the participants whose samples were tested in this study (85% of TBI subjects were male). The cmTBI participants were somewhat older and had shorter mean duration of post-traumatic amnesia (PTA) than that which was recorded for the cohort of msTBI Table 2). Median PTA was 4 days in the mTBI group, indicating that many of these patients would be classified as having moderate TBI (modTBI) using alternate TBI classification schemes. Approximately one half of the cmTBI patients recovered to a GOSE of 8 by 6 months. There was no difference between controls and TBI subjects with respect to age, but the fraction of females was significantly higher in controls than in TBI subjects (49% vs. 15%).
Table 2.
Demographics of the Study Subjects
| TBI patients (n = 34) | Complicated mTBI GCS 13–15 (n = 21) | Moderate-severe TBI GCS 3–12 (n = 13) | Controls (n = 69) | |
|---|---|---|---|---|
| Age, yrs. median (IQR) | 39 (23–52) | 46 (29–58) | 24 (21–40) | 45 (31–52) |
| Gender (% male) | 85% male | 85% male | 85% male | 51% male |
| PTA, days (median, IQR) | 7 (1–22) | 4 (0–7) | 58 (8–145) | n/a |
| GCS (median, IQR) | 13 (3–15) | 15 (14–15) | 3 (3–3) | n/a |
| GOSE 180 days (median, IQR) | 7 (3–8) | 8 (5–8) | 5 (3–7) | n/a |
TBI, traumatic brain injury; mTBI, mild TBI; GCS, Glasgow Coma Scale; IQR, interquartile range; PTA, post-traumatic amnesia; GOSE, Glasgow Outcome Scale Extended.
Temporal profiles of GFAP, tau, and Aβ42 in TBI cohort compared with controls
Figure 1 shows the results for GFAP (Fig. 1a), tau (Fig. 1b), and Aβ42 (Fig. 1c) for the controls as well as for the TBI patients on the day of injury and 30 and 90 days later. GFAP was increased at Day 0 (median 17.60 pg/mL; interquartile range [IQR] 3.88–129.6 pg/mL), Day 30 (1.330 [0.8–2.21] pg/mL) and Day 90 (1.350 [0.8870–2.280] pg/mL) after TBI compared with controls (0.80 [0.8–1.070] pg/mL), (p < 0.0001 for all comparisons).
FIG. 1.

Median and interquartile rate for glial fibrillary acidic protein (GFAP) (A), tau (B), and amyloid β peptide (Aβ42) (C) for controls and for traumatic brain injury (TBI) patients at Days 0, 30, and 90. Color image is available online at www.liebertpub.com/neu
Tau was similarly increased at Day 0 (9.560 [5.895–17.10] pg/mL), Day 30 (6.665 [4.705–9.163] pg/mL), and Day 90 (5.720 [3.850–7.180] pg/mL) compared with controls (4.340 [2.745–5.128] pg/mL), (p < 0.0001, p < 0.0001, and p = 0.0044, respectively). Aβ42 was increased at Day 0 (8.3 [3.3–14] pg/mL), Day 30 (15.80 [8.67–24] pg/mL), and Day 90 (11.99 [2.7–22.3] pg/mL) compared with controls (4.1 [2.3–5.6] pg/mL), (p < 0.001, p < 0.0001, and p < 0.0203, respectively).
ROC analysis performed for each biomarker at the three time points, compared with the uninjured controls revealed excellent sensitivity and specificity for discrimination between uninjured controls and cmTBI cases at Day 0 for both GFAP and tau (AUC 0.9360 and 0.9013, correspondingly). At the later time points, the plasma level for GFAP and tau decreased; however, it remained moderately elevated at days 30 and 90 after injury, and their ability to discriminate cmTBI from controls became lower. In contrast, Aβ42 poorly discriminated between TBI subjects and controls at days 0 and 90, but provided moderate to excellent discrimination at Day 30 (AUC 0.8362), (Table 3).
Table 3.
AUC for ROC Analysis for Plasma GFAP, Tau, and Aβ42 in Complicated Mild TBI at Three Time Points After the Injury versus Controls
| Biomarker | Day 0 | Day 30 | Day 90 |
|---|---|---|---|
| GFAP | 0.9360** | 0.6425 | 0.7482^ |
| Tau | 0.9013** | 0.8225** | 0.6671 |
| Aβ42 | 0.6887^ | 0.8362* | 0.5543 |
p < 0.0001, *p < 0.001, ^p < 0.05.
AUC, area under curve; ROC, receiver operating characteristic; GFAP, glial fibrillary acidic protein; Aβ42, amyloid β peptide; TBI, traumatic brain injury.
Analysis of temporal biomarker levels of a subset (n = 21 for tau and GFAP and n = 13 for Aβ42) of patients who had data available at all three time points revealed that GFAP levels were the highest within 24 h of injury (Fig. 2a), and in almost all cases, decreased by 30 and 90 days, while still being elevated compared with the controls. The Friedman test revealed that the difference among the three time points for GFAP was significant (p < 0.0001). On the other hand, tau levels decreased in some patients, but were stable or even increased in others at the later testing dates. The Friedman test showed a difference (p < 0.0001) for tau levels among the three time points. Importantly, Aβ42 levels increased over the 1st month after injury (Fig. 2c), and then remained elevated in a number of subjects. The Friedman test did not reach statistical significance of Aβ42 (p = 0.292).
FIG. 2.
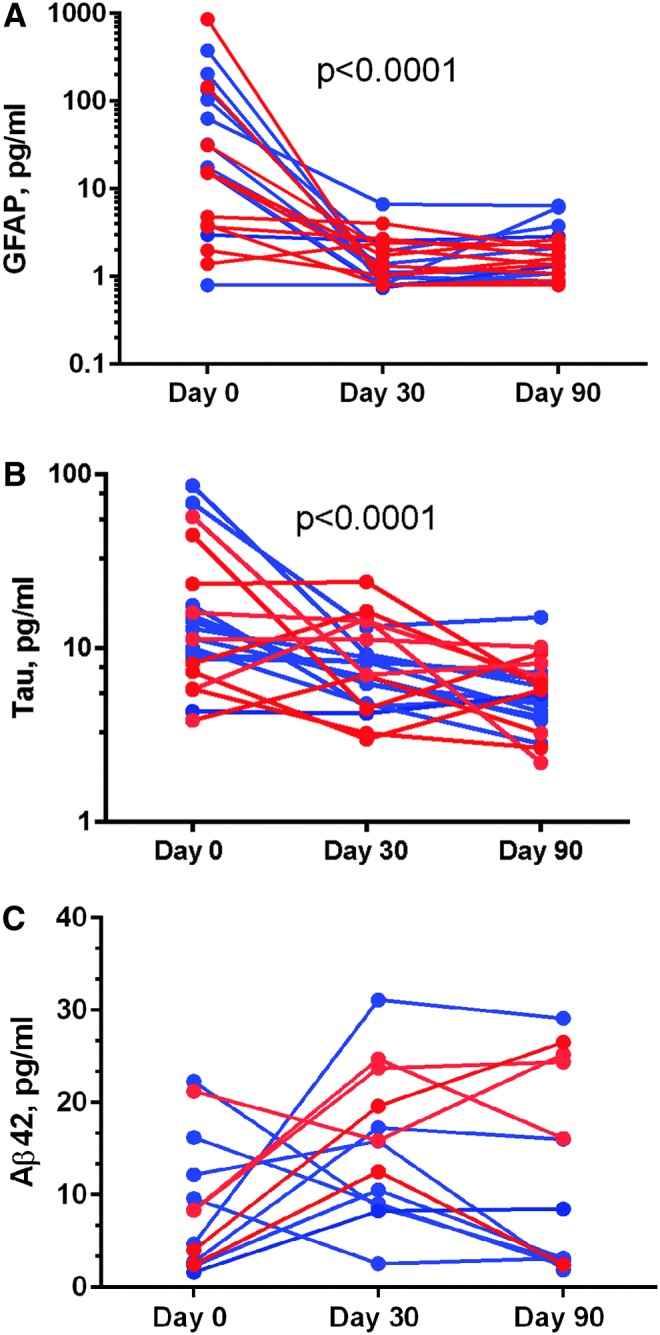
Temporal profiles (red represents complicated mild traumatic brain injury [TBI] patients and blue represents moderate to severe TBI for glial fibrillary acidic protein (GFAP) (A), tau (B), and amyloid β peptide (Aβ42) (C) at Days 0, 30, and 90. The Friedman test showed that biomarker levels changed significantly over 90 days for GFAP and tau (p < 0.0001) but not for Ab42 (p = 0.292). Aβ42 levels showed bimodal distribution at Day 90. Color image is available online at www.liebertpub.com/neu
Similar temporal biomarker profiles were observed when GFAP, tau, and Aβ42 levels were analyzed separately for complicated mild and moderate TBI cases for which three time point data were available (GFAP and tau [10 cmTBI and 11 modTBI] and Aβ42 [5 cmTBI and 8 modTBI]). There as a difference among three time points for GFAP for cmTBI (p < 0.0002). There was no difference among three time points for modTBI (p < 0.06). No differences in temporal profiles for all three time points in Aβ42 levels in cmTBI or modTBI cases were found.
There were no differences in plasma levels of GFAP or Aβ42 among participants by TBI severity (cmTBI compared with moderate to severe) at any testing time (days 0, 30, or 90). There was, however, a difference in the trajectory of plasma tau at Day 30 (Fig. 3). For cmTBI subjects, plasma tau levels declined between days 0 and 30, and remained low at Day 90, whereas for moderate or severe TBI subjects, tau remained equally elevated at Days 0 and 30 and only declined modestly by Day 90. There was no difference in the trajectory of plasma Aβ42 levels by TBI injury severity grade.
FIG. 3.
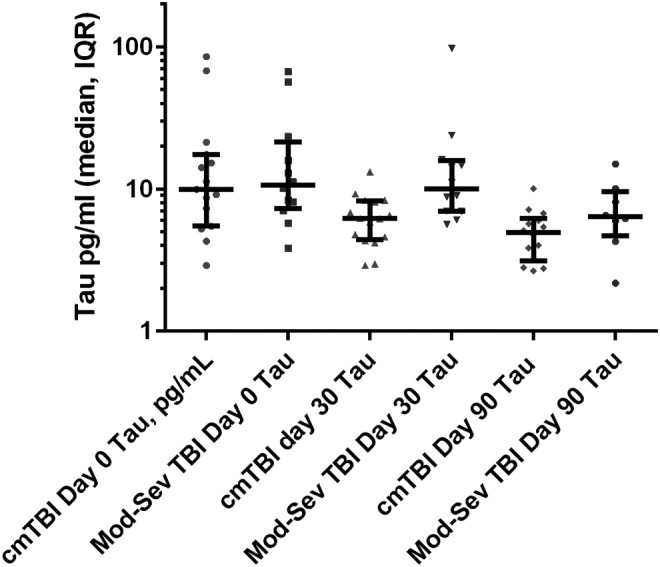
Trajectory of plasma tau levels for complicated mild traumatic brain injury (TBI) (Glasgow Coma Scale [GCS] score 13–15 with abnormal CT), compared with moderate to severe TBI (GCS score 3–12) on 0, 30, and 90 days after injury. The difference between groups for day 30 tau was significant (Mann–Whitney test, p = 0.0031).
Correlations of GFAP, tau, and Aβ42 with clinical and radiological measures of TBI severity
There was no relationship between GFAP and tau levels at any time point and functional outcome 180 days after injury as measured by GOSE. However, there was a relationship between Day 30 Aβ42 levels and GOSE at 180 days, after adjustment for age (standardized β −0.486, p = 0.042).
PTA, a recognized measure of injury severity, correlated with plasma tau levels at Day 30 (Spearman's r = 0.40; 95% CI 0.0003–0.60; p = 0.044). Tau levels at Day 30 also weakly correlated with injury severity as measured radiologically. Spearman's correlation coefficient between Day 30 tau levels and the Marshall CT grade on admission was 0.41 (95% CI 0.04–0.68, p = 0.027). However, tau levels at Day 0 or 90 were not correlated to either measure of injury severity. There was no relationship between GFAP or Aβ42 levels and Marshall CT grade or PTA duration at any of the time points evaluated.
GFAP and tau levels were moderately correlated (Spearman's r = 0.526; 95% CI 0.170 – 0.760; p = 0.005). There was no correlation between GFAP and Aβ42 levels or between tau and Aβ42 levels.
DCA for all three biomarkers at all three time points significantly differentiated controls from the cmTBI cohort. Wilks λ, representing a multivariate test of significance, was significant for all three time points; however, it was better for Day 0 and Day 30 biomarker combinations. Particularly, DCA for Day 0 and Day 30 combination of GFAP, tau, and Aβ42 showed excellent discrimination between cmTBI and controls (Table 4).
Table 4.
DCA of Combined Plasma GFAP, Tau, and Aβ42 at Three Time Points After the Injury Showing Ability to Discriminate Complicated Mild TBI from Controls
| % Correctly classified | Canonical correlation | Wilks' lambda | Chi square | p | |
|---|---|---|---|---|---|
| Day 0 | 91.1% | 0.576 | 0.668 | 21.184 | <0.0001 |
| Day 30 | 89.7% | 0.740 | 0.453 | 43.216 | <0.0001 |
| Day 90 | 85.5% | 0.534 | 0.715 | 17.255 | 0.001 |
DCA, discriminant component analysis; GFAP, glial fibrillary acidic protein; Aβ42, amyloid β peptide; TBI, traumatic brain injury.
Discussion
Our study shows that the ultrasensitive Simoa assay provides measurable values for GFAP in most of the TBI cases. Previous studies demonstrated increased GFAP in the acute period (1–24 h) after TBI, 10,11,16 but up to 23% of acute TBI had levels below LLOD compared with only 3% in this study. For example, in a study of 79 moderately and severely injured TBI subjects, GFAP levels were increased, serving as a predictor of death (GOSE 1) or an unfavorable outcome (GOSE 1–4).16 In a study of predominantly mTBI, GFAP levels were able to differentiate patients with TBI from uninjured controls with a high level of sensitivity and specificity (AUC of 0.90). Further, GFAP was able to discriminate TBI severity, between those with intracranial lesions on CT scanning and those without CT lesions (AUC 0.79).33 Similarly, in our study using Simoa methodology, we noted excellent discrimination of cmTBI from controls by GFAP at Day 1 (AUC 0.936). Further, our study is the first to show that even though GFAP levels decrease over the first several weeks after injury, they remain elevated compared with uninjured controls for up to 3 months in a subset of patients. These findings overall indicate that GFAP may have value as a brain-specific diagnostic biomarker in the subacute period after TBI.
Standard immunoassays for peripheral tau, because of their relatively high LLOD, are insufficiently sensitive to intracranial TBI pathology, including the inability to differentiate controls from those with cmTBI.28,34 More recent publications using the more sensitive technology, Simoa, have demonstrated elevations of plasma tau after hypoxic-ischemic brain injury and sports concussion.8,20 A recent study in boxers showed increased plasma tau immediately after a bout with head blows, but no loss of consciousness (LOC) followed by decrease in plasma tau to the pre-bout level after rest.20 A most recent TBI study using Simoa showed increased plasma tau in service members who had sustained a TBI during military deployment compared with controls.28 In this study, total tau was significantly increased in the TBI group (mean level, 1.13 pg/mL), compared with the controls (0.63 pg/mL), and increased levels were associated with increasing severity of the initial brain injury, increasing numbers of TBIs, and increasing severity of post-concussive symptoms.28 Our results confirm those earlier findings, and extend them by demonstrating that in cmTBI, tau levels remained elevated up to 90 days after TBI. These results support the hypothesis that plasma tau may be useful as a diagnostic biomarker in the subacute period, and may also find utility as a prognostic biomarker, because elevation at 90 days is associated with increased TBI symptom burden. Further, as anti-tau therapies are developed, plasma tau may be useful as a predictive and pharmacodynamic biomarker to select those likely to respond to the therapy and as a marker of therapy response.
Aβ42 (amyloid β 1–42) is a peptide produced by proteolytic cleavage of amyloid precursor protein (APP) and is deposited in plaques in AD.35 There is an association between decreased CSF Aβ42 and autosomal dominant AD.36 In previous studies, CSF Aβ42 increased after msTBI, with a peak on Day 6 after the injury,37 but without a detectable increase in peripheral blood.12,38 Similar findings were recently reported by another group using Simoa,39 when measurable Aβ42 levels were detected in plasma of severe TBI subjects and in controls (17.02 [14.75–28.59] pg/mL and 7.289 [6.126–8.668] pg/mL), and also in CSF.39 The study showed an increase of Aβ42 levels in plasma after TBI compared with controls (p < 0.001), with concomitant decrease of Aβ42 in CSF in TBI subjects compared with controls.39 The study also showed persistently reduced levels of CSF Aβ42 and elevated levels of plasma A4β2 over time, supporting that Aβ42 reflects pathophysiological processes after TBI.39 Our study supports this observation and extends Aβ42 increase to the subacute and chronic phase after TBI.
Compared with GFAP and tau temporal profiles in our TBI cohort, our results demonstrated a distinct temporal pattern for Aβ42 peptide. Although the levels are elevated compared with uninjured controls in the 1st day after injury, in a number of subjects there is an increase in circulating peptide levels over the first several months after injury. There is apparently a bimodal distribution (Fig. 1c) for plasma Aβ42 after TBI, indicating that some but not all patients may show a deficit in amyloid clearance pathways after TBI and potentially may be at risk for long-term TBI-related neurodegeneration. Notably, similar bimodal distribution was noted for patients with cmTBI and msTBI, indicating that elevation of Aβ42 is not primarily dependent upon the severity of TBI. It is interesting that prolonged elevation of peripheral Aβ42 was noted in a subset of patients with hypoxia after cardiac arrest, and that this was correlated with unfavorable outcome.27 Experimental data indicate that TBI in triple transgenic AD model mice causes accumulation and aggregation of Aβ oligomers in the brain, which may contribute to developing AD later in life.40 Genetic factors, such as presence of an ApoE4 allele44 or neprilysin gene transfer (GT) repeat polymorphism45 may potentially explain the bimodal distribution of Aβ42 after TBI. Testing such a hypothesis will require a larger sample size, as the number of patients in our cohort was too small to allow adequately powered allelic association studies. Further, extracerebral production of A4β2 by platelets may contribute to increase of plasma Aβ42.41
TBI is a pleotropic condition with multiple mechanisms of secondary injury and recovery. As a result, a single biomarker is unlikely to reflect the extent and diversity of neural tissue damage, aid in monitoring mechanism-directed therapies, or reliably forecast outcomes. Accordingly, the different trajectories observed among our three biomarkers may reflect different biological processes triggered by TBI, and suggest the potential for assessing multiple biomarkers at several specified time points to adequately reflect potential inter-individual differences in pathophysiology and associated biomarker elevation profiles.11
In this study, TBI severity was determined basing on clinical (GCS, LOC, and PTA) and CT imaging variables. It is widely accepted that assessment of TBI severity based on GCS, LOC, and PTA is imprecise.42 Further, scoring of GCS may be compromised in patients who are intubated and sedated, and in an acute setting has lower inter-rater reliability.43 These factors may explain why we did not observe differences in tau levels by injury severity. However, our study showed a relationship of tau levels with injury severity by demonstrating a correlation between duration of PTA and Day 30 tau, and between injury severity by Marshall Grade and tau levels at Day 30.
Our study has several limitations. The most important is the small sample size, which made it impossible to assess the contribution of confounders such as age, gender, injury mechanism, and their effects on recovery after TBI. Previous studies showed increased GFAP in modTBI compared with mTBI.11 This study was based on the larger sample (206 TBI patients) with the majority (83%, 170) being mTBI patients, and 43% (73 patients) diagnosed with cmTBI, which is comparable to our cmTBI cohort (67%, 46 patients). The number of moderate and severe TBI subgroups in TRACK TBI was lower than in our study: 17% (35 TBI) versus 33% (24 TBI) patients. Second, even the mTBI patients in our study were “complicated mTBI” meaning that they had intracranial trauma-related abnormalities noted on CT scan, whereas most mTBI patients in TRACK-TBI had normal CT scans.11 The sample size in our study was substantially smaller than in TRACK-TBI study; therefore, we have less power to detect statistically significant but small differences, which is relevant to later time points as well. Because only CT imaging was available for these subjects, precise characterization of important injury mechanisms such as diffuse axonal injury and diffuse vascular injury, and their association with individual plasma biomarkers, was also not possible. Finally, our study focuses on only three biomarkers, and it is likely that additional biomarkers will be needed to fully characterize a complex injury such as TBI.
Conclusions
Using the ultrasensitive Simoa technology, plasma levels of GFAP, tau, and Aβ42 are all increased up to 90 days after TBI compared with controls. The levels are maximal at Day 0 for GFAP and tau and at Day 30 for Aβ42. Total tau levels at Day 30 correlated with clinical and radiological variables of TBI severity. Day 30 plasma Aβ42 correlated with clinical outcome. Combination of all three biomarkers at Day 0 and Day 30 can be used to differentiate between controls and cmTBI populations. Tau, GFAP, and Aβ42 measured by Simoa may be useful as biomarkers of TBI in both the acute and subacute phases.
Acknowledgments
This work was supported by the Center for Neuroscience and Regenerative Medicine (CNRM), GE/NFL TBI Award, U01 HD 42652.
Author Disclosure Statement
D. Wilson, Y. Chen, D. Hanlon, A. Jeromin, and L. Song are employed by Quanterix, Inc. Other authors do not report competing financial interests.
References
- 1.Corso P., Finkelstein E., Miller T., Fiebelkorn I., and Zaloshnja E. (2006). Incidence and lifetime costs of injuries in the United States. Inj. Prev. 12, 212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lesko L.J., and Atkinson A.J., Jr (2001). Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu. Rev. Pharmacol. Toxicol. 41, 347–366 [DOI] [PubMed] [Google Scholar]
- 3.Izumi K., Lin W.J., Miyamoto H., et al. (2014). Outcomes and predictive factors of prostate cancer patients with extremely high prostate-specific antigen level. J. Cancer Res. Clin. Oncol. 140, 1413–1419 [DOI] [PubMed] [Google Scholar]
- 4.Wang T.J., Gona P, Larson M.G., Tofler G.H., Levy D., Newton-Cheh C., Jacques P.F., Rifai N., Selhub J., Robins S.J., Benjamin E.J., D'Agostino R.B., and Vasan R.S. (2006). Multiple biomarkers for the prediction of first major cardiovascular events and death. N. Engl. J. Med. 355, 2631–2639 [DOI] [PubMed] [Google Scholar]
- 5.Strathmann F.G., Schulte S., Goerl K., and Petron D.J. (2014). Blood-based biomarkers for traumatic brain injury: evaluation of research approaches, available methods and potential utility from the clinician and clinical laboratory perspectives. Clin. Biochem. 47:876–888 [DOI] [PubMed] [Google Scholar]
- 6.Berger R.P., Hayes R.L., Richichi R., Beers S.R., and Wang K.K. (2012). Serum concentrations of ubiquitin C-terminal hydrolase-L1 and alphaII-spectrin breakdown product 145 kDa correlate with outcome after pediatric TBI. J. Neurotrauma 29, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatson J.W., Barillas J., Hynan L.S., Diaz–Arrastia R., Wolf S.E., and Minei J.P. (2014). Detection of neurofilament–H in serum as a diagnostic tool to predict injury severity in patients who have suffered mild traumatic brain injury. J. Neurosurg. 121, 1232–1238 [DOI] [PubMed] [Google Scholar]
- 8.Shahim P., Tegner Y., Wilson D.H., Randall J., Skillbäck T., Pazooki D., Kallberg B., Blennow K., and Zetterberg H. (2014). Blood biomarkers for brain injury in concussed professional ice hockey players. J.A.M.A. Neurol. 71, 684–692 [DOI] [PubMed] [Google Scholar]
- 9.Management of Concussion/m TBI Working Group (2009). VA/DoD Clinical Practice Guideline for Management of Concussion/Mild Traumatic Brain Injury. J. Rehabil. Res. Dev. 46, CP1–68 [PubMed] [Google Scholar]
- 10.Bohmer A.E., Oses J.P., Schmidt A.P., et al. (2011). Neuron-specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury. Neurosurgery 68, 1624–1630 [DOI] [PubMed] [Google Scholar]
- 11.Diaz–Arrastia R., Wang K.K., Papa L., et al. (2014). Acute biomarkers of traumatic brain injury: relationship between plasma levels of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein. J. Neurotrauma 31, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franz G., Beer R., Kampfl A., Engelhardt K., Schmutzhard E., Ulmer H., and Deisenhammer F. (2003). Amyloid beta 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology 60, 1457–1461 [DOI] [PubMed] [Google Scholar]
- 13.Gabbita S.P., Scheff S.W., Menard R.M., Roberts K., Fugaccia I., and Zemlan F.P. (2005). Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J. Neurotrauma 22, 83–94 [DOI] [PubMed] [Google Scholar]
- 14.Bazarian J.J., Zemlan F.P., Mookerjee S., and Stigbrand T. (2006). Serum S-100B and cleaved-tau are poor predictors of long–term outcome after mild traumatic brain injury. Brain Inj. 20, 759–765 [DOI] [PubMed] [Google Scholar]
- 15.Rosengren L.E., Wikkelso C., and Hagberg L. (1994). A sensitive ELISA for glial fibrillary acidic protein: application in CSF of adults. J. Neurosci. Methods 51, 197–204 [DOI] [PubMed] [Google Scholar]
- 16.Vos P.E., Jacobs B., Andriessen T.M., Lamers K.J., Borm G.F., Beems T., Edwards M., Rosmalen C.F., and Vissers J.L. (2010). GFAP and S100B are biomarkers of traumatic brain injury: an observational cohort study. Neurology 75, 1786–1793 [DOI] [PubMed] [Google Scholar]
- 17.Buonora J.E., Yarnell A.M., Lazarus R.C., Mousseau M., Latour L.L., Rizoli S.B., Baker A.J., Rhind S.G., Diaz-Arrastia R., and Mueller G.P. (2015). Multivariate analysis of traumatic brain injury: development of an assessment score. Front. Neurol. 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson J.M., Hampton D.W., Patani R., et al. (2008). Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain 131, 1736–1748 [DOI] [PubMed] [Google Scholar]
- 19.Ost M., Nylen K., Csajbok L., Ohrfelt A.O., Tullberg M., Wikkelsö C., Nellgård P., Rosengren L., Blennow K., and Nellgård B. (2006). Initial CSF total tau correlates with 1–year outcome in patients with traumatic brain injury. Neurology 67, 1600–1604 [DOI] [PubMed] [Google Scholar]
- 20.Neselius S., Zetterberg H., Blennow K., Randall J., Wilson D., Marcusson J., and Brisby H. (2013). Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj. 27, 425–433 [DOI] [PubMed] [Google Scholar]
- 21.Irizarry M.C. (2004). Biomarkers of Alzheimer disease in plasma. NeuroRx 1, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentleman S.M., Greenberg B.D., Savage M.J., Noori M., Newman S.J., Roberts G.W., Griffin W.S., and Graham D.I. (1997). A beta 42 is the predominant form of amyloid beta–protein in the brains of short–term survivors of head injury. Neuroreport 8, 1519–1522 [DOI] [PubMed] [Google Scholar]
- 23.Johnson V.E., Stewart W., and Smith D.H. (2012). Widespread tau and amyloid–beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong Y.T., Veenith T., Dewar D., Outtrim J.G., Mani V., Williams C., Pimlott S., Hutchinson P.J., Tavares A., Canales R., Mathis C.A., Klunk W.E., Aigbirhio F.I., Coles J.P., Baron J.C., Pickard J.D., Fryer T.D., Stewart W., and Menon D.K. (2014). Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. J.A.M.A. Neurol. 71, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson D.H., Rissin D.M., Kan C.W., Fournier D.R., Piech T., Campbell T.G., Meyer R.E., Fishburn M.W., Cabrera C., Patel P.P., Frew E., Chen Y., Chang L., Ferrell E.P., von Einem V., McGuigan W., Reinhardt M., Sayer H, Vielsack C., and Duffy D.C. (2015). The Simoa HD–1 Analyzer: a novel fully automated digital immunoassay analyzer with single–molecule sensitivity and multiplexing. J. Lab. Autom. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 26.Randall J., Mortberg E., Provuncher G.K., et al. (2013). Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation 84, 351–356 [DOI] [PubMed] [Google Scholar]
- 27.Zetterberg H., Mortberg E., Song L., Zetterberg H., Mörtberg E., Song L., Chang L., Provuncher G.K., Patel P.P., Ferrell E., Fournier D.R., Kan C., Campbell T.G., Meyer R., Rivnak A.J., Pink B.A., Minnehan K.A., Piech T., Rissin D.M., Duffy D.C., Rubertsson S., Wilson D.H., and Blennow K. (2011). Hypoxia due to cardiac arrest induces a time–dependent increase in serum amyloid beta levels in humans. PloS one 6, e28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivera A., Lejbman N., Jeromin A., French L.M., Kim H.S, Cashion A., Mysliwiec V., Diaz-Arrastia R., and Gill J. (2015). Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. J.A.M.A. Neurol. 72, 1109–1116 [DOI] [PubMed] [Google Scholar]
- 29.Zafonte R.D., Bagiella E., Ansel B.M., et al. (2012). Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). J.A.M.A. 308, 1993–2000 [DOI] [PubMed] [Google Scholar]
- 30.Rissin D.M., Kan C.W., Campbell T.G., et al. (2010). Single–molecule enzyme–linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 28, 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshall L.F., Marshall S.B., Klauber M.R., et al. (1991). A new classification of head injury based on computerized tomography. Special Suppl. 75, Suppl. 1, S14–S20 [Google Scholar]
- 32.Wilson J.T., Pettigrew L.E., and Teasdale G.M. (1998). Structured interviews for the Glasgow Outcome Scale and the extended Glasgow Outcome Scale: guidelines for their use. J. Neurotrauma 15, 573–585 [DOI] [PubMed] [Google Scholar]
- 33.Papa L., Lewis L.M., Falk J.L., et al. (2012). Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann. Emerg. Med. 59, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kavalci C., Pekdemir M., Durukan P., Ilhan N., Yildiz M., Serhatlioglu S., and Seckin D. (2007). The value of serum tau protein for the diagnosis of intracranial injury in minor head trauma. Am. J. Emerg. Med. 25, 391–395 [DOI] [PubMed] [Google Scholar]
- 35.Blennow K., de Leon M.J., and Zetterberg H. (2006). Alzheimer's disease. Lancet 368, 387–403 [DOI] [PubMed] [Google Scholar]
- 36.Quiroz Y.T., Schultz A.P., Chen K., Protas H.D., Brickhouse M., Fleisher A.S., Langbaum J.B., Thiyyagura P., Fagan A.M., Shah A.R., Muniz M., Arboleda-Velasquez J.F., Munoz C., Garcia G., Acosta-Baena N., Giraldo M., Tirado V., Ramírez D.L., Tariot P.N., Dickerson B.C., Sperling R.A., Lopera F., and Reiman E.M. (2015). Brain imaging and blood biomarker abnormalities in children with autosomal dominant alzheimer disease: a cross-sectional study. J.A.M.A. Neurol. 72, 912–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsson A., Csajbok L., Ost M., et al. (2004). Marked increase of beta–amyloid(1–42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J. Neurol. 251, 870–876 [DOI] [PubMed] [Google Scholar]
- 38.Emmerling M.R., Morganti–Kossmann M.C., Kossmann T., Stahel P.F., Watson M.D., Evans L.M., Mehta P.D., Spiegel K., Kuo Y.M., Roher A.E., and Raby C.A. (2000). Traumatic brain injury elevates the Alzheimer's amyloid peptide A beta 42 in human CSF. A possible role for nerve cell injury. Ann. N.Y. Acad. Sci. 903, 118–122 [DOI] [PubMed] [Google Scholar]
- 39.Mondello S., Buki A., Barzo P., Randall J., Provuncher G., Hanlon D., Wilson D., Kobeissy F., and Jeromin A. (2014). CSF and plasma amyloid–beta temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci. Rep. 4, 6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Washington P.M., Morffy N., Parsadanian M., Zapple D.N., and Burns M.P. (2014). Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid–beta in an Alzheimer's disease mouse model. J. Neurotrauma 31, 125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehta P.D., Pirttila T., Patrick B.A., Barshatzky M., and Mehta S.P. (2001). Amyloid beta protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci. Lett. 304, 102–106 [DOI] [PubMed] [Google Scholar]
- 42.Sherer M., Struchen M.A., Yablon S.A., Wang Y., and Nick T.G. (2008). Comparison of indices of traumatic brain injury severity: Glasgow Coma Scale, length of coma and post-traumatic amnesia. J. Neurol. Neurosurg. Psychiatry 79, 678–685 [DOI] [PubMed] [Google Scholar]
- 43.Bledsoe B.E., Casey M.J., Feldman J., Johnson L., Diel S., Forred W., andGorman C. (2015). Glasgow Coma Scale scoring is often inaccurate. Prehosp. Disaster Med. 30, 46–53 [DOI] [PubMed] [Google Scholar]
- 44.Lawrence D.W., Comper P., Hutchison M.G., Sharma B. (2015). The role of apolipoprotein E episilon (epsilon)-4 allele on outcome following traumatic brain injury: A systematic review. Brain injury 29, 1018–1031 [DOI] [PubMed] [Google Scholar]
- 45.Johnson V.E., Stewart W., Graham D.I., Stewart J.E., Praestgaard A.H., Smith D.H. (2009). A neprilysin polymorphism and amyloid-beta plaques after traumatic brain injury. J Neurotrauma 26, 1197–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]