

Abstract
After experimental traumatic brain injury (TBI), calcineurin is upregulated; blocking calcineurin is associated with improved outcomes. In humans, variation in the calcineurin A-gamma gene (PPP3CC) has been associated with neuropsychiatric disorders, though any role in TBI recovery remains unknown. This study examines associations between PPP3CC genotype and mortality, as well as gross functional status assessed at admission using the Glasgow Coma Scale (GCS) and at 3, 6, and 12 months after severe TBI using the Glasgow Outcome Score (GOS). The following tagging single nucleotide polymorphisms (tSNPs) in PPP3CC were genotyped: rs2443504, rs2461491, rs2469749, and rs10108011. The rs2443504 AA genotype was univariately associated with GCS (p = 0.022), GOS at 3, 6, and 12 months (p = 0.002, p = 0.034, and p = 0.004, respectively), and mortality (p = 0.007). In multivariate analysis controlling for age, sex, and GCS, the AA genotype of rs2443504 was associated with GOS at 3 (p = 0.02), and 12 months (p = 0.01), with a trend toward significance at 6 months (p = 0.05); the AA genotype also was associated with mortality in the multivariate model (p = 0.04). Further work is warranted to better understand the role of calcineurin, as well as the genes encoding it and their relevance to outcomes after brain injury.
Keywords: : cognitive function, outcome measures, recovery, traumatic brain injury
Introduction
Calcineurin is a highly conserved calcium/calmodulin-dependent phosphatase responsive to intracellular calcium fluctuations.1,2 Calcineurin is comprised of a 57–61 kDa catalytic subunit (CnA) and 19 kDa regulatory subunit (CnB).1,3,4 The CnA subunit consists of three isozymes: alpha (calna1), beta (calna2) and gamma (calna3), each of which is encoded by a unique gene. The gamma isozyme encoded by the PPP3CC gene is well categorized5,6 and associated with neurological disorders similar to traumatic brain injury (TBI).7–20 CnA subunit isoforms play a critical role in initiating and maintaining long-term potentiation (LTP)21–23 and are involved in the structural plasticity of cortical circuits.24,25 Over-expression of CnA19 is associated with apoptosis,26–29 mitochondrial dysfunction,30 neuro-inflammation,29,31–33 and CNS pathology in the context of axotomy,34 nerve constriction,35 and ischemia.36 In neural tissue, calcineurin is involved in synaptic release of gamma-Aminobutyric acid and glutamate,37 neuronal excitability via N-methyl-D-aspartate (NMDA) receptor modulation,38 and regulation of axonal outgrowth.20,39
Experimental brain injury increases calcineurin,40–43 which is associated with regulation of synaptic plasticity and dendritic spine alterations.44 In this context, blocking calcineurin is neuroprotective45–48 and attenuates axonal injury.49,50 Calcineurin inhibitors such as FK-506 and cyclosporine A (CsA) are protective after TBI by attenuating cortical damage,30 reducing seizure activity,51 and promoting LTP.45–48 Notably, the beneficial effects of CsA may not be calcineurin-mediated, but rather may be due to the effect of CsA on the mitochondrial transition pore.52–54 A recent study sought to examine the effect of blockade of the astrocytic (calcineurin-dependent transcription factor Nuclear Factor of Activated T cells [CN/NFAT]) pathway in rats exposed to controlled cortical impact; in this study, CN/NFAT blockade resulted in normalization of synaptic function and plasticity within the hippocampus.55 Still, targeting calcineurin after TBI has not led to clinical translation,56–59 and existing evidence is inconclusive.30,56–60 A 2008 clinical trial reported dose-dependent associations with favorable neurological outcomes with no increase in mortality or adverse effects, compared with placebo.61
Variation in genes encoding calcineurin may impact TBI outcomes, although this has never been tested. Several human single nucleotide polymorphisms (SNPs) exist for genes encoding components of the calcineurin molecule; variation in the gamma isozyme, encoded by PPP3CC on chromosome 8 (Fig. 1),5,6,62,63 is associated with neuropsychiatric problems common to TBI (e.g., executive function, attention, social interaction, motor function).7–18,64–68 In pre-clinical models, knocking out PPP3CC leads to deficits in working memory, reduced social behavior, and increased locomotor activity.69,70 Variation in PPP3CC has been found in a large genome-wide study to be associated with therapy (e.g., antidepressant) response via its effects on immune cell signaling,71 suggesting possible pharmacogenomic applications. Understanding the role of endogenous calcineurin expression (and related genes) is an important early step in the effort to target this pathway therapeutically. In preparing this manuscript, no human study was found exploring the association of calcineurin polymorphisms with TBI outcomes, though such associations have been hypothesized.72,73 This study used data and biological samples from a larger parent study to examine the association between four tagging single nucleotide polymorphisms (tSNPS) in PPP3CC and gross functional status (e.g. Glasgow Coma Scale [GCS], Glasgow Outcome Score [GOS], and mortality) out to 1 year after severe brain injury.
FIG. 1.

Idiogram depicting the location of each examined tagging single nucleotide polymorphism (tSNP) gene, as well as graphical display of allele and genotype frequencies in our sample. For all tSNPs examined, the variant allele was found in at least 25% of participants in the sample.
Methods
Patient enrollment
Institutional review board approval was obtained prior to study onset. Initial participant recruitment occurred between May 2000 and August 2014 in a neurointensive care unit through the University of Pittsburgh Brain Trauma Research Center. Due to the altered cognitive status of severe TBI patients, informed consent was obtained from the injured individual's legal representative prior to data collection. If a participant's status improved to a level that (s)he could consent, continued participation in the study was contingent upon assent. Following consent, participants were included in the parent study if they met the following criteria: 1) sustained a severe TBI (GCS ≤8); 2) aged 16–80 at time of injury; and 3) possessed an indwelling ventriculostomy catheter. Individuals were excluded if they had: 1) penetrating TBI; 2) cardiac and/or respiratory arrest; or 3) a pre-existing neurological deficit, defined as a pre-TBI history of a serious neurological condition (e.g. mental retardation; stroke; dementia) that may affect scoring on the chosen measures (e.g., GCS, GOS) and could confound study findings. Demographic and initial injury severity data were collected from medical records. Cerebrospinal fluid (CSF) was collected from the indwelling catheter, and blood was collected from an intravenous/intra-arterial catheter; biological samples were stored using methods known to promote DNA stability until extraction.
A total of 480 individuals were genotyped; however, not all were included in the final analysis for various reasons as summarized below and depicted in Figure 2. After elimination of those individuals who lacked genotypic data on at least one tSNP and phenotypic data on at least one dependent variable, 463 cases remained. Beyond the aforementioned inclusion criteria from the parent study, the sample was further refined based on two exclusion criteria to address potential confounders. First, 63 samples from non-Caucasian participants were excluded to control for population stratification and differences in allele frequencies across ancestral groups, as reported in public databases (e.g., dbSNP; HapMap). In our sample, there was an insufficient number of participants in any of the non-Caucasian subgroups to warrant a subgroup analysis and allelic frequency differences negated considering all non-Caucasians together as a single subgroup for analysis. Finally, 20 individuals ages 16–17 years were excluded because brain development continues during adolescence, which may confound recovery.74 The final analysis was restricted to 380 participants who had genotype and GCS data at baseline. Importantly, there was attrition in our sample across the 3-, 6-, and 12-month data collection time-points. Specifically, by 3 months 31 participants were lost to follow-up (n = 349), an additional 3 were lost between the 3 and 6 month assessment (n = 346), and 20 more participants were lost between the 6 and 12 month assessment (n = 326). The sample remaining at 12 months was 85.7% of the original 380 participants included at baseline. Missing data was not imputed due to the complexity of TBI and the fact that loss to follow-up may have been associated with change in status.
FIG. 2.

Flowchart depicting the inclusion, exclusion, and loss of participants in this sample. There were a total of 480 genotyped participants from the parent study that were screened for inclusion in this study. Of those screened, 380 met the criteria for inclusion in this study. An additional 54 participants were lost to follow-up during the 12 month study (31 before the 3-month follow-up, three before the 6-month follow-up, and 20 before the 12-month follow-up).
Genotyping
DNA was extracted from CSF using a commercially-available kit (Qiagen, Valencia, CA) or from blood using a salting out procedure. Selection of tSNPs was based on data from a Caucasian population in HapMap Build 36. To be included, tSNPs were required to have an r2 ≥ 0.80 and a minor allele frequency ≥0.20. A TaqMan® system (ABI 700) and associated SDS 2.0 software (Applied Biosystems, Foster City, CA) was used to genotype each tSNP using allelic discrimination assays obtained from a single commercial supplier (Life Technologies, Carlsbad, CA). The assays for three of the tSNPs (rs10108011, rs2461491, and rs2469749) were readily available commercially, while the fourth (rs2443504) was a custom assay. Relevant details of the custom assay include the forward primer sequence (TTGATGCAAACCCGATGAACAAAAA), reverse primer sequence (ACTATTCCTCTCTTTTTCTCTTTGTGATTTGAT), reporter 1 sequence (CTTTTTATCTTTTGTGTATCTAC), and reporter 2 sequence (CTTTTTATCTTTTGTATATCTAC). All genotype assignments were double called in a blinded manner and any discrepancies were rectified using raw data or re-genotyped.
Measures
Clinical measures employed were overall mortality, admission GCS, and GOS at 3, 6, and 12 months. GCS was dichotomized as GCS 3–5 versus GCS 6–8. Similarly, GOS was analyzed dichotomously (1–2 vs. 3–5).75 When possible, GOS assessments were completed face-to-face; otherwise, a phone interview was used. Whether face-to-face or by-phone, all interviews were performed by a trained technician supervised by a neuropsychiatric or neurosurgery attending physician. All individuals involved in collecting data were blinded to PPP3CC genotypes.
Statistical analysis
Statistical analysis was performed using SPSS version 23 (Chicago, IL). For each continuous variable, summary statistics were obtained, including: range, mean, standard error of the mean, and standard deviation. Chi-square analysis was used to determine associations for sex with tSNP genotype and measures of functional status (GCS, GOS, mortality). Similarly, univariate associations between tSNP genotype and injury severity assessments (GCS, GOS) were determined by chi-square analysis and, when appropriate, Fisher's exact test. Associations between age with tSNP genotype and GOS at 3-, 6-, and 12- months were determined using logistic regression. In the second phase of analysis, multivariate analysis was used to determine tSNP associations with outcome, while controlling for covariates chosen based on either significant association with GOS (p < 0.05; e.g., age, GCS) or clinical relevancy (e.g., sex) based on published literature. Ultimately, a multiple logistic regression model was used to explore associations between genotype and outcome while controlling for age, sex, and GCS score.
Results
Population demographics
Table 1 shows demographic data for the 380 cases included at baseline, as well as the results from univariate analysis, associating demographics with our dependent variables. The age range was 18–78 years, with a mean of 39.9 years, a median of 37.0 years, and a standard deviation of 16.8. The sample was mostly male (79%). At admission, the majority of the subjects (66%) had a GCS score falling into the higher category (6–8); still, it is important to note that scores in the higher (6–8) category still meet criteria for classification as severe TBI. The overall mortality during the 12-month study was 33.3%, with the majority of the deaths occurring before the 3- month data collection time-point. Older age and lower GCS scores were associated with poorer outcome at all three time-points (p < 0.0005), as well as with overall mortality (p < 0.0005). Sex was not significantly associated with outcome at any time-point.
Table 1.
Demographics and Univariate Analysis to Identify Significant Predictors of Outcome
| 3-month GOS | 6-month GOS | 12-month GOS | Overall mortality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Sample | 1–2 | 3–5 | p | 1–2 | 3–5 | p | 1–2 | 3–5 | p | No | Yes | p |
| Age (years) | |||||||||||||
| N (total) | N = 380 | 129 | 220 | <0.0005 *** | 134 | 212 | <0.0005 *** | 128 | 198 | <0.0005 *** | 205 | 121 | <0.0005 *** |
| Mean | 39.9 | 48.9 | 35.5 | 47.9 | 34.9 | 50.0 | 34.2 | 34.3 | 50.6 | ||||
| (SD) | (16.8) | (17.5) | (14.5) | (17.2) | (15.0) | (17.2) | (14.3) | (14.3) | (17.0) | ||||
| Range | 18–78 | 18–78 | 18–74 | 18–78 | 18–74 | 18–78 | 18–74 | 18–74 | 18–78 | ||||
| Sex | |||||||||||||
| N (total) | N = 380 | N = 129 | N = 220 | 0.24 | N = 134 | N = 212 | 0.33 | N = 128 | N = 198 | 0.49 | N = 205 | N = 121 | 0.27 |
| n Male | n = 302 | n = 106 | n = 169 | n = 109 | n = 163 | n = 102 | n = 152 | n = 158 | n = 99 | ||||
| (%) | (79%) | (82%) | (77%) | (81%) | (77%) | (80%) | (77%) | (77%) | (82%) | ||||
| n Female | n = 78 | n = 23 | n = 51 | n = 25 | n = 49 | n = 26 | n = 46 | n = 47 | n = 22 | ||||
| (%) | (21%) | (18%) | (23%) | (19%) | (23%) | (20%) | (23%) | (23%) | (18%) | ||||
| Admission GCS | |||||||||||||
| N | N = 380 | N = 129 | N = 220 | <0.0005 *** | N = 134 | N = 212 | <0.0005 *** | N = 128 | N = 198 | <0.0005 *** | N = 205 | N = 121 | <0.0005 *** |
| n GCS 3–5 | n = 129 | n = 70 | n = 49 | n = 70 | n = 49 | n = 69 | n = 48 | n = 51 | n = 66 | ||||
| (%) | (34%) | (54%) | (22%) | (52%) | (23%) | (54%) | (24%) | (245%) | (55%) | ||||
| nGCS 6–8 | n = 251 | n = 59 | n = 171 | n = 64 | n = 163 | n = 59 | n = 150 | n = 154 | n = 55 | ||||
| % GCS 6–8 | (66%) | (46%) | (78%) | (48%) | (77%) | (46%) | (76%) | (75%) | (45%) | ||||
Our sample was 79% male with a mean age of 39.9 years and a median of 37 years (SD = 16.8). All participants had severe traumatic brain injury (defined as GCS ≤8), with the majority (66%) falling in the range of GCS 6–8, suggesting at least some degree of eye, verbal, and/or motor response. Age and admission GCS were significantly associated with GOS at 3, 6, and 12 months, along with overall mortality; sex was not associated with outcome. *p < 0.05, **p < 0.01, ***p < 0.0005.
GOS, Glasgow Outcome Score; SD, standard deviation; GCS, Glasgow Coma Scale.
Genetic associations with demographic and clinical variables
For each of the four tSNPs, associations with genotype were examined, comparing homozygous wildtype individuals with individuals either heterozygous or homozygous for the variant allele. Genetic associations were associated with the following independent variables: age, sex, mechanism of injury (automobile/motorcycle vs. fall/jump vs. other), GCS, length of stay in hospital, Injury Severity Score, and the presence of specific neurological differences. Specifically, the neurological injuries considered included: subdural hemorrhage, diffuse axonal injury, epidural hematoma, contusion, intraventricular hemorrhage, intracranial hemorrhage, and subarachnoid hemorrhage.
As summarized in Table 2, none of the 13 aforementioned demographic and clinical variables were significantly associated (p < 0.05) with genotype on any of the four tSNPs.
Table 2.
Association of Genotype for the 4 tSNPs with Demographic and Clinical Variables
| rs10108011 | rs2469749 | rs2443504 | rs2461491 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype for each tSNP Demographic variables | AÂ | AG/GG | p | CĈ | CT/TT | p | GĜ | AG/AA | p | AÂ | AG/GG | p |
Age (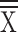 years ± SD) years ± SD) |
39.17 ± 16.89 | 40.09 ± 16.73 | 0.61 | 38.88 ± 16.33 | 40.37 ± 17.03 | 0.40 | 40.23 ± 17.46 | 39.42 ± 16.02 | 0.65 | 40.79 ± 17.40 | 39.37 ± 16.68 | 0.47 |
| Sex (n # and % male) | n = 106 80.0% | n = 193 79.0% | 0.69 | n = 123 (79.0%) | n = 166 (81.0%) | 0.62 | n = 116 (76.0%) | n = 177 (82.0%) | 0.16 | n = 80 (78.0%) | n = 214 (80.0%) | 0.72 |
| Mechanism of injury | 0.86 | 0.25 | 0.58 | 0.50 | ||||||||
| Automobile/motorcycle | n = 75 (56.4%) | n = 136 (55.7%) | n = 86 (55.1%) | n = 112 (54.6%) | n = 86 (56.6%) | n = 121 (56.0%) | n = 59 (57.8%) | n = 114 (53.9%) | ||||
| Fall/jump | n = 25 (18.8%) | n = 53 (21.7%) | n = 27 (17.3) | n = 48 (23.4%) | n = 34 (22.4%) | n = 41 (19.0%) | n = 23 (22.5%) | n = 55 (20.6%) | ||||
| Other | n = 33 (24.8%) | n = 55 (22.5%) | n = 43 (27.6%) | n = 45 (22.0%) | n = 32 (21.1%) | n = 54 (25.0%) | n = 20 (19.6%) | n = 68 (25.5) | ||||
| GCS (median) | 6 | 6 | 0.58 | 6 | 6 | 0.53 | 6 | 6 | 0.44 | 6 | 6 | 0.14 |
Length of stay (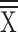 days ± SD) days ± SD) |
18.61 ± 11.95 | 20.44 ± 13.60 | 0.20 | 19.23 ± 11.82 | 20.00 ± 12.79 | 0.56 | 20.26 ± 13.60 | 19.35 ± 12.82 | 0.52 | 19.47 ± 10.95 | 19.56 ± 12.83 | 0.95 |
Injury Severity Score (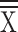 ± SD) ± SD) |
30.95 ± 9.82 | 32.61 ± 11.56 | 0.160 | 32.23 ± 10.36 | 31.82 ± 11.59 | 0.73 | 31.19 ± 11.02 | 32.42 ± 11.08 | 0.30 | 31.34 ± 11.46 | 32.28 ± 10.92 | 0.47 |
| Neurological injury type (%) | ||||||||||||
| SDH | n = 79 (59.4%) | n = 141 (58.0%) | 0.79 | n = 91 (58.7%) | n = 121 (59.0%) | 0.95 | n = 83 (54.6%) | n = 133 (61.9%) | 0.16 | n = 60 (58.8%) | n = 155 (58.3%) | 0.92 |
| DAI | n = 25 (18.9%) | n = 39 (16.0%) | 0.80 | n = 37 (24.0%) | n = 24 (11.7%) | 0.06 | n = 28 (18.4%) | n = 35 (16.4%) | 0.61 | n = 18 (17.6%) | n = 47 (17.7%) | 0.98 |
| EDH | n = 22 (16.8%) | n = 24 (9.9%) | 0.48 | n = 19 (12.3%) | n = 23 (11.2%) | 0.76 | n = 12 (7.9%) | n = 32 (15.0%) | 0.06 | n = 8 (7.8%) | n = 35 (13.2%) | 0.15 |
| Contusion | n = 76 (57.1%) | n = 159 (65.4%) | 0.13 | n = 98 (63.2%) | n = 130 (63.4%) | 0.52 | n = 97 (63.8%) | n = 134 (62.3%) | 0.68 | n = 69 (67.6%) | n = 164 (61.7%) | 0.49 |
| IVH | n = 36 (27.1%) | n = 62 (25.5%) | 0.74 | n = 41 (26.5%) | n = 53 (25.9%) | 0.90 | n = 39 (25.7%) | n = 56 (26.0%) | 0.93 | n = 26 (25.5%) | n = 70 (26.3%) | 0.87 |
| ICH | n = 122 (91.7%) | n = 236 (97.1%) | 0.07 | n = 145 (93.5%) | n = 197 (96.1%) | 0.27 | n = 143 (94.1%) | n = 205 (95.3%) | 0.59 | n = 98 (96.1%) | n = 251 (94.4%) | 0.51 |
| SAH | n = 85 (63.9%) | n = 170 (70.0%) | 0.23 | n = 112 (72.3%) | n = 131 (63.9%) | 0.09 | n = 100 (65.8%) | n = 148 (68.8%) | 0.54 | n = 64 (62.7%) | n = 185 (69.5%) | 0.21 |
There were no associations between genotype and outcome in this sample. *p < 0.05 (there were no significant differences based on genotype for any of the variables included).
tSNP, tagging single nucleotide polymorphism; SD, standard deviation; GCS, Glasgow Coma Scale; SDH, subdural hematoma; DAI, diffuse axonal injury; EDH, epidural hematoma; IVH, intraventricular hemorrhage; ICH, intracranial hemorrhage; SAH, subarachnoid hemorrhage.
Sample tSNP allele frequencies, genotype frequencies, and univariate analysis
Minor allele frequency and genotypic frequencies for each tSNP are presented in Tables 3 and 4, respectively. Each of the four tSNPs was in Hardy-Weinberg equilibrium, suggesting the genotype proportions in our sample were consistent with what is expected in the population. Univariate analysis revealed no significant associations between variant allele presence (vs. absence) on any of the four tSNPs examined with either age, sex or GCS; similarly, no association between genotype and either age, sex, or GCS was detected. Moreover, there was no significant univariate associations between rs10108011, rs2461491, and rs2469749 and GCS or GOS at any time-point. Univariate chi-square analysis revealed the AA genotype (i.e., two copies of the minor allele) of rs2443504 was associated with GCS (p = 0.022), GOS at 3 months (p = 0.002), GOS at 6 months (p = 0.034), GOS at 12 months (p = 0.004), as well as overall mortality (p = 0.007). When analysis was based on variant allele presence (vs. absence), there were no significant associations for any of the functional status variables examined. Additional analysis revealed that participants with the rs2443504 AA genotype had a significantly lower mean GCS score then the rest of the cohort (5.44 ± 1.50 for AA vs. 6.02 ± 1.54 for AG, and 5.83 ± 1.50 for GG; p = 0.045). Figure 3 shows the univariate relationship between the genotypes for each of the four tSNPs and the percentage of participants with poorer outcome (GOS 1–2) using chi-square analysis.
Table 3.
Results from Multivariate Analysis Exploring Associations of tSNP Minor (i.e., Variant) Allele Presence with Patient Outcomes, While Controlling for Age, Sex, and GCS on Admission
| 3-month GOS | 6-month GOS | 12-month GOS | Overall mortality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant allele presence | Allele frequency (%) | OR | 95% CI | p | OR | 95% CI | p | OR | 95%CI | p | OR | 95% CI | p |
| rs10108011 | 42.5% | 1.23 | 0.73–2.08 | 0.44 | 0.96 | 0.57–1.61 | 0.87 | 1.76 | 1.00–3.08 | 0.05 | 0.59 | 0.433–1.04 | 0.07 |
| Variant (G) present | |||||||||||||
| rs2469749 | 32.1% | 1.19 | 0.71–2.01 | 0.51 | 1.20 | 0.72–2.01 | 0.48 | 1.33 | 0.77–2.31 | 0.31 | 0.86 | 0.49–1.51 | 0.61 |
| Variant (T) present | |||||||||||||
| rs2443504 | 36.4% | 0.75 | 0.44–1.26 | 0.28 | 0.75 | 0.44–1.27 | 0.28 | 0.72 | 0.42–1.26 | 0.25 | 1.19 | 0.68–2.08 | 0.55 |
| Variant (A) present | |||||||||||||
| rs2461491 | 48.2% | 0.96 | 0.54–1.70 | 0.88 | 0.74 | 0.42–1.31 | 0.31 | 0.95 | 0.52–1.75 | 0.87 | 0.97 | 0.52–1.80 | 0.93 |
| Variant (G) present | |||||||||||||
Possessing one or two copies of the variant allele was not significantly associated with 3-, 6-, or 12-month GOS or mortality for any of the four tSNPs examined. *p < 0.05, **p < 0.01, ***p < 0.0005.
tSNP, tagging single nucleotide polymorphism; GCS, Glasgow Coma Scale; GOS, Glasgow Outcome Score; OR, odds ratio; CI, confidence interval.
Table 4.
Results From Multivariate Analysis Exploring Associations of tSNP Genotype with GOS and Mortality, While Controlling for Age, Sex, and GCS
| GOS @ 3 months: 1–2 vs. 3–5 | GOS @ 6 months: 1–2 vs. 3–5 | GOS @ 12 months: 1–2 vs. 3–5 | Overall mortality | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Frequency (%) | OR | 95% CI | p | OR | 95% CI | p | OR | 95% CI | p | OR | 95% CI | p |
| rs10108011 | |||||||||||||
| GG | n = 76 (20.2%) | 0.65 | 0.32–1.32 | 0.23 | 0.78 | 0.39–1.59 | 0.50 | 0.52 | 0.24–1.11 | 0.09 | 1.64 | 0.76–3.52 | 0.21 |
| AG | n = 168 (44.6%) | 0.91 | 0.52–1.60 | 0.74 | 1.19 | 0.68–2.07 | 0.54 | 0.60 | 0.33–1.09 | 0.09 | 1.70 | 0.92–3.14 | 0.09 |
| AÂ | n = 133 (35.2%) | ||||||||||||
| rs2469749 | |||||||||||||
| TT | n = 27 (7.5%) | 1.61 | 0.58–4.46 | 0.36 | 1.48 | 0.54–4.01 | 0.45 | 1.19 | 0.41–3.41 | 0.75 | 0.86 | 0.29–.2.54 | 0.79 |
| CT | n = 178 (49.3%) | 0.76 | 0.44–1.31 | 0.32 | 0.76 | 0.45–1.30 | 0.32 | 0.70 | 0.39–1.24 | 0.22 | 1.21 | 0.68–2.17 | 0.52 |
| CĈ | n = 156 (43.2%) | ||||||||||||
| rs2443504 | |||||||||||||
| AA | n = 52 (14.1%) | 2.64 | 1.20–5.82 | 0.02* | 2.16 | 0.99–4.73 | 0.05 | 2.98 | 1.30–6.81 | 0.01* | 0.41 | 0.18–0.94 | 0.04* |
| AG | n = 164 (44.6%) | 1.06 | 0.60–1.86 | 0.85 | 1.13 | 0.64–1.98 | 0.67 | 1.06 | 0.59–1.91 | 0.85 | 1.09 | 0.60–2.00 | 0.78 |
| GĜ | n = 152 (41.3%) | ||||||||||||
| rs2461491 | |||||||||||||
| GG | n = 89 (24.1%) | 1.08 | 0.53–2.18 | 0.83 | 1.27 | 0.62–2.57 | 0.51 | 1.24 | 0.58–2.65 | 0.57 | 0.87 | 0.40–1.88 | 0.73 |
| AG | n = 178 (48.2%) | 1.02 | 0.55–1.88 | 0.96 | 1.34 | 0.73–2.45 | 0.35 | 0.94 | 0.49–1.79 | 0.85 | 1.17 | 0.60–2.25 | 0.65 |
| AÂ | n = 102 (27.6%) | ||||||||||||
The AA genotype of the rs2443504 tSNP (vs. reference GG genotype) was associated with lower GOS (score ≤2) and higher mortality in univariate analysis (as depicted in Fig. 2), as well as multivariate analysis (shown in Table 3) after controlling for age, sex, and GCS. No other tSNP was associated with outcome in the multivariate analysis. *p < 0.05, **p < 0.01, ***p < 0.0005. ^Homozygous wildtype genotype.
tSNP, tagging single nucleotide polymorphism; GOS, Glasgow Outcome Scale; GCS, Glasgow Coma Scale; OR, odds ratio; CI, confidence interval.
FIG. 3.

Graph depicting the percent of patients falling into the poor outcome categories (Glasgow Outcome Score [GOS] 1–2 and overall mortality) in univariate chi-square analysis. Significant association between the AA genotype of rs2443504 and percent of poor outcomes at 3 months (p = 0.002), 6 months (p = 0.034), and 12 months (p = 0.004), along with overall mortality (p = 0.007). (A) Three-month GOS by genotype; (B) 6-month GOS by genotype; (C) 12-month GOS by genotype; (D) overall mortality at 12 months by genotype. *p < 0.05, **p < 0.01, ***p < 0.0005.
Multivariate associations between PPP3CC tSNP minor allele presence and outcome
Table 3 shows the results from multiple logistic regressions used to examine the odds of having unfavorable outcomes (GOS = 1–2 and overall mortality) by minor allele presence. In this analysis, we controlled for age, sex, and GCS. There were no significant associations between minor allele presence for any tSNP and odds of unfavorable outcome (all, p ≥ 0.05) at any time-point. To further explore the relationship between the PPP3CC gene and severe TBI outcomes, the analysis was repeated using full genotype, which allowed us to explore whether or not a double dosage of the variant allele for a given tSNP was associated with unfavorable outcome after severe TBI.
Multivariate associations between PPP3CC genotype and outcome
Multivariate analyses examined PPP3CC genetic associations with GOS 3, 6, and 12 months after TBI, controlling for sex, age, and initial GCS as summarized in Table 4. There were no significant associations between three of the four tSNPs (rs10108011, rs2469749, and rs2461491) and outcome. For rs2443504, participants with the AA genotype had poorer outcomes when compared to those with the GG genotype at 3 months (p = 0.02) and 12 months (p = 0.04), with a trend toward significance at 6 months (p = 0.05). Notably, the significant findings when genotype was considered, as opposed to variant allele presence, suggest that two copies of the variant allele (AA) were necessary to show significant outcome differences from the homozygous wild type genotype (GG). Moreover, heterozygous individuals (AG) on rs2443504 did not have significantly poorer outcomes than GG individuals. As was true in the univariate tests, age and initial GCS score were significantly associated with GOS at 3, 6, and 12 months in all four tSNP multivariate models (all, p < 0.05).
Discussion
In this exploratory pilot study, only one of the four SNPs investigated in the PPP3CC gene was associated with any outcome examined. Specifically, a homozygous variant genotype 0for the PPP3CC gene SNP rs2443504 was associated with poorer outcome (assessed via GOS) following severe TBI. Individuals with two copies of the variant allele had lower GOS at 3 months and 12 months, with a trend toward significance at 6 months; this genotype also was associated with higher mortality. In this sample, the heterozygous variant genotype group was not significantly associated with outcomes. Interestingly, little is known about rs2443504; to date, this tSNP has not been associated with any health condition. It also is worth acknowledging that all tSNPs in this analysis were intronic tSNPS with no known direct impact on the final calcineurin molecule. However, the tSNPs may play a regulatory role by impacting the amount of available mature functional protein. Moreover, this SNP tags a region of linkage disequilibrium. Using publicly-available resources (e.g., HapMap, Haploview 4.2, UCSC Genome Browser), we discovered that rs2443504 was in linkage disequilibrium with rs10088686, an intronic SNP in SORBS3, which encodes an SH3 domain-adaptor protein implicated in many important cellular processes, including cytoskeletal structure, adhesion, migration, and signaling. Additional research should further explore the mechanism. Consistent with published literature, older participants and those with lower GCS score had poorer outcome and higher mortality.76–78 Notably, despite published evidence suggesting females have better recovery after TBI than males,79,80 this study failed to find any associations between sex and GCS score, GOS, or mortality. Our sample was characterized by an over-representation of males; however, this is consistent with national data suggesting that males outnumber females 3:1 in the TBI population.81
This exploratory pilot study is limited in that it lacks statistical correction for multiple testing. Considering some of these associations are close to the 0.05 threshold for statistical significance, it is not anticipated that the associations detected in this pilot would have remained significant if such a correction were applied. However, the fact that the same SNP was associated with outcomes on admission and over the course of recovery warrants further examination in a larger sample.
A second important limitation of this study surrounds the homogenous sample included in the final analysis. The sample was limited to Caucasian adults from a limited geographic region, which limits the generalizability of the findings. The rationale was the low non-Caucasian recruitment in the parent study, which is common in the western region of Pennsylvania where this work was conducted. Moreover, there are known allelic frequency differences across different ethnicities that could have affected the results of this study, especially since the non-Caucasian subsample was not sufficiently large to warrant a subgroup analysis. Ultimately, this limits the generalizability of these findings and necessitates future efforts to replicate these findings in more diverse samples. The sample is further limited in that it included only adults, with the rationale being that brain development is ongoing throughout adolescence,74 which may have affected the recovery profile and ultimately the results of this study. Further, as mentioned above, there was an overrepresentation of males, although this higher incidence in males is consistent with what is observed in the clinical TBI population for every age group.81 Still, replicating these results a more diverse sample with respect to sex, as well as age and race, would strengthen the evidence. An additional limitation in this study is the reliance on GCS and GOS data that were dichotomized instead of left in their original ordinal scale. The rationale for dichotomization was twofold: many GCS and GOS values had low numbers of participants, and to balance the groups compared for analysis. Future studies with larger samples should use the GCS and GOS in their original scales and also include additional measures with clinical relevance to TBI survivors (e.g., cognitive testing).
A third limitation surrounds the sole focus on the gamma isozyme. This study did not examine other loci relevant to calcineurin or related proteins that share a biological pathway with calcineurin or get acted on by this phosphatase. This limits the ability to interpret the results of this study. Additional evidence exploring more genetic loci, as well as using additional methodologies and measures, will enhance the knowledge base with evidence that may lead to elucidation of an underlying mechanism.
That said, the study is strengthened by the fact that it takes a tagging SNP approach, enabling us to consider all the variability in the gene of interest. Future studies will be strengthened by inclusion of participants with less severe injuries, as well as more complex outcome measures (e.g., memory). Additional research also should explore the relationship between PPP3CC gene variation and protein levels (e.g., calcineurin regulatory and catalytic subunits), the downstream targets of the active phosphatase, and other markers in linkage disequilibrium. Moreover, analysis of tissue samples (e.g., autopsy) will help to better elucidate the relationship between calcineurin and gross functional status after TBI.
In summary, this exploratory study examined the association between four tSNPs in the PPP3CC gene and outcomes following severe TBI; the homozygous variant genotype of rs2443504 was found to be significantly associated with poorer outcomes assessed using GOS (at 3 and 12 months, with a trend toward significance at 6 months), as well as overall mortality. Further research is needed to confirm these observations in a larger sample size and to hone in on the specific locus or loci responsible for driving the association with outcomes that were observed.
Acknowledgments
This work was generously supported by several grants, including: PPG Grant 30315, P50NS030318, VA RR&D#B6761R, NIH R01NR008424, R01NR13342, F31NR014957, and T32NR009759. Additional support for this work was provided by grants from the following foundations and professional societies: the Pittsburgh Foundation, Sigma Theta Tau International Eta Chapter, the International Society for Nurses in Genetics, and the American Association of Neuroscience Nursing/Neuroscience Nursing Foundation. The authors would like to thank Marilyn K. Farmer, Amanda Savarese, and Lan Pham for their continued editorial support and Michael D. Farmer for his assistance with the figures.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Rusnak F. and Mertz P. (2000). Calcineurin: form and function. Physiol. Rev. 80, 1483–521 [DOI] [PubMed] [Google Scholar]
- 2.Bales J.W., Ma X., Yan H.Q., Jenkins L.W., and Dixon C.E. (2010). Expression of protein phosphatase 2B (calcineurin) subunit A isoforms in rat hippocampus after traumatic brain injury. J. Neurotrauma 27, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klee C.B., Crouch T.H., and Krinks M.H. (1979). Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. U. S. A. 76, 6270–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groth R.D., Dunbar R.L., and Mermelstein P.G. (2003). Calcineurin regulation of neuronal plasticity. Biochem. Biophys. Res. Commun. 311, 1159–1171 [DOI] [PubMed] [Google Scholar]
- 5.Strausberg R.L., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Zeeberg B., Buetow K.H., Schaefer C.F., Bhat N.K., Hopkins R.F., Jordan H., Moore T., Max S.I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A.A., Rubin G.M., Hong L., Stapleton M., Soares M.B., Bonaldo M.F., Casavant T.L., Scheetz T.E., Brownstein M.J., Usdin T.B., Toshiyuki S., Carninci P., Prange C., Raha S.S., Loquellano N.A., Peters G.J., Abramson R.D., Mullahy S.J., Bosak S.A., McEwan P.J., McKernan K.J., Malek J.A., Gunaratne P.H., Richards S., Worley K.C., Hale S., Garcia A.M., Gay L.J., Hulyk S.W., Villalon D.K., Muzny D.M., Sodergren E.J., Lu X., Gibbs R.A., Fahey J., Helton E., Ketteman M., Madan A.A., Rodrigues S., Sanchez A., Whiting M., Madan A.A., Young A.C., Shevchenko Y., Bouffard G.G., Blakesley R.W., Touchman J.W., Green E.D., Dickson M.C., Rodriguez A.C., Grimwood J., Schmutz J., Myers R.M., Butterfield Y.S.N., Krzywinski M.I., Skalska U., Smailus D.E., Schnerch A., Schein J.E., Jones S.J.M., and Marra M.A. (2002). Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. U. S. A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ota T., Suzuki Y., Nishikawa T., Otsuki T., Sugiyama T., Irie R., Wakamatsu A., Hayashi K., Sato H., Nagai K., Kimura K., Makita H., Sekine M., Obayashi M., Nishi T., Shibahara T., Tanaka T., Ishii S., Yamamoto J., Saito K., Kawai Y., Isono Y., Nakamura Y.Y., Nagahari K., Murakami K., Yasuda T., Iwayanagi T., Wagatsuma M., Shiratori A., Sudo H., Hosoiri T., Kaku Y., Kodaira H., Kondo H., Sugawara M., Takahashi M., Kanda K., Yokoi T., Furuya T., Kikkawa E., Omura Y., Abe K., Kamihara K., Katsuta N., Sato K., Tanikawa M., Yamazaki M.M., Ninomiya K., Ishibashi T., Yamashita H., Murakawa K., Fujimori K., Tanai H., Kimata M., Watanabe M.M., Hiraoka S., Chiba Y., Ishida S., Ono Y., Takiguchi S., Watanabe S., Yosida M., Hotuta T., Kusano J., Kanehori K., Takahashi-Fujii A., Hara H., Tanase T., Nomura Y., Togiya S., Komai F., Hara R., Takeuchi K., Arita M., Imose N., Musashino K., Yuuki H., Oshima A., Sasaki N., Aotsuka S., Yoshikawa Y., Matsunawa H., Ichihara T., Shiohata N., Sano S., Moriya S., Momiyama H., Satoh N., Takami S., Terashima Y., Suzuki O., Nakagawa S., Senoh A., Mizoguchi H., Goto Y., Shimizu F., Wakebe H., Hishigaki H., Watanabe T., Sugiyama A., Takemoto M., Kawakami B., Yamazaki M., Watanabe K., Kumagai A., Itakura S., Fukuzumi Y., Fujimori Y., Komiyama M., Tashiro H., Tanigami A., Fujiwara T., Ono T., Yamada K., Fujii Y., Ozaki K., Hirao M., Ohmori Y., Kawabata A., Hikiji T., Kobatake N., Inagaki H., Ikema Y., Okamoto S., Okitani R., Kawakami T., Noguchi S., Itoh T., Shigeta K., Senba T., Matsumura K., Nakajima Y., Mizuno T., Morinaga M., Sasaki M., Togashi T., Oyama M., Hata H., Watanabe M., Komatsu T., Mizushima-Sugano J., Satoh T., Shirai Y., Takahashi Y., Nakagawa K., Okumura K., Nagase T., Nomura N., Kikuchi H., Masuho Y., Yamashita R., Nakai K., Yada T., Nakamura Y., Ohara O., Isogai T., and Sugano S. (2004). Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 36, 40–45 [DOI] [PubMed] [Google Scholar]
- 7.Gerber D.J., Hall D., Miyakawa T., Demars S., Gogos J.A., Karayiorgou M., and Tonegawa S. (2003). Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proc. Natl. Acad. Sci. U. S. A. 100, 8993–8998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y.L., Fann C.S.J., Liu C.M., Chang C.C., Yang W.C., Hung S.I., Yu S.L., Hwang T.J., Hsieh M.H., Liu C.C., Tsuang M.M., Wu J.Y., Jou Y.S., Faraone S. V, Tsuang M.T., Chen W.J., and Hwu H.-G. (2007). More evidence supports the association of PPP3CC with schizophrenia. Mol. Psychiatry 12, 966–974 [DOI] [PubMed] [Google Scholar]
- 9.Eastwood S.L., Salih T., and Harrison P.J. (2005). Differential expression of calcineurin A subunit mRNA isoforms during rat hippocampal and cerebellar development. Eur. J. Neurosci. 22, 3017–3024 [DOI] [PubMed] [Google Scholar]
- 10.Kinoshita Y., Suzuki T., Ikeda M., Kitajima T., Yamanouchi Y., Inada T., Yoneda H., Iwata N., and Ozaki N. (2005). No association with the calcineurin a gamma subunit gene (PPP3CC) haplotype to Japanese schizophrenia. J. Neural Transm. 112, 1255–1262 [DOI] [PubMed] [Google Scholar]
- 11.Mathieu F., Miot S., Etain B., El Khoury M.A., Chevalier F., Bellivier F., Leboyer M., Giros B., and Tzavara E.T. (2008). Association between the PPP3CC gene, coding for the calcineurin gamma catalytic subunit, and bipolar disorder. Behav. Brain Funct. 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egry E.Y., Salum M.J., and Castellanos B.E. (1991). Participant research: reinterpreting the roads traversed. Rev. Paul. Enferm. 10, 13–20 [PubMed] [Google Scholar]
- 13.Horiuchi Y., Ishiguro H., Koga M., Inada T., Iwata N., Ozaki N., Ujike H., Muratake T., Someya T., and Arinami T. (2007). Support for association of the PPP3CC gene with schizophrenia. Mol. Psychiatry 12, 891–893 [DOI] [PubMed] [Google Scholar]
- 14.Kvajo M., McKellar H., and Gogos J.A. (2010). Molecules, signaling, and schizophrenia. Curr. Top. Behav. Neurosci. 2010, 629–656 [DOI] [PubMed] [Google Scholar]
- 15.Kyogoku C., Yanagi M., Nishimura K., Sugiyama D., Morinobu A., Fukutake M., Maeda K., Shirakawa O., Kuno T., and Kumagai S. (2011). Association of calcineurin A gamma subunit (PPP3CC) and early growth response 3 (EGR3) gene polymorphisms with susceptibility to schizophrenia in a Japanese population. Psychiatry Res. 185, 16–19 [DOI] [PubMed] [Google Scholar]
- 16.Mammis A., Agarwal N., and Mogilner A.Y. (2014). Alternative treatment of intracranial hypotension presenting as postdural puncture headaches using epidural fibrin glue patches: two case reports. Int. J. Neurosci. 124, 863–866 [DOI] [PubMed] [Google Scholar]
- 17.Shi J., Gershon E.S., and Liu C. (2008). Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr. Res. 104, 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wockner L.F., Noble E.P., Lawford B.R., Young R.M., Morris C.P., Whitehall V.L.J., and Voisey J. (2014). Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4, e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asai A., Qiu J.H., Narita Y., Chi S., Saito N., Shinoura N., Hamada H., Kuchino Y., and Kirino T. (1999). High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 274, 34450–34458 [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto T., Kawamata T., Saito N., Sasaki M., Nakai M., Niu S., Taniguchi T., Terashima A., Yasuda M., Maeda K., and Tanaka C. (1998). Isoform-specific redistribution of calcineurin A alpha and A beta in the hippocampal CA1 region of gerbils after transient ischemia. J. Neurochem. 70, 1289–98 [DOI] [PubMed] [Google Scholar]
- 21.Wang J.H. and Kelly P.T. (1996). The balance between postsynaptic Ca(2+)-dependent protein kinase and phosphatase activities controlling synaptic strength. Learn. Mem. 3, 170–181 [DOI] [PubMed] [Google Scholar]
- 22.Kayyali U.S., Zhang W., Yee A.G., Seidman J.G., and Potter H. (1997). Cytoskeletal changes in the brains of mice lacking calcineurin A alpha. J. Neurochem. 68, 1668–1678 [DOI] [PubMed] [Google Scholar]
- 23.Winder D.G., and Sweatt J.D. (2001). Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2, 461–474 [DOI] [PubMed] [Google Scholar]
- 24.Victor R.G., Thomas G.D., Marban E., and O'Rourke B. (1995). Presynaptic modulation of cortical synaptic activity by calcineurin. Proc. Natl. Acad. Sci. U. S. A. 92, 6269–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakel J.L. (1997). Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol. Sci. 18, 124–134 [DOI] [PubMed] [Google Scholar]
- 26.Wang H.G., Pathan N., Ethell I.M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T.F., and Reed J.C. (1999). Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284, 339–343 [DOI] [PubMed] [Google Scholar]
- 27.Springer J.E., Azbill R.D., Nottingham S.A., and Kennedy S.E. (2000). Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J. Neurosci. 20, 7246–7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anantharam V., Lehrmann E., Kanthasamy A.A.G., Yang Y., Banerjee P., Becker K.G., Freed W.J., and Kanthasamy A.A.G. (2007). Microarray analysis of oxidative stress regulated genes in mesencephalic dopaminergic neuronal cells: relevance to oxidative damage in Parkinson's disease. Neurochem. Int. 50, 834–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morioka M., Hamada J., Ushio Y., and Miyamoto E. (1999). Potential role of calcineurin for brain ischemia and traumatic injury. Prog. Neurobiol. 58, 1–30 [DOI] [PubMed] [Google Scholar]
- 30.Scheff S.W. and Sullivan P.G. (1999). Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J. Neurotrauma 16, 783–792 [DOI] [PubMed] [Google Scholar]
- 31.Manalan A.S. and Klee C.B. (1983). Activation of calcineurin by limited proteolysis. Proc. Natl. Acad. Sci. U. S. A. 80, 4291–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H.-Y., Tomizawa K., Oda Y., Wei F.Y., Lu Y.F., Matsushita M., Li S.T., Moriwaki A., and Matsui H. (2004). Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem. 279, 4929–4940 [DOI] [PubMed] [Google Scholar]
- 33.Fernandez A.M., Fernandez S., Carrero P., Garcia-Garcia M., and Torres-Aleman I. (2007). Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci. 27, 8745–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cavallucci V., Bisicchia E., Cencioni M.T., Ferri A., Latini L., Nobili A., Biamonte F., Nazio F., Fanelli F., Moreno S., Molinari M., Viscomi M.T., and D'Amelio M. (2014). Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death Dis. 5, e1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miletic G., Sullivan K.M., Dodson A.M.K., Lippitt J.A., Schneider J.A., and Miletic V. (2011). Changes in calcineurin message, enzyme activity and protein content in the spinal dorsal horn are associated with chronic constriction injury of the rat sciatic nerve. Neuroscience 188, 142–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brait V.H., Martin K.R., Corlett A., Broughton B.R.S., Kim H.A., Thundyil J., Drummond G.R., Arumugam T. V., Pritchard M.A., and Sobey C.G. (2012). Over-Expression of DSCR1 Protects against Post-Ischemic Neuronal Injury. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang R.Q. and Dillon G.H. (1998). Maintenance of recombinant type A gamma-aminobutyric acid receptor function: role of protein tyrosine phosphorylation and calcineurin. J. Pharmacol. Exp. Ther. 286, 243–255 [PubMed] [Google Scholar]
- 38.Tong G., Shepherd D., and Jahr C.E. (1995). Synaptic desensitization of NMDA receptors by calcineurin. Science 267, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 39.Yang S.A. and Klee C.B. (2000). Low affinity Ca2+-binding sites of calcineurin B mediate conformational changes in calcineurin A. Biochemistry 39, 16147–16154 [DOI] [PubMed] [Google Scholar]
- 40.Kurz J.E., Parsons J.T., Rana A., Gibson C.J., Hamm R.J., and Churn S.B. (2005). A significant increase in both basal and maximal calcineurin activity following fluid percussion injury in the rat. J. Neurotrauma 22, 476–490 [DOI] [PubMed] [Google Scholar]
- 41.Kurz J.E., Hamm R.J., Singleton R.H., Povlishock J.T., and Churn S.B. (2005). A persistent change in subcellular distribution of calcineurin following fluid percussion injury in the rat. Brain Res. 1048, 153–160 [DOI] [PubMed] [Google Scholar]
- 42.Wu P., Zhao Y., Haidacher S.J., Wang E., Parsley M.O., Gao J., Sadygov R.G., Starkey J.M., Luxon B.A., Spratt H., Dewitt D.S., Prough D.S., and Denner L. (2013). Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J. Neurotrauma 30, 30, 775–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bales J., Ma X., Yan H., Jenkins L., and Dixon C. (2010). Regional calcineurin subunit B isoform expression in rat hippocampus following a traumatic brain injury. Brain Res. 1358, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell J.N., Low B., Kurz J.E., Patel S.S., Young M.T., and Churn S.B. (2012). Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury. J. Neurotrauma 29, 218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butcher S.P., Henshall D.C., Teramura Y., Iwasaki K., and Sharkey J. (1997). Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J. Neurosci. 17, 6939–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friberg H., Ferrand-Drake M., Bengtsson F., Halestrap A.P., and Wieloch T. (1998). Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J. Neurosci. 18, 5151–5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okonkwo D.O., Büki A., Siman R., and Povlishock J.T. (1999). Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport 10, 353–358 [DOI] [PubMed] [Google Scholar]
- 48.Okonkwo D.O., Melon D.E., Pellicane A.J., Mutlu L.K., Rubin D.G., Stone J.R., and Helm G.A. (2003). Dose-response of cyclosporin A in attenuating traumatic axonal injury in rat. Neuroreport 14, 463–466 [DOI] [PubMed] [Google Scholar]
- 49.Singleton R.H., Stone J.R., Okonkwo D.O., Pellicane A.J., and Povlishock J.T. (2001). The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J. Neurotrauma 18, 607–614 [DOI] [PubMed] [Google Scholar]
- 50.Suehiro E., Singleton R.H., Stone J.R., and Povlishock J.T. (2001). The immunophilin ligand FK506 attenuates the axonal damage associated with rapid rewarming following posttraumatic hypothermia. Exp. Neurol. 172, 199–210 [DOI] [PubMed] [Google Scholar]
- 51.Campbell J.N., Gandhi A., Singh B., and Churn S.B. (2014). Traumatic brain injury causes a tacrolimus-sensitive increase in non-convulsive seizures in a rat model of post-traumatic epilepsy. Int. J. Neurol. Brain Disord. 1, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Readnower R.D., Pandya J.D., McEwen M.L., Pauly J.R., Springer J.E., and Sullivan P.G. (2011). Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J. Neurotrauma 28, 1845–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokobori S., Mazzeo A.T., Gajavelli S., and Bullock M.R. (2013). Mitochondrial neuroprotection in traumatic brain injury: rationale and therapeutic strategies. CNS Neurol. Disord. Drug Targets 13, 606–619 [DOI] [PubMed] [Google Scholar]
- 54.Sullivan P.G., Rabchevsky A.G., Waldmeier P.C., and Springer J.E. (2005). Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J. Neurosci. Res. 79, 231–239 [DOI] [PubMed] [Google Scholar]
- 55.Furman J.L., Sompol P., Kraner S.D., Pleiss M.M., Putman E.J., Dunkerson J., Mohmmad Abdul H., Roberts K.N., Scheff S.W., and Norris C.M. (2016). Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci. 36, 1502–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kay J.E., Doe S.E., and Benzie C.R. (1989). The mechanism of action of the immunosuppressive drug FK-506. Cell. Immunol. 124, 175–181 [DOI] [PubMed] [Google Scholar]
- 57.Wiederrecht G., Lam E., Hung S., Martin M., and Sigal N. (1993). The mechanism of action of FK-506 and cyclosporin A. Ann. N. Y. Acad. Sci. 696, 9–19 [DOI] [PubMed] [Google Scholar]
- 58.Fujita M., Oda Y., Wei E.P., and Povlishock J.T. (2011). The combination of either tempol or FK506 with delayed hypothermia: implications for traumatically induced microvascular and axonal protection. J. Neurotrauma 28, 1209–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mbye L.H.A.N., Singh I.N., Carrico K.M., Saatman K.E., and Hall E.D. (2009). Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J. Cereb. Blood Flow Metab. 29, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dileonardi A.M., Huh J.W., and Raghupathi R. (2012). Differential effects of FK506 on structural and functional axonal deficits after diffuse brain injury in the immature rat. J. Neuropathol. Exp. Neurol. 71, 959–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hatton J., Rosbolt B., Empey P., Kryscio R., and Young B. (2008). Dosing and safety of cyclosporine in patients with severe brain injury. 109, 699–707 [DOI] [PMC free article] [PubMed]
- 62.Muramatsu T. and Kincaid R.L. (1992). Molecular cloning and chromosomal mapping of the human gene for the testis-specific catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin A). Biochem. Biophys. Res. Commun. 188, 265–271 [DOI] [PubMed] [Google Scholar]
- 63.National Center for Biotechnology Information. (2015). PPP3CC protein phosphatase 3, catalytic subunit, gamma isozyme [ Homo sapiens (human) ]. Available at: www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=5533 Accessed May25, 2016
- 64.Finnanger T.G., Skandsen T., Andersson S., Lydersen S., Vik A., and Indredavik M. (2013). Differentiated patterns of cognitive impairment 12 months after severe and moderate traumatic brain injury. Brain Inj. 27, 1606–1616 [DOI] [PubMed] [Google Scholar]
- 65.Weddell R., Oddy M., and Jenkins D. (1980). Social adjustment after rehabilitation: a two year follow-up of patients with severe head injury. Psychol Med 10, 257–263 [DOI] [PubMed] [Google Scholar]
- 66.Janusz J.A., Kirkwood M.W., Yeates K.O., and Taylor H.G. (2002). Social problem-solving skills in children with traumatic brain injury: long-term outcomes and prediction of social competence. Child Neuropsychol. 8, 179–94 [DOI] [PubMed] [Google Scholar]
- 67.Vasa R.A., Suskauer S.J., Thorn J.M., Kalb L., Grados M.A., Slomine B.S., Salorio C.F., and Gerring J.P. (2015). Prevalence and predictors of affective lability after paediatric traumatic brain injury. Brain Inj. 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Backeljauw B. and Kurowski B.G. (2014). Interventions for attention problems after pediatric traumatic brain injury: what is the evidence? PM R. 6, 814–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takao K. and Miyakawa T. (2008). Investigating genes-to-behavior pathways in psychiatric diseases: an approach using a comprehensive behavioral test battery on genetically engineered mice. Nihon Shinkei Seishin Yakurigaku Zasshi 28, 135–142 [PubMed] [Google Scholar]
- 70.Takao K. and Miyakawa T. (2006). Investigating gene-to-behavior pathways in psychiatric disorders: The use of a comprehensive behavioral test battery on genetically engineered mice. Ann. N. Y. Acad. Sci. 1086, 144–159 [DOI] [PubMed] [Google Scholar]
- 71.Fabbri C., Marsano A., Albani D., Chierchia A., Calati R., Drago A., Crisafulli C., Calabrò M., Kasper S., Lanzenberger R., Zohar J., Juven-Wetzler A., Souery D., Montgomery S., Mendlewicz J., and Serretti A. (2014). PPP3CC gene: a putative modulator of antidepressant response through the B-cell receptor signaling pathway. Pharmacogenomics J. 14, 463–472 [DOI] [PubMed] [Google Scholar]
- 72.Davidson J., Cusimano M.D., and Bendena W.G. (2015). Post-traumatic brain injury: genetic susceptibility to outcome. Neuroscientist 21, 424–441 [DOI] [PubMed] [Google Scholar]
- 73.Schaart D.R., Clarijs M.C., and Bos A.J. (2001). On the applicability of the AAPM TG-60/TG-43 dose calculation formalism to intravascular line sources: proposal for an adapted formalism. Med. Phys. 28, 638–653 [DOI] [PubMed] [Google Scholar]
- 74.Dahl R.E. (2004). Adolescent brain development: a period of vulnerabilities and opportunities. Keynote address. Ann. N. Y. Acad. Sci. 1021, 1–22 [DOI] [PubMed] [Google Scholar]
- 75.McHugh G.S., Butcher I., Steyerberg E.W., Lu J., Mushkudiani N., Marmarou A., Maas A.I.R., and Murray G.D. (2007). Statistical approaches to the univariate prognostic analysis of the IMPACT database on traumatic brain injury. J. Neurotrauma 24, 251–258 [DOI] [PubMed] [Google Scholar]
- 76.Shekhar C., Gupta L.N., Premsagar I.C., Sinha M., and Kishore J. (2015). An epidemiological study of traumatic brain injury cases in a trauma centre of New Delhi (India). J. Emerg. Trauma. Shock 8, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davis K.C., Slomine B.S., Salorio C.F., and Suskauer S.J. (2015). Time to follow commands and duration of posttraumatic amnesia predict GOS-E Peds scores 1 to 2 years after TBI in children requiring inpatient rehabilitation. J. Head Trauma Rehabil. 31, E39–E47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hukkelhoven C.W.P.M., Steyerberg E.W., Rampen A.J.J., Farace E., Habbema J.D.F., Marshall L.F., Murray G.D., and Maas A.I.R. (2003). Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg 99, 666–673 [DOI] [PubMed] [Google Scholar]
- 79.Ratcliff J.J., Greenspan A.I., Goldstein F.C., Stringer A.Y., Bushnik T., Hammond F.M., Novack T.A., Whyte J., and Wright D.W. (2007). Gender and traumatic brain injury: do the sexes fare differently? Brain Inj. 21, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 80.Slewa-Younan S., Green A.M., Baguley I.J., Gurka J.A., and Marosszeky J.E. (2004). Sex differences in injury severity and outcome measures after traumatic brain injury. Arch. Phys. Med. Rehabil. 85, 376–379 [DOI] [PubMed] [Google Scholar]
- 81.Faul M., Xu L., Wald M.M., and Coronado V.G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Centers for Disease Control and Prevention: Atlanta, GA [Google Scholar]