

Abstract
As an outcome of the 2010 Asian Pacific Conference for Chromatography and Mass Spectrometry in Hong Kong, a collaborative working group was formed to promote the harmonisation of mass spectrometry methods. The Mass Spectrometry Harmonisation Working Group resides under the combined auspices of the Asia-Pacific Federation for Clinical Biochemistry and Laboratory Medicine (APFCB) and the Australasian Association of Clinical Biochemists (AACB). A decision was made to initially focus attention on serum steroids due to the common interest of members in this area; with the first steroid to assess being testosterone.
In principle, full standardisation with traceability should be achievable for all steroids as they are small compounds with defined molecular weight and structure. In order to achieve this we need certified reference materials, reference methods, reference laboratories, reference intervals and external quality assurance programs; each being an important pillar in the process. When all the pillars are present, such as for serum testosterone, it is feasible to fully standardise the liquid chromatography – tandem mass spectrometry (LC-MS/MS) methods. In a collaborative process with interested stakeholders, we commenced on a pathway to provide ongoing assessment and seek opportunities for improvement in the LC-MS/MS methods for serum steroids. Here we discuss the outcomes to date and major challenges related to the accurate measurement of serum steroids with a focus on serum testosterone.
Introduction
The history of mass spectrometry in medical testing now spans over fifty years, with the majority of this period seeing its confinement to specialist laboratories.1 The breakthrough of coupling the liquid phase to the mass spectrometer provided the stimulus for this methodology to expand its reach into routine laboratories.2 The broad implementation of LC-MS/MS in clinical diagnostic laboratories now encompasses a range of techniques from expanded new born screening programs, to toxicology screening, therapeutic drug monitoring, and quantification of biogenic amines, vitamins and hormones; all of which relate to small molecules with defined molecular weights and structures.3 In addition, more recently methods for larger molecular weight compounds have emerged in translational research laboratories; and are likely to move broadly into the clinical diagnostic area in the near future.4 Irrespective of the analytical group, accurate quantification is essential to ensure appropriate clinical interpretation.
Initially, LC-MS/MS entered the clinical arena with resounding accolades of being the new “gold standard” as the problems of immunoassay sensitivity and accuracy would be solved. Many LC-MS/MS techniques in the early period of the 2000s were simply “dilute and shoot” and sample cleanup / chromatographic separation was minimal. In memories of the second AACB Chromatography Mass Spectrometry meeting (themed “Coming together to separate”) held in Sydney in 2007 these ideas were still being discussed.5 However also around the mid 2000s the literature was consistently reporting limitations in methods.6,7 This led to questions of accuracy with differences seen between the home brew methods. This disillusionment culminated in many ways with the now infamous retraction of LC-MS/MS vitamin D results by a major laboratory in 2008.8 Hence by the time of the next regional Chromatography Mass Spectrometry conference in Hong Kong in 2010 there was significant discussion about how we could ensure the reliability of our LC-MS/MS assays as they expanded into the repertoire of methods offered by an increasing number of diagnostic laboratories.9
As an outcome of the 2010 conference in Hong Kong a collaborative working group was formed; the Mass Spectrometry Harmonisation Working Group (MSHWG).9 The group resides under the combined auspices of the APFCB and the AACB.10,11 The goal of the MSHWG is to promote harmonisation, and where practicable, standardisation of mass spectrometry methods through a consensus approach with laboratories; principally in the Asia and Pacific area. A decision was made to initially focus attention on serum steroids due to the common interest of members in this area.12
In principle, full standardisation with traceability should be achievable for all steroids as they are small defined molecular weight compounds. In order to achieve this, certified reference materials, reference methods, reference laboratories, reference intervals and external quality assurance programs are required; each being an important pillar in the process.13,14 The Joint Committee for Traceability in Laboratory Medicine (JCTLM) was established to support this process worldwide through the development of a database to recognise primary reference materials, methods and laboratories. Currently some (e.g. serum cortisol, estradiol, progesterone and testosterone) but not all steroids (e.g. serum 17-hydroxyprogesterone, androstenedione, cortisone and dihydrotestosterone) measured routinely by mass spectrometry have complete listings in the JCTLM database; see Table 1 for detail.15 When all the JCTLM pillars are present, such as for serum testosterone, it is feasible to fully standardise our LC-MS/MS methods.
Table 1.
Measurands routinely determined by Mass Spectrometry (primarily LC-MS/MS) as reported in the end of cycle RCPAQAP report mid 2015 compared to the JCTLM listing of certified reference materials, procedures and laboratories for non-peptide hormones as of 2nd July 2015.
| Steroids included in the RCPAQAP Endocrine Program in the first half of 2015 | Total number of participants at end of cycle 43 | Total number of MS participants at end of cycle 43 (percentage) | JCTLM listed Material | JCTLM listed Measurement Procedure | JCTLM listed Laboratories |
|---|---|---|---|---|---|
| 25-OH Vitamin D3* | 91 | 8 (9%) | x | Yes | Yes |
| 17 Hydroxy Progesterone | 28 | 7 (25%) | x | Yes | Yes |
| Aldosterone | 25 | 2 (8%) | x | Yes | Yes |
| Androstenedione | 24 | 6 (25%) | x | x | x |
| Cortisol | 131 | 1 (<1%) | Yes | Yes | Yes |
| DHEAS | 78 | 1 (1%) | x | x | x |
| DHT | 5 | 3 (60%) | x | x | x |
| Estradiol | 185 | 0 | Yes | Yes | Yes |
| Estriol - unconjugated | 5 | 0 | x | Yes | Yes |
| Progesterone | 180 | 0 | Yes | Yes | Yes |
| Testosterone | 118 | 6 (5%) | Yes | Yes | Yes |
25 OH Vitamin D3 is listed under the vitamin JCTLM listing. NIST SRM 972 was previously listed on the JCTLM database, but was delisted due to depletion of stock.
The terms ‘standardisation’ and ‘harmonisation’ when related to laboratory medicine define two distinct, albeit closely linked concepts. Yet both are based on traceability principles described in the International Organization for Standardization (ISO) standard 17511,16 in which the term ‘standardisation’ is used when results for a measurand are equivalent, and the results are traceable to the International System of Units (SI) through a high-order primary reference material and/or a reference measurement procedure (RMP).17 By contrast, the term ‘harmonisation’ is generally used when results are equivalent, being either traceable to a reference material or based on a consensus approach, namely in agreement with the mean values obtained with different methods, but neither a suitable high-order primary reference material nor a RMP is available. However, the term “harmonisation” can also be used more broadly to relate to the overall testing process, encompassing the pre-analytical phase, methods of analysis, calibration materials, reporting units and reference intervals. Harmonisation in this broader sense can be applied to the critical aspects that should be aligned to promote agreement. The development of implementation guidelines and best practice statements form part of this process. In relation to steroids, we use the term harmonisation in this broader context.
In a collaborative process with interested stakeholders, we commenced on a pathway to provide ongoing assessment and seek opportunities for improvement in the LC-MS/MS methods for serum steroids. Here we discuss the outcomes to date and major challenges related to the accurate measurement of serum steroids with a focus on serum testosterone.
Materials and Processes to Support Harmonisation
Harmonisation of the total testing process, and where practicable standardisation of the analytical method with established trueness, is fundamental to the delivery of quality pathology. Whilst this goal is not new, advances in information technology, the move towards the electronic health record and the recognition of patients as part of the global village, have led to the appreciation that discordance in results between laboratories and between methods is no longer acceptable practice. There are many different aspects to harmonisation which encompass the total testing process. Often the first step in the recognition of discordance between laboratory results is through assessment against an External Quality Assurance (EQA) scheme. As such, EQA is recognised as the fifth pillar and in many aspects is central to this process.18 In this section we discuss some of the strategies used to gain a further understanding of method performance and improve agreement.
Participation in Harmonisation Process
Eight laboratories, including four laboratories from Australia, two laboratories in Hong Kong, one laboratory from Austria and the National Measurement Institute of Australia (NMIA) participated in this process. In addition, the group included scientific staff from the RCPAQAP.
In order to have ongoing evaluation of performance, participation in an EQA program provides a basis for objective peer comparison. Hence, a mandatory requirement to participate in this activity is to enrol and submit results to a common EQA program. In Australasia, the Royal College of Pathologists of Australasia Quality Assurance Programs (RCPAQAP) Endocrine Program is the obvious common denominator (hence the mutually agreed program) for these and future initiatives for the harmonisation of serum steroids as it provides an ongoing mechanism to objectively assess analytical performance.19 Participation in this process was (and still is) by open invitation based on the analytical expertise and interest in overall method improvement. Of note there were a minority of laboratories (Australian research based) who did not wish to participate in this common peer review process (i.e. submit results to the RCPAQAP) and hence were deemed ineligible for inclusion.
Establishing RCPAQAP Quality Specifications
The RCPAQAP Endocrine material consists of six linearly related levels; each level is analysed twice in a cycle. There are two cycles per annum. This material is lyophilised and during the manufacturing process the base material may be charcoal stripped and supplemented with analytes of interest. The program uses analytical performance goals to assess the quality of results. These goals, called Allowable Limits of Performance (ALP), are quality standards which allow participating laboratories to assess their performance and respond accordingly.
For analytes in the Endocrine program, these ALP goals are set using the internationally agreed hierarchy and biological variability is the highest level applicable.20,21 These goals can be applied for monitoring and or diagnosis of a patient and in the latter case both imprecision and bias are included in the calculation. When monitoring is the aim, as for testosterone, the calculation is based on imprecision. Professor Callum Fraser’s fitness for purpose definitions are then applied to fine tune the biological variation:
Then the ALP is calculated as two times the CVa set for the program. The level set for the program of minimum, desirable or optimal is based on at least 80% of participants being able achieve the performance. The ALP for testosterone is based on the intra-individual biological variation data of 9.25% obtained from the Ricos database23 and interpreted against the minimum CVa is therefore 6.9375% (i.e. 9.25% × 0.75 = 6.9375%). This is then multiplied by two for the 95% range (uncertainty of measurement) and finally rounded to ±15%. In practice the RCPAQAP ALP for testosterone is applied as +/−0.4 nmol/L up to 2.7 nmol/L, then ±15%.13
Target values are considered preferable to the use of medians, particularly when there are significant differences between method or instrument groups; such as mass spectrometry compared to immunoassay methods. Target values are ideally set by higher order methods with established traceability and trueness. Practically, target values of the RCPAQAP material are assigned for at least levels 2, 4 and 6 and the other values are then determined by linear regression by one or more reference laboratories. This process can vary slightly depending on whether level 1 is part of the linear range and the cost involved in the setting process and the data returned to the RCPAQAP. In 2012 as an example, target values for serum testosterone material were assigned by WEQAS for levels 2, 4 and 6 followed by linear regression to obtain the targets for levels 1, 3 and 5.
Method Questionnaire
The RCPAQAP collects method details from participants based on method principle, sample preparation technique (where relevant), instrument brand and calibrator source as part of the enrolment process. Previously, members of this group have successfully developed and utilised participant questionnaires to provide further insight into the analytical methods used by RCPAQAP participants.24–26 Hence in 2013, through the RCPAQAP, a detailed questionnaire was developed and sent to participating laboratories to look more closely at the serum steroid methods with a focus on testosterone. The basis for the development of this questionnaire was information obtained from an initial questionnaire generated from all group participants in 2011 (unpublished). It addressed the pre-analytical, analytical and post-analytical components of each laboratory’s serum testosterone method and was designed to understand in more detail the similarities and differences that may exist between LC-MS/MS serum testosterone methods.
Reference Materials Prepared by NMIA
At the top of the traceability chain are primary certified reference materials (CRM). These materials are often made by metrology institutes and have stated uncertainty of measurement. This provides the anchor for trueness provided commutability is established. In Australia, the NMIA makes a number of steroid reference materials; as do other metrology institutes around the world. This includes a variety of CRM for steroids including testosterone (NMIA M914B).27
Reference Methods Prepared by NMIA
For the investigation of serum testosterone harmonisation, NMIA developed two reference methods as anchors for the studies conducted; an Ultra-High Performance LC-MS/MS method and a gas chromatography high resolution mass spectrometry (GC-HRMS) method. This was a significant undertaking requiring many months to fully validate the approach. These methods are listed in the BIPM metrology data base28 and once published will be submitted to the JCTLM database. Briefly, the methods consisted of the following (details provided by Dr Veronica Vamathevan from NMIA):
Calibration
The NMIA M914B material was obtained from the Chemical Reference Materials Facility at NMIA and was certified with a purity of 99.7% ± 1.7%. Stock solutions of testosterone were prepared using this certified pure substance reference material. Working standard solutions of testosterone were prepared at concentrations of approximately 0.1, 0.4, 1.0, 1.8, 4.8, 16, 35 and 55 ng/g in methanol. Deuterated (D3−) testosterone (NMIA D644) was also obtained from NMIA. Internal standard solutions of deuterated testosterone were prepared gravimetrically in 20% methanol/water at similar concentrations to the native testosterone solutions. Calibration and internal standard solutions were stored at −20°C in the dark.
NMIA Sample Preparation
Sample and calibration blends were prepared as follows for isotope dilution analysis.
Gravimetric preparation: Sample blends were prepared gravimetrically by combining the desired mass of the serum sample (∼1 g) with an aliquot of the corresponding D3-testosterone internal standard solution. The amount of internal standard added to samples was matched to the amount of native testosterone present in the sample to achieve a gravimetric amount ratio of approximately one. Calibration blends were prepared gravimetrically by combining aliquots of testosterone calibration standard solutions and D3-testosterone internal standard solutions. The amounts of native and deuterated testosterone present in the calibration blends were matched to the corresponding amounts present in sample blends. After preparation, blends were gently mixed and equilibrated for at least an hour prior to solvent extraction.
Liquid-Liquid Extraction (LLE): Testosterone in serum samples was extracted twice using 5 mL aliquots of hexane/ethyl acetate (3:2). The hexane/ethyl acetate extracts were evaporated to dryness at 50°C under nitrogen gas and the dried extracts were reconstituted in 1 mL of 20% methanol/water in preparation for solid-phase extraction (SPE) clean-up. Calibration blends were analysed directly.
(SPE) Clean-Up: Serum sample extracts were purified using Waters Oasis HLB SPE cartridges (200 mg, 6 mL). Sample extracts were loaded in 20% methanol/water. The cartridges were then washed with water and then 60% methanol/water. Adsorbed testosterone was eluted with 100% methanol. The eluates collected were evaporated to dryness at 50°C under nitrogen gas and then reconstituted with 60% methanol/water (40 µL–150 µL) for LC-MS/MS analysis.
LC-MS/MS Reference Method
Samples were prepared as described above. Testosterone in the sample and calibration blends was separated from matrices using two-dimensional Ultra-High Performance LC-MS/MS (Thermo TSQ Vantage/TLX1). A Waters CSH Phenyl-Hexyl column with an acetonitrile/formic acid (0.02%, aqueous) mobile phase was employed in the first dimension and coupled with a Waters BEH Shield column and a methanol/formic acid (0.02% formic, aqueous) mobile phase in the second dimension. A narrow window containing testosterone was transferred from the first dimension to the second dimension by means of a dual valve switching system for additional separation of compounds in the sample extract by chromatography. The mass spectrometer was operated in the positive ion mode with electrospray ionisation and multiple reaction monitoring (MRM) of fragment ions. The MRM transitions monitored were 289.2 > 109.1 and 289.2 > 97.1m/z for native testosterone and 292.2 > 109.1 and 292.2 > 100.2 m/z for deuterated testosterone.
Confirmatory Analysis Using GC-HRMS
A second reference measurement procedure was developed for confirmatory analysis. Serum samples were prepared and extracted as described above and subjected to preparative HPLC clean-up. A C18 Alltima column (4 × 250 mm, 5 um, Grace) was used with a mobile phase of acetonitrile/water. The elution of testosterone during chromatography was monitored using a UV-Visible detector at a wavelength of 245 nm. A fraction containing testosterone was collected in a glass tube and evaporated to dryness at 50°C under nitrogen. The dried extracts were derivatised with trimethylsilyl iodosilane (TMIS) reagent and then analysed by GC-HRMS analysis (Finnigan MAT95). Samples were chromatographed on an Agilent VF-17MS column (0.25 mm × 30 m, 0.25 um film thickness). The GC-HRMS was operated in the Multiple Ion Detection (MID) mode at a resolution of approximately 3000.
Trial of a Common Calibrator
Many LC-MS/MS laboratories currently gravimetrically prepare their own calibrators and purchase primary materials to check their prepared standards.19 On a small scale this works and provides an excellent foundation for trueness, but as serum steroid MS methods become more common, it would be more practical (and probably more robust) to use a secondary commercial calibrator.
There are advantages as a group to use a common calibrator for harmonisation and peer support. Certainly laboratories have been doing this in an ad hoc fashion for many years as it also provides leverage for trouble shooting issues. Therefore, as a group we decided to investigate the first secondary calibrator for MS based serum steroid methods commercially available (i.e. that could be purchased independently, as distinct from being part of a full mass spectrometry method kit) to see if it was a potential candidate as a secondary calibrator.
Selection of a Common Calibrator
Following an extensive search and subsequent discussions, an established commercial calibrator was selected to trial as a “Common Calibrator” (CC) for this project. This calibrator is supplied as a seven level set (containing 17 steroids), which is in line with the published 2011 recommendations from Honour.29 The AbsoluteIDQ® Steroid Calibrators (lot number 388421, BIOCRATES Life Sciences AG, Innsbruck, Austria) were supplied as lyophilised materials (calibrator 1–7, separate calibrator matrix for reconstitution). The Steroid Calibrator set (labelled as “Research Use Only”) is designed for LC-MS/MS based analysis of steroid hormones. In addition to testosterone, the CC set contains the following steroid hormones: aldosterone, androstenedione, androsterone, corticosterone, cortisol, cortisone, 11-deoxycorticosterone, 11-deoxycortisol, dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEAS), dihydrotestosterone (DHT), 17β-estradiol (E2), estrone (E1), etiocholanolone, 17α-hydroxyprogesterone (17OHP), and progesterone.30
Values for these calibrators are routinely assigned by weighing in pure steroids purchased from Sigma-Aldrich. The density of the Biocrates calibrator is 1.006 kg/m3. These calibrators have a proficiency test certificate awarded on a quarterly-basis (based on their use with the Biocrates kit steroid method) for the accredited proficiency test program HM (hormone group 1, testosterone, aldosterone, cortisol, 17β-estradiol, progesterone, DHEAS, and 17α-hydroxyprogesterone) of the German Reference Institute for Bioanalytic (RfB -Referenzinstitut für Bioanalytik) under the umbrella of the Deutsche Vereinte Gesellschaft für klinische Chemie und Laboratoriumsmedizin (DGKL).31 For testosterone the average relative bias through 11 proficiency tests, each with two test samples, was −1.5%. It means that the average accuracy of 22 reported values against target values was 98.5%. The standard deviation of these measurements accuracies was 4.7%.
For the Common Calibrator (CC) study, indicative values for testosterone were assigned by NMIA using the methods described above.
CC Study Protocol
Two common calibrator sets, one EQA sample set (2012 RCPAQAP material) and two sets of de-identified, “Unknown”, fresh frozen human serum samples (one male and one female) were sent to each laboratory. The two Unknown serum samples were collected from two project team members (a male 36 years and a female 24 years) and stored in 1 ml frozen aliquots. Samples were analysed in duplicate by each laboratory’s (n=8) routine LC-MS/MS method on two separate occasions. As there was only one set of EQA material distributed these samples were frozen after the first analysis and then thawed for the second analytical run. An outline of the protocol is provided in Figure 1.
Figure 1:

- On receipt of the material immediately store as per the individual instructions
- Immediately after preparation of the RCPAQAP endocrine material vials on the first day of analysis, an aliquot of each level should be stored at −20 degrees C until the second day of analysis
- Analyse the materials on two separate days one month apart in duplicate as per your patient samples i.e. run and report all results with you usual calibrators
- Report results for all steroids in your analytical run to one more decimal place than you would report your patient samples
- The second day of analysis should be four weeks (plus or minus one week) after the first day of analysis
- Submit the excel file with your results to the RCPAQAP by email.
For the routine diagnostic laboratories, the RCPAQAP and CC material were reconstituted as per the manufacturer’s instructions on the day of analysis. The CC was supplied with a separate lyophilised matrix vial and reconstituted with 10 mL high-purity water (Milli-Q water). This matrix was then used to reconstitute the seven levels of the lyophilised CC; each with 1.2 mL of matrix solution. The “Unknown” fresh frozen serum samples were analysed as supplied by each laboratory.
Being one of the eight laboratories, target or indicative values for the RCPAQAP, CC and Unknown material were determined by NMIA’s LC-MS/MS two dimensional method. As proof of concept (i.e. when sufficient sample volume was available to achieve sensitivity) the target values were cross-checked against the NMIA’s GC-HRMS serum testosterone method. GC-MS has the advantage of not suffering from the potential matrix effects from phospholipids that can confound LC-MS/MS serum analysis.32 All target values provided by NMIA for this project were by weight (denominator in grams) and then converted to volume (denominator in litres) for the clinical laboratory comparison. To ensure alignment with other harmonisation initiatives NIST SRM971, which is a serum matrix matched commutable material, was used as a QC material.
For NMIA a more rigorous approach was applied for the reconstitution of the material as follows:
The RCPAQAP freeze-lyophilised serum samples were reconstituted with fresh MilliQ water prior to analysis according to the reconstitution protocol provided. Approximately 5 g of water was added to the sample bottle through the rubber septum using a Pasteur pipette. The exact mass of water added for reconstitution was determined by gravimetry. The bottle was allowed to stand for at least 20 minutes and then gently inverted and swirled to completely dissolve the contents. The sample was allowed to stand for a further 10 minutes prior to sub-sampling for analysis.
The CC was supplied as lyophilised materials to be reconstituted with a solution of calibrator matrix. The exact mass of water added was determined by gravimetry. Approximately 1.2 g of reconstituted calibrator matrix was added to each steroid calibrator vial. The exact mass of calibrator matrix solution added to each vial was determined gravimetrically.
The Unknown fresh frozen serum samples were brought to room temperature, mixed and analysed as supplied.
Evaluation Tools to Define Acceptable Performance
Determination of Target and Indicative Values by NMIA
The process of value assignment by metrology institutes is an extensive, exact and time consuming task. Target values are assigned in mass units by metrology institutes. Density is then approximated for the material in order to calculate the concentration in volume units. The volume used by NMIA for determination depends on the expected approximate concentration of the measurand. In this process the metrology institute aims to analyse a consistent mass and hence will vary the sample volume used in the process of value assignment. In many instances four replicates are performed for each measurement. This is a higher order approach to that used by the clinical diagnostic laboratory.
The process of reference and indicative values for the RCPAQAP, CC and Unknown samples serves as an example of the process used by metrology (i.e. NMIA).
Target values for testosterone in the RCPAQAP samples were determined from multiple mass fraction determinations made on four different bottles of each sample. At least two sub-samples were analysed from each bottle and analysed in independent experiments performed on different days. As the results of LCMS/MS and GC-HRMS analysis were in excellent agreement, the testosterone mass fractions determined using both reference measurement procedures were used in the calculation of reference values. Measurement uncertainties in the reference values were estimated as described in ISO/IEC Guide 98-3.33 The associated absolute and relative expanded uncertainties in the reference values were determined at a level of confidence of 95%.
Information (i.e. indicative) values were determined for the mass fractions of testosterone in the seven CC solutions and the Unknown fresh human sera samples. Due to their limited sample volume, only two mass fraction determinations were possible on these samples and thus the target values provided for these samples are information values only. The two mass fraction determinations were performed in two separate experiments conducted on different days. The indicative values are the average mass fractions of the two determinations made on each sample. For CC levels 1 and 2, only one mass fraction determination was possible. The two vials of CC levels 1 and 2 supplied were combined following reconstitution to enable approximately 2.4 g of sample to be used for analysis. This was necessary due to the low testosterone concentrations present in these samples. These indicative values did not have their uncertainty determined.
Statistical Analysis of Data
Comparison of method details: The questionnaire was informative in nature. Results were collated and summarised using a Microsoft Excel spreadsheet. Interpretation of the data was qualitative.
Comparison of methods using the common calibrator: The values determined by NMIA were utilised as the assigned value of the common calibrator set, RCPAQAP material and Unknown samples.
To assess if the adjustment of results with the CC was statistically significant for an individual laboratory an unpaired two tailed t-test was used to compare the results returned for each sample (RCPAQAP levels and Unknown samples) pre and post adjustment with the CC. Consistent standard deviation was not assumed and the statistical significance was determined using the Holm-Sidak method, with alpha=5.000%.
To determine if there was a statistically significant change for the group as a whole pre and post adjustment with the CC, results were compared for the group of laboratories by 2 way ANOVA with p<0.05 indicating a statistical significant difference for the group. Bland Altman Difference Plots were also developed to visually characterise the percentage difference across all levels compared to the target value and compared to desirable total allowable error (see below).
Microsoft Excel and GraphPad Prism version 6 software were used for assessment of the data.34
Biological variation and fitness for purpose: To assess the limits of performance in the CC trial, biological variation data and the desirable specification for fitness for purpose compared with the NMIA assigned values were used as the criteria for acceptance of results.23
The desirable imprecision can be determined based on:
where CVa is the analytical imprecision and CVi is the intra-individual biological variation.
The desirable bias (Ba) can be determined based on:
where CVg is the between subject biological variation. Hence the total allowable error (TEa) will be the combination of the CVa and the Ba calculated from the desirable specifications using the equation
Given the assignment of values was made by NMIA, bias was assumed to be zero for the CC trial; hence imprecision was the major error of measurement considered. With regard to serum testosterone, the Ricos biological variation data base for desirable specifications gives the CVi and CVg as 9.25% and 22.05% respectively. This is used to calculate the CVa as 4.63%, Ba as 5.98%, and TEa as 13.61%. Table 2 provides the current biological variation data available for all steroids.23
Table 2.
Biological Variation data of serum steroid hormones compared to the desirable specifications of fitness for purpose listed in the RCPAQAP Endocrine program. Progesterone, dihydrotestosterone and 25 hydroxyvitamin D are not listed in this database. 11-Desoxycortisol is listed in the database but is not currently offered by the RCPAQAP Endocrine Program.19
| Analyte measured in serum (no. of references)* | Biological Variation % | Desirable specifications % | |||
|---|---|---|---|---|---|
| CVI | CVg | CVa | Ba | TEa | |
| 11-Desoxycortisol (2) | 21.3 | 31.5 | 10.7 | 9.5 | 27.1 |
| 17-Hydroxyprogesterone (2) | 19.6 | 50.4 | 9.8 | 13.5 | 29.7 |
| Aldosterone (2) | 29.4 | 40.1 | 14.7 | 12.4 | 36.7 |
| Androstenedione (2) | 15.8 | 38.8 | 7.9 | 10.47 | 23.51 |
| Cortisol (3) | 15.2 | 38.1 | 7.6 | 10.26 | 22.8 |
| DHEAS (3) | 6.35 | 30.70 | 3.188 | 7.84 | 13.08 |
| Estradiol (5) | 22.5 | 24.4 | 11.25 | 8.3 | 26.86 |
| Testosterone (7) | 9.25 | 22.05 | 4.63 | 5.98 | 13.61 |
This is the number of references cited in the database which were used to compose the biological variation information.
Comparison of results over time: In 2010 the RCPAQAP commenced target setting of the serum testosterone material. From 2010 to 2015 the RCPAQAP end of cycle reports were compared for the number of participating laboratories, LCMS/MS peer group median bias and also median imprecision of the participants. Trends in imprecision and bias (from 2010) were used to assess between laboratory performance over time.19
Findings of the Harmonisation Project
Whilst there are some significant differences between the LCMS/MS methods for the RCPAQAP participants, there are also areas of commonality demonstrated from the questionnaire. In brief, all participants responded to this questionnaire, i.e. a return rate of 100%. Eight laboratories (including NMIA) returned nine sets of results; with NMIA reporting results for two methods (LC-MS/MS and GC-HRMS). Most laboratories prepared samples by liquid-liquid extraction (LLE) and one used solid phase extraction (SPE). Consistent MRMs were seen (289>109 and 289>97) for the testosterone quantifier and qualifier, respectively. The source of the laboratory’s calibrator, number of calibration levels and deuterated sites on the internal standard differed (Table 3).
Table 3.
Summary of methods of participating laboratories, based on responses to the questionnaire distributed to the group participants who measured serum testosterone by mass spectrometry. This approach was based on the previous work of two of the group members; KH and RG.54
| Question Topic | Responses |
|---|---|
| Calibration | |
| Standard Material Source | Sigma (3), NMI (1), WEQAS (2), Lipomed (1), Steraloids (1). All purity > >98%. |
| Working/Secondary Calibrator Matrix | Stripped serum (6), Calibrants in solvent (1), Biocrates matrix (1) |
| Lowest Calibrator (nmol/L) | Median 0.14, Range LOD - 0.80 |
| Highest Calibrator (nmol/L) | Median 39, Range 10–50 |
| Internal Standard Material | |
| Isotope-labelled testosterone | D2 (4), D3 (2), D5 (2) |
| Labelling Position | 1,2 (4), 19,19,19 (1), 16,16,17 (1), 2,2,4,6,6 (2), |
| Sample Preparation | |
| Volume Used | 100µL (5), 200µL (1), 500µL (1), 1000µL (1) |
| Preparation | Liquid/liquid extraction (5), Protein precipitation followed by liquid/liquid extraction (2), SPE (1) |
| Extraction Solvent | methyl tert butyl ether (5), SPE (1), ethyl acetate (1) |
| LC-MS/MS Analysis | |
| LC-MS/MS Instrument | Waters Quattro Premier XE (2), Waters Xevo TQD (2), Waters Quattro Micro (1), Agilent 6490 Jetstream (1), Thermo TSQ Vantage (1), Thermo (Ultimate 3000 UHPLC + TSQ Vantage) (1) |
| Mobile Phase in Various Gradient Methods | Ammonium acetate / formic acid / methanol (3), Water / methanol / formic acid (2), Water / acetonitrile / formic acid (1), Acetonitrile / formic acid / methanol (1), Acetonitrile / methanol / water (1) |
| Run Time (min) | Median 8, Range 3–32 |
| MRM for Quantification (m/z) | 289 > 97 (6), 289 > 109 (1), 289 > 97, 289 > 109 (1) |
| MRM for Confirmation (m/z) | 289 >109 (6), 289 > 97 (1), 289 > 97, 289 > 109 (1) |
| Method Validation Parameters | |
| LOQ (nmol/L) [CV=20%] | Median 0.3, Range 0.06 – 0.5 Median 0.1, Range 0.02 – 0.5 |
| LOQ (nmol/L) [S/N = 10] | |
| % Recovery | Median 98.6%, Range 90–102% |
| Internal QC | BioRad - Liquichek Immunoassay Plus (4), BioRad - Lyphochek Immunoassay Plus (1), Biocrates QC (1), In-house (1), NIST SRM 971 & ERM DA346a (1) |
| Reporting concentration range (nmol/L) | 0.3 – 40 (3), <0.2 to 20 (1), 0.2 – 50 (1), 0.8 – 19.2 (1), 0.03 – 34.6 (1), 0.5 – 50 (1) |
| ? Separate epi-testosterone | Yes (5), Unknown (2), No (1) |
| Post-analytical | |
| Reference female intervals (nmol/L) | <1.7 (2), <2.0 (2), 0.3– 3.8 (2) |
| Reference male intervals (nmol/L) | >8 (1),8 – 35 (1), 8.3 – 33 (1), 9.0 – 35 (1) |
The NMIA methods and materials were used to assign target values to the RCPAQAP, and provide indicative values for the CC and Unknown samples throughout all studies. Initially the 2010 RCPAQAP Endocrine material’s target values were obtained from another metrology institute (WEQAS). In 2012 the WEQAS target values were compared to the target values obtained by NMIA for the RCPAQAP material; good agreement was demonstrated between NMIA and WEQAS targets (based on Bland-Altman difference plots where the 95% confidence interval included zero); results not provided due to confidentiality. NMIA also assigned targets for the seven levels of the CC material and the values demonstrated good agreement with the gravimetric values supplied by the manufacturer of the CC. (Table 4).
Table 4.
Reference values, with their associated uncertainties, and indicative values (without uncertainty) for serum testosterone standardisation. Reference and Indicative values supplied by NMIA for testosterone in the CC and the Unknown human sera samples were applied as the agreed “true” values for the CC, RCPAQAP and Unknown samples.
| Sample | Values provided by company (with associated uncertaintya) | Values provided by NMIA (with associated uncertainty a,b) | |||
|---|---|---|---|---|---|
| Expected ng/mL | Measured ng/mL | Expected nmol/Lc | Mass fraction ng/g | Molar Concentration nmol/L | |
| RCPAQAP Level 1 | N/A | 0.228 ± 0.021 | 0.804 ± 0.072 (9.0%) | ||
| RCPAQAP Level 2 | 1.206 ± 0.055 | 4.27 ± 0.19 (4.6%) | |||
| RCPAQAP Level 3 | 2.33 ± 0.11 | 8.27 ± 0.40 (4.9%) | |||
| RCPAQAP Level 4 | 3.35 ± 0.18 | 11.89 ± 0.65 (5.4%) | |||
| RCPAQAP Level 5 | 4.34 ± 0.18 | 15.45 ± 0.65 (4.2%) | |||
| RCPAQAP Level 6 | 5.38 ± 0.18 | 19.16 ± 0.63 (3.3%) | |||
| Common Calibrator 1 | 0.01 | 0.011 ± 0.002 | 0.035 | 0.009 | 0.033 |
| Common Calibrator 2 | 0.04 | 0.038 ± 0.003 | 0.139 | 0.035 | 0.123 |
| Common Calibrator 3 | 0.20 | 0.20 ± 0.02 | 0.693 | 0.190 | 0.676 |
| Common Calibrator 4 | 0.8 | 0.80 ± 0.03 | 2.77 | 0.738 | 2.609 |
| Common Calibrator 5 | 3.0 | 3.0 ± 0.2 | 10.4 | 2.86 | 10.16 |
| Common Calibrator 6 | 6.0 | 6.0 ± 0.2 | 20.8 | 5.61 | 20.00 |
| Common Calibrator 7 | 10.0 | 10.0 ± 0.4 | 34.7 | 9.38 | 32.21 |
| Unknown 1 (adult male) | N/A | 4.67 | 15.98 | ||
| Unknown 2 (adult female) | 0.163 | 0.570 | |||
Note: Measurement uncertainties are expanded uncertainties determined at a level of confidence of 95%.
The density of the Common Calibrator is 1.006.
The average molecular weight of testosterone is 288.424 Da was used to convert the expected value from ng/mL to nmol/L (ChemSpider ID 5791 - http://www.chemspider.com/)
The values determined by NMIA were used as the assigned value for the CC trial. To determine if there was a statistically significant difference in results pre and post adjustment with the CC, results for the group of laboratories were compared by ANOVA and found to be statistically significant (p<0.05). ANOVA was also applied to the recalculation of the human testosterone samples against the CC which did demonstrate a significant change (p<0.05) in the group results for the male serum (NMIA assigned value 16.19 nmol/L) whereas the change for the female serum (NMIA assigned value 0.57 nmol/L) was not statistically significant for the group. The Bland-Altman plots pre and post adjustment with the common calibrator did not appear to tighten performance for the group as a whole, however the group results did improve for the Unknown Female sample in terms of overall percentage difference (Figure 2). Additional detail of the results from the CC pilot is provided in the Appendix for the interested reader. This lack of significant change in the CC pilot is important, as it indicates that the different calibrators employed, how they are prepared and used, all lead to the same results i.e. as performance was not improved by the CC. This is an area of strength for the current laboratory practice and calibrator quality overall.
Figure 2:
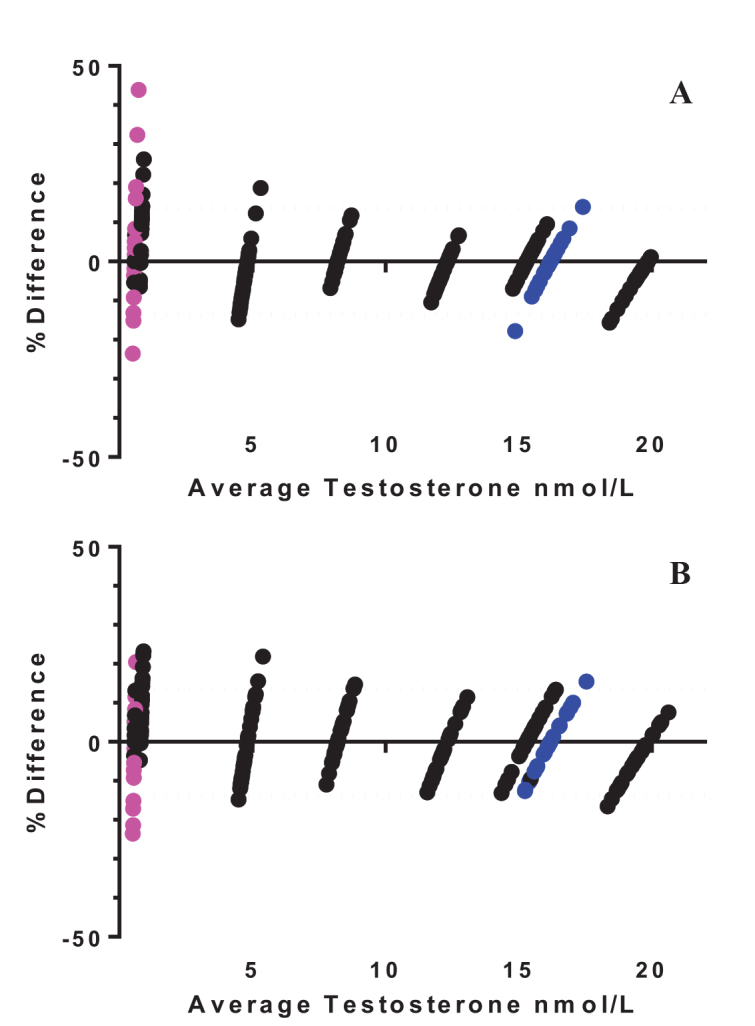
 ) are female human serum and blue dots (
) are female human serum and blue dots (
 ) are male human serum. Dashed line (…….) indicates the desirable total allowable error of 13.61% for serum testosterone taken from the Ricos Biological Variation database23
) are male human serum. Dashed line (…….) indicates the desirable total allowable error of 13.61% for serum testosterone taken from the Ricos Biological Variation database23
- Uncorrected results returned by each laboratory presented as a percentage difference compared to the NMIA assigned values.
- Laboratory results recalculated against the CC presented as a percentage difference compared to the NMIA assigned values.
For the first time in cycle 43 (first half of 2015) the median imprecision for the LC-MS/MS group (median CV of 4.2%) met the desirable imprecision for testosterone; i.e. >50% of LC-MS/MS participants achieved the biological variation imprecision target of 4.63%. This imprecision group median is also the best performing analytical method group in the RCPAQAP; out-performing immunoassay methods. In addition, the median high level bias (based on RCPAQAP level 6, i.e. adult male concentration level) for the LC-MS/MS group has demonstrated at least desirable performance since cycle 35. Regression analysis has also improved for the LC-MS/MS group with the linear regression for cycle 43 in 2015 being y=0.99x +0.314. The visual comparison of the median bias and median imprecision for the LC-MS/MS testosterone method group over time also demonstrates an overall improvement in performance. (Figure 3).
Figure 3:
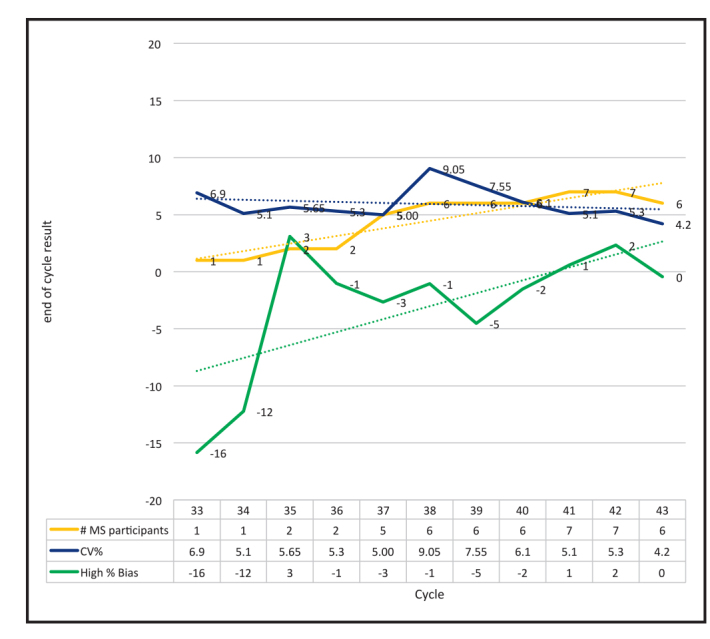
RCPAQAP End of Cycle report results for serum testosterone by LC-MS/MS. The first laboratory to report serum testosterone was in the second cycle for 2009, i.e. cycle 32. From the start of 2010 i.e. cycle 33, target values were assigned to the RCPAQAP material for serum testosterone; providing clear data on bias and imprecision of the LC-MS/MS assays. In cycle 43, representing the first half of 2015 the number of laboratories has increased and the overall median CV has decreased; indicating that at least 50% of LC-MS/MS methods now meet the desirable imprecision target of 4.63% based on biological variation. Median bias at the high end has also decreased. The low end bias (not shown for clarity of the graphed statistics) continues to fluctuate which may be due to the increased uncertainty at the lower concentration points.
Discussion of Harmonisation Initiative: Advancements and Challenges
There has been a flurry of LC-MS/MS methods in the peer review published literature over the past decade which attest to the accurate and precise analysis of serum steroids, particularly testosterone.30,35–50 LC-MS/MS technology does offer a number of significant advantages compared to immunoassay methods for many small molecular weight measurands. Some of these advantages include the simultaneous analysis of multiple steroids in the same run as well as improved specificity and sensitivity of the target analytes. A purported disadvantage is the current level of technical expertise required to run and interpret the data generated, however this is likely to present less of a problem in the near future as we move towards improved sample processing and reach agreement, with evidence, on best laboratory practice. The broad implementation of this technique into routine clinical biochemistry laboratories is now at hand and it is timely to ensure the methods are harmonised and where practicable fully standardised to safeguard their optimum clinical use.51,52
In this review we have provided a practical snapshot of the current routine LC-MS/MS serum testosterone methods used by medical testing laboratories in the Asia-Pacific Region. To provide ongoing comparison of method performance it is essential for laboratories to participate in a common EQA scheme; and here we have highlighted a number of benefits with the RCPAQAP Endocrine Program serving as an example. The review of participant methods, through the distribution of a questionnaire, determined that there are areas of commonality but also significant areas of difference. Even so, the group as a whole has seen an improvement in imprecision and bias for the group in the last five years. This demonstrates the practical application of working together to improve harmonisation.
As part of the traceability chain, primary reference methods are required to provide the link to the working methods. Mass spectrometry is generally considered a superior technique compared to immunoassay for steroid hormones and as such used as a reference anchor. LC-MS/MS methods also need this traceability anchor for steroids. GC-MS / GC-MS/MS / GC-HRMS verification provides a separate methodological platform for this purpose. This approach is ideal for steroids (which can be made volatile) as they often demonstrate improved separation by GC, and do not suffer from the significant problem of ion alteration due to phospholipid interference. GC analysis also has some disadvantages, namely the potential alteration of the target analyte during the derivatisation process, which is necessary for making the steroids “volatile”. When there is agreement between the result generated by LC-MS/MS and GC-MS a solid anchor is provided for method harmonisation. The development of a GC-MS/MS method to complement the LC-MS/MS methods by NMIA demonstrates the value of this approach to support “trueness” of results.
Even with the aid of primary reference materials and methods, the employment of a common secondary calibrator only made a significant improvement in the distribution of the female Unknown human results compared to the indicative values provided by NMIA. The lack of change with the CC (other than the female sample) can be seen as a good thing, in that there is already good quality being delivered; indicating that value assignment of calibrators, commutability and calibrator preparation and handling were done well. This is important as it indicates that the different calibrators in use, and how they are prepared and used, all lead to the same results (i.e. not improved by a common calibrator) and hence is a statement of strength for current laboratory and calibrator quality.
Trials to improve the standardisation of methods through the application of a common calibrator have been reported previously.53–55 The results from our CC pilot were consistent with these earlier studies. The change of bias did vary between laboratories. As expected imprecision was not affected. Hence we postulate that the routine methods themselves (as distinct from the calibrator) may be influencing the imprecision and bias; which are likely to include the areas of difference observed in our participant questionnaire. In particular we speculate that assay imprecision could be affected by: 1) Instrument maintenance, especially of the ion source, and in rare occasions also the cross-talk in the collision cell; 2) phospholipid interference may have contributed to this variation for some laboratories; and 3) the choice of isotope-labelled internal standard.6 Additional information and problems raised from our investigations related to the biological variation data and the practical approach to maintaining ongoing traceability.
To determine if the bias and imprecision in results were acceptable, biological variation data was used to establish clinically significant differences.23
In determining the acceptability of these new “recalculated” values, the acceptable limits were determined against the desirable total allowable error from the Ricos biological variation database.22,23 This data is however potentially problematic for serum testosterone as it is primarily generated from studies conducted on adult males.56–62 There is currently no biological variation data for serum testosterone available for children. There is however one study that provides combined information for adult male (n=13) and female (n=13) plasma testosterone levels analysed by immunoassay which demonstrates the intra-individual and inter-individual CV for plasma testosterone to be 12.6% and 40.8% respectively.63 Hence the use of the biological variation database may not be appropriate to determine acceptability of analytical performance for serum testosterone levels in women and children. Further studies are therefore needed to determine the CVi and CVg in women and children using assays that are appropriate for the task.
Other harmonisation initiatives have also shown that LC-MS/MS methods do not fully meet the desirable imprecision, bias and total allowable error performance criteria. In a study of the certified assays in the Centres for Disease Control and Prevention (CDC) HoSt program, five “certified” laboratories (four LC-MS/MS and one immunoassay) were challenged with 40 specimens that had assigned testosterone values based on the CDC reference method. These laboratories were compared over a one year period against the CDC reference method. Biological variation data was used to determine acceptable performance. None of the LC-MS/MS laboratories achieved desirable imprecision or bias 100% of the time; with only one lab showing 100% desirable imprecision for males. Only one LC-MS/MS laboratory met the desirable TEa 100% of the time. Even when the minimum performance criteria was applied, only one out of four LC-MS/MS labs met the minimum imprecision level and two out of four labs met the bias and TEa performance criteria.64 “As has been pointed out previously, simply implementing a mass spectrometry-based assay does not equate with accuracy and precision; it is essential that all assays are rigorously validated”.64 Having an appreciation of the methods used by these laboratories would provide insight into the similarities and differences that may be influencing LC-MS/MS method performance.
The choice of isotope labelling for the internal standard for LC-MS/MS analysis of serum testosterone can potentially have a significant influence on the results. The outcome of our method questionnaire demonstrated that the choice of internal standard varied between laboratories; with four (50%) laboratories using D2. It is generally considered better to use a D3 or higher deuterium-labelled internal standard as there are less isotope effects compared with D2.29 A study by Owen and colleagues comparing D2 with D5 and also C13 internal standards for testosterone demonstrates the influence on patient results based on the choice of internal standard; with this variability being consistent in the male and female serum testosterone range.65 In Owen’s study, the D2 results were reported to “give results close to the reference method using conditions described here, but this may not give the best results using different sample clean-up procedures and chromatography columns”.65 The D5 and C13 results (compared to D2) were generally lower.65 The selection of D2 is not ideal as it is only two additional daltons from the target analyte which may lead to interference from the target analyte at high concentrations due to the presence of 13C2 isotopomers of the target.66 On the other hand, stable isotopes can only compensate for ion alteration effects if they co-elute with the compound and hence are present in the ion source at the same time. The greater the number of deuterated atoms, the less likely the internal standard will co-elute; however D5 is usually acceptable for low resolution chromatography. C13-labelled testosterone is considered a more stable and acceptable, albeit more expensive, alternative to deuterium labelled internal standards and is now commercially available for testosterone and overall may be a better alternative to support serum testosterone harmonisation.
The collaborative process of harmonisation of LC-MS/MS measurement of serum testosterone highlights the advantages and also issues related to establishing and maintaining standardisation with traceability. These background studies also provide a mechanism to generate initial recommendations and associated gaps in knowledge related to the LC-MS/MS analysis of serum testosterone; which are presented in Table 5.
Table 5.
Recommendations and major gaps identified related to the measurement of serum testosterone.
| Section | Recommendations | Gap Analysis |
|---|---|---|
| Pre-analytical: |
|
|
| Analytical – LC-MS/MS sample preparation: |
|
|
| Analytical – LC-MS/MS analysis: |
|
|
| Analytical - Standardisation: |
|
|
| Post-analytical: |
|
|
The determination of commutability of the calibrators and EQA material is vital and in this study we have made a presumption of commutability based on the studies with the two Unknown samples compared to the RCPAQAP and CC material based on the observed slopes (Appendix). In addition, the data generated from the RCPAQAP Liquid Serum Chemistry Program also supports commutability of the material for the MS based methods (data not shown). Even so, there is more work to be performed in this area, which could include the sharing of more native samples to: (a) demonstrate the between-method assay performance and (b) validate (or not) the commutability of the EQA material for LC-MS/MS methods. However, the validation of each laboratory’s calibrator through sample sharing is not an easy process. Hence, the formal validation of commutability of the RCPAQAP (and other EQA program) material is essential to monitor and interpret data related to harmonisation.
Future Directions
The findings of our harmonisation project provide directions for further harmonisation of mass spectrometry based serum steroid methods. Even with these initial recommendations in place there is still need to address a number of other issues, which include:
Sample preparation and extraction procedure: A more detailed look at the individual sample preparations would help to determine which assays could be compromised by co-extracted lipid matrix components, such as phospholipids. The extent of this problem would vary depending on the sample preparation protocol used and cannot be totally avoided. Therefore, it is very important to chromatographically separate this lipid fraction from the target analytes. This can be routinely monitored through the phospholipid common MRM of 184>184 and 104>104 for positive electrospray ionisation.29
Establishment of commutability of all calibrators and EQA material: Commutability needs to be established experimentally for primary and secondary calibrators, and EQA materials to compare between methods.52,62,67,68 Commutability was first used to describe the “ability of an enzyme material to show inter-assay activity changes comparable to those of the same enzyme in human serum”.69 The Clinical and Laboratory Standards Institute provides a practical definition to encompass all measurands specific to laboratory medicine being the “property of a reference material, demonstrated by the equivalence of the mathematical relationships among the results of different measurement procedures for a reference material and for representative samples of the type intended to be measured.”67 Establishing the commutability of a reference material for a specific measurand confirms that it is suitable for use as a calibrator and has potential to be employed in robust target setting in an EQA program. Ideally to assess commutability a range of samples of at least 40 patients is used for assessment. Calibrators and EQA material should be assessed in this manner along with the patient samples. The results are compared between two laboratories by comparing one laboratory on the ordinate (y) axis against the other on the abscissa (x) axis. If all material is commutable then the slopes of each of the materials will be consistent between each sample type. Defining limits of acceptance is more difficult and there are varying approaches in the literature.62 To look further at this, the AACB has recently formed a Commutability Working Party to develop assessment protocols and decision criteria to advance this work.
Common Reference Intervals and Decision Limits: There are now a number of publications in the recent peer reviewed literature promoting mass spectrometry based serum testosterone reference intervals.41–45,47–49 However there are differences in the reference intervals generated between these publications. Once methods are harmonised/standardised, common reference intervals and decision limits can be recommended for testosterone and the other commonly measured serum steroids. Harmonisation of reference intervals for serum steroids within our group is planned for the future, with serum testosterone being the trial analyte considered in the first instance.
Reflection
Since 2010, when we commenced the formal collaborative process to improve agreement of our LC-MS/MS methods, we have now achieved an overall group imprecision for serum testosterone that meets the desirable specifications based on biological variation. Whilst the selection of serum testosterone as the ‘test case’ model to apply to this collaborative process was based on the common interest of the founding group membership, it also proved to be the ideal steroid to trial. This was because there was already clear information related to testosterone in the JCTLM and biological variation databases and that the common EQA program used traceable targets. Unfortunately, this is not the case for a number of other steroids listed in the RCPAQAP Endocrine program. Hence, alternative processes will also need to be considered to aid the progression of harmonisation of all common steroids measured by LC-MS/MS.
Conclusions
Obtaining between laboratory agreements of results is a challenging process. Here we have highlighted the current challenges of standardisation for serum steroids measured by LC-MS/MS focusing on serum testosterone. This is the first such report to fully characterise the LC-MS/MS methods for serum testosterone in common use by clinical diagnostic laboratories. In all aspects the participation in a common EQA program is essential to ensure ongoing agreement of results and the activities presented here provide a practical demonstration of the central importance of EQA in this process. It is gratifying to know that over time, as a group, performance in the common EQA scheme has significantly improved in terms of both imprecision and bias.
Acknowledgments
First and foremost we wish to thank Ms Danny Sampson for her impetus and encouragement for the formation of this working group. We wish to thank Drs Veronica Vamathevan, John Murby and Lindsey Mackay from the National Measurement Institute in North Ryde, NSW for their tireless work in promoting traceability and developing the serum testosterone reference methods. In relation to this project, there were many additional people who have supported this work and we gratefully extend our thanks to the following individuals: Mr Michael Rennie from PM Separations for his coordination of distribution of the CC study materials to each participating laboratory; Mr Ian Farrance for his input into the statistical analysis; Dr Christa Cobbaert for her insights into the practical assessment of commutability; Ms Jill Tate for her vast unassuming knowledge and support of this project as chair of the AACB Harmonisation Committee; and Dr Leslie Lai for his unyielding support for this project as President of the APFCB.
Appendix
Table:
Comparison of the slopes obtained be each laboratory (y axis) compared to the NMIA testosterone target value (x axis). The slope (a) and intercept (b) are calculated by y=ax +b. These results were obtained using the laboratories own calibrator and are therefore note corrected in any way i.e. prior to recalculation against the CC. All labs (except lab 4) used 1/x weighting for their calibration curve. At the time of this study lab 4 used no weighting - as a result of this study lab 4 moved to 1/x weighting. All samples were run in duplicate and the run was repeated one month later. Results were NOT averaged for the purpose of this comparison. A two tailed paired t-test demonstrated the difference between the means of the slopes were not significantly different i.e. all comparisons had a p>0.05.
| Laboratory | RCPAQAP Material Slope | Common Calibrator Material Slope | Unknown Fresh frozen serum Slope |
|---|---|---|---|
| 1* | 0.986 | 0.985 | 1.0399 |
| 2* | 0.9784 | 0.9667 | 0.9989 |
| 3** | 0.8949 | 0.9369 | 0.8915 |
| 4** | 0.9196 | 0.9706 | 0.9406 |
| 5* | 0.9961 | 1.0268 | 0.992 |
| 6*** | 0.9252 | 1.0834 | 0.9539 |
| 7* | 1.0251 | 1.019 | 1.0286 |
| 8* | 1 | 1 | 0.9621 |
| Summary of slopes | |||
| Minimum slope | 0.895 | 0.937 | 0.892 |
| Maximum slope | 1.025 | 1.083 | 1.040 |
| Mean slope | 0.966 | 0.999 | 0.976 |
| Median slope | 0.982 | 0.993 | 0.977 |
| SD of slope | 0.046 | 0.045 | 0.049 |
| Minus 2SD of slope | 0.873 | 0.908 | 0.878 |
| Plus 2SD of slope | 1.058 | 1.089 | 1.074 |
| Potential overall difference (95% range) between slopes | 19% | 18% | 20% |
R2 was >0.99 for all materials
R2 was >0.99 for Common Calibrator & Fresh frozen serum, but < 0.99 for the RCPAQAP material assessment (R2= 0.988 for lab 3) and (R2 = 0.9771 for lab 4)
R2 was >0.99 for the RCPAQAP material, but < 0.99 for the Common Calibrator (R2 = 0.9891) and the Fresh frozen serum (R2 = 0.9883)
Note: Laboratory 8 was the reference laboratory, i.e. NMIA LC-MS/MS method.
Figure.

comparison of CC weighed in values compared to NMIA values assigned by RMP. The NMIA values were taken as the “true” concentration for each level of the common calibrator group comparison.
Footnotes
Competing Interests: None declared (RFG,CSH, KEH, JJ, JPG, BCM, CF, YPI, BRC, CB, HTP, LMJ). TK and HTP are employees of Biocrates Life Sciences AG.
References
- 1.Shackleton C. Clinical steroid mass spectrometry: a 45-year history culminating in HPLC-MS/MS becoming an essential tool for patient diagnosis. J Steroid Biochem Mol Biol. 2010;121:481–90. doi: 10.1016/j.jsbmb.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 2.The Nobel Prize in Chemistry. 2002. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2002/ (Accessed 21 October 2015)
- 3.Pitt JJ. Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin Biochem Rev. 2009;30:19–34. [PMC free article] [PubMed] [Google Scholar]
- 4.Adaway JE, Keevil BG, Owen LJ. Liquid chromatography tandem mass spectrometry in the clinical laboratory. Ann Clin Biochem. 2015;52:18–38. doi: 10.1177/0004563214557678. [DOI] [PubMed] [Google Scholar]
- 5.AACB Chromatography and Mass Spectrometry Meeting: Coming Together to Separate.; Royal Prince Alfred Hospital, Sydney, Australia. 16–18th July 2007. [Google Scholar]
- 6.Annesley TM. Ion suppression in mass spectrometry. Clin Chem. 2003;49:1041–4. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 7.Koal T, Deigner HP. Challenges in mass spectrometry based targeted metabolomics. Curr Mol Med. 2010;10:216–26. doi: 10.2174/156652410790963312. [DOI] [PubMed] [Google Scholar]
- 8.Pollack A. Quest Acknowledges Errors in Vitamin D Tests. New York Times. Jan 7–8, 2009. http://www.nytimes.com/2009/01/08/business/08labtest.html?_r=0 (Accessed 18 September 2015)
- 9.Asian Pacific Conference of Chromatography and Mass Spectrometry: New Horizons in Clinical Chemistry”.; Hong Kong SAR, China.. 14–16 January 2010; http://www.medicine.org.hk/hkscc/communication/2010/Mass%20Spectrometry.pdf (Accessed 21 October 2015) [Google Scholar]
- 10.Asian and Pacific Federation of Clinical Biochemistry and Laboratory Medicine. (APFCB) Mass Spectrometry Harmonisation Working Group. http://www.apfcb.org/scientific-activities.html (Accessed 21 September 2015)
- 11.Australasian Association of Clinical Biochemists (AACB) Harmonisation Committee. http://www.aacb.asn.au/professionaldevelopment/harmonisation (Accessed 21 September 2015)
- 12.Greaves RF, Sampson D, Yakamora K, Ho CS, Introducing the Mass Spectrometry Harmonisation Working Group 12th Asia Pacific Congress of Clinical Biochemistry; Seoul South Korea.. 3–7 October 2010. [Google Scholar]
- 13.Jones GR, Sikaris K, Gill J. ‘Allowable Limits of Performance’ for External Quality Assurance Programs - an Approach to Application of the Stockholm Criteria by the RCPA Quality Assurance Programs. Clin Biochem Rev. 2012;33:133–9. [PMC free article] [PubMed] [Google Scholar]
- 14.Panteghini M. Implementation of standardization in clinical practice: not always an easy task. Clin Chem Lab Med. 2012;50:1237–41. doi: 10.1515/cclm.2011.791. [DOI] [PubMed] [Google Scholar]
- 15.Joint Committee for Traceability in Laboratory Medicine. 2002. Appendix III, The JCTLM Framework: A Framework for the international recognition of available higher-order reference materials, available higher-order reference measurement procedures and reference measurement laboratories for laboratory medicine. http://www.bipm.org/utils/en/pdf/jctlm_framework.pdf (Accessed 17 August 2015)
- 16.International Organization for Standardization. In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values assigned to calibrators and control materials. ISO. 17511:2003. http://infostore.saiglobal.com/store/Details.aspx?ProductID=259933&gclid=CMygs5uN8cgCFYmUvQodPaMAJw (Accessed 2 November 2015) [Google Scholar]
- 17.The International System of Units (SI) http://www.bipm.org/en/si/.http://www.bipm.org/en/si/ (Accessed 2 November 2015)
- 18.Greaves RF. Using QAP to support standardisation efforts. Clin Biochem Rev. 2013;34:S10. [Google Scholar]
- 19. Chemical Pathology Programs RC. Endocrine Program. http://www.rcpaqap.com.au/chempath (Accessed 27 September 2015)
- 20.Hyltoft Petersen P, Fraser CG, Kallner A, Kenny D. Strategies to set global analytical quality specifications in laboratory medicine. Scand J Clin Lab Invest. 1999;59:475–585. [PubMed] [Google Scholar]
- 21.Sandberg S, Fraser CG, Horvath AR, Jansen R, Jones G, Oosterhuis W, et al. Defining analytical performance specifications: Consensus Statement from the 1st Strategic Conference of the European Federation of Clinical Chemistry and Laboratory Medicine. (CCLM) Clin Chem Lab Med. 2015;53:833–5. doi: 10.1515/cclm-2015-0067. [DOI] [PubMed] [Google Scholar]
- 22.Fraser CG. Biological Variation: From Principle to Practice. Washington DC: AACC Press; 2001. ISBN 1-890883-49-2. [Google Scholar]
- 23.Ricós C, Alvarez V, Cava F, García-Lario JV, Hernández A, Jiménez CV, et al. Current databases on biological variation: pros, cons and progress. Scand J Clin Lab Invest. 1999;59:491–500. doi: 10.1080/00365519950185229. http://www.westgard.com/biodatabase1.htm (Accessed 27 September 2015). [DOI] [PubMed] [Google Scholar]
- 24.Greaves R, Jolly L, Woollard G, Hoad K. Serum vitamin A and E analysis: comparison of methods between laboratories enrolled in an external quality assurance programme. Ann Clin Biochem. 2010;47:78–80. doi: 10.1258/acb.2009.009116. [DOI] [PubMed] [Google Scholar]
- 25.Hoad KE, Johnson LA, Woollard GA, Walmsley TA, Briscoe S, Jolly LM, et al. Vitamin B1 and B6 method harmonization: comparison of performance between laboratories enrolled in the RCPA Quality Assurance Program. Clin Biochem. 2013;46:772–6. doi: 10.1016/j.clinbiochem.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Coakley J, Scott S, Mackay R, Greaves R, Jolly L, Massie J, et al. Sweat testing for cystic fibrosis: standards of performance in Australasia. Ann Clin Biochem. 2009;46:332–7. doi: 10.1258/acb.2009.009023. [DOI] [PubMed] [Google Scholar]
- 27.Sports Doping Control Reference Material Catalogue (Steroids, Steroid Metabolites and Stimulants). Australian Government National Measurement Institute 2014 Oct; http://www.measurement.gov.au/Services/Documents/SportsDopingCatalogue.pdf (Accessed 21 October 2015) [Google Scholar]
- 28.Bureau International des Poids et Mesures (BIPM) Key Comparisons Database. http://kcdb.bipm.org/ (Accessed 21 September 2015) [DOI] [PubMed]
- 29.Honour JW. Development and validation of a quantitative assay based on tandem mass spectrometry. Ann Clin Biochem. 2011;48:97–111. doi: 10.1258/acb.2010.010176. [DOI] [PubMed] [Google Scholar]
- 30.Koal T, Schmiederer D, Pham-Tuan H, Röhring C, Rauh M. Standardized LC-MS/MS based steroid hormone profile-analysis. J Steroid Biochem Mol Biol. 2012;129:129–38. doi: 10.1016/j.jsbmb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 31.German Reference Institute for Bioanalytic. (Referenzinstitut für Bioanalytik, RfB,). http://www.dgkl-rfb.de/ (Accessed 21 October 2015)
- 32.Sanchez-Guijo A, Hartmann MF, Wudy SA. Chapter 3 Introduction to gas chromatograph-mass spectrometry. In: Wheeler MJ, editor. Hormone Assays in Biological Fluids. Second Edition. Humana Press; 2013. Methods in Molecular Biology, volume 1065. [DOI] [Google Scholar]
- 33.International Organization for Standardization. Uncertainty of measurement — Part 3: Guide to the expression of uncertainty in measurement (GUM:1995). http://webstore.iec.ch/preview/info_isoiecguide98-3%7Bed1.0%7Den.pdf (Accessed 21 October 2015)
- 34. GraphPad Prism. http://www.graphpad.com/scientific-software/prism/ (Accessed 2 November 2015)
- 35.Cawood ML, Field HP, Ford CG, Gillingwater S, Kicman A, Cowan D, et al. Testosterone measurement by isotope-dilution liquid chromatography-tandem mass spectrometry: validation of a method for routine clinical practice. Clin Chem. 2005;51:1472–9. doi: 10.1373/clinchem.2004.044503. [DOI] [PubMed] [Google Scholar]
- 36.Kushnir MM, Rockwood AL, Roberts WL, Pattison EG, Bunker AM, Fitzgerald RL, et al. Performance characteristics of a novel tandem mass spectrometry assay for serum testosterone. Clin Chem. 2006;52:120–8. doi: 10.1373/clinchem.2005.052167. [DOI] [PubMed] [Google Scholar]
- 37.Kushnir MM, Rockwood AL, Roberts WL, Pattison EG, Owen WE, Bunker AM, et al. Development and performance evaluation of a tandem mass spectrometry assay for 4 adrenal steroids. Clin Chem. 2006;52:1559–67. doi: 10.1373/clinchem.2006.068445. [DOI] [PubMed] [Google Scholar]
- 38.Rauh M, Gröschl M, Rascher W, Dörr HG. Automated, fast and sensitive quantification of 17 alpha-hydroxyprogesterone, androstenedione and testosterone by tandem mass spectrometry with on-line extraction. Steroids. 2006;71:450–8. doi: 10.1016/j.steroids.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 39.Magnisali P, Dracopoulou M, Mataragas M, Dacou-Voutetakis A, Moutsatsou P. Routine method for the simultaneous quantification of 17alpha-hydroxyprogesterone, testosterone, dehydroepiandrosterone, androstenedione, cortisol, and pregnenolone in human serum of neonates using gas chromatography-mass spectrometry. J Chromatogr A. 2008;1206:166–77. doi: 10.1016/j.chroma.2008.07.057. [DOI] [PubMed] [Google Scholar]
- 40.Shiraishi S, Lee PW, Leung A, Goh VH, Swerdloff RS, Wang C. Simultaneous measurement of serum testosterone and dihydrotestosterone by liquid chromatography-tandem mass spectrometry. Clin Chem. 2008;54:1855–63. doi: 10.1373/clinchem.2008.103846. [DOI] [PubMed] [Google Scholar]
- 41.Soldin OP, Sharma H, Husted L, Soldin SJ. Pediatric reference intervals for aldosterone, 17α-hydroxyprogesterone, dehydroepiandrosterone, testosterone and 25-hydroxy vitamin D3 using tandem mass spectrometry. Clin Biochem. 2009;42:823–7. doi: 10.1016/j.clinbiochem.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulle AE, Riepe FG, Melchior D, Hiort O, Holterhus PM. A novel ultrapressure liquid chromatography tandem mass spectrometry method for the simultaneous determination of androstenedione, testosterone, and dihydrotestosterone in pediatric blood samples: age- and sex-specific reference data. J Clin Endocrinol Metab. 2010;95:2399–409. doi: 10.1210/jc.2009-1670. [DOI] [PubMed] [Google Scholar]
- 43.Kushnir MM, Blamires T, Rockwood AL, Roberts WL, Yue B, Erdogan E, et al. Liquid chromatography-tandem mass spectrometry assay for androstenedione, dehydroepiandrosterone, and testosterone with pediatric and adult reference intervals. Clin Chem. 2010;56:1138–47. doi: 10.1373/clinchem.2010.143222. [DOI] [PubMed] [Google Scholar]
- 44.Fanelli F, Belluomo I, Di Lallo VD, Cuomo G, De Iasio R, Baccini M, et al. Serum steroid profiling by isotopic dilution-liquid chromatography-mass spectrometry: comparison with current immunoassays and reference intervals in healthy adults. Steroids. 2011;76:244–53. doi: 10.1016/j.steroids.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 45.Kyriakopoulou L, Yazdanpanah M, Colantonio DA, Chan MK, Daly CH, Adeli K. A sensitive and rapid mass spectrometric method for the simultaneous measurement of eight steroid hormones and CALIPER pediatric reference intervals. Clin Biochem. 2013;46:642–51. doi: 10.1016/j.clinbiochem.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Botelho JC, Shacklady C, Cooper HC, Tai SS, Van Uytfanghe K, Thienpont LM, et al. Isotope-dilution liquid chromatography-tandem mass spectrometry candidate reference method for total testosterone in human serum. Clin Chem. 2013;59:372–80. doi: 10.1373/clinchem.2012.190934. [DOI] [PubMed] [Google Scholar]
- 47.Søeborg T, Frederiksen H, Mouritsen A, Johannsen TH, Main KM, Jørgensen N, et al. Sex, age, pubertal development and use of oral contraceptives in relation to serum concentrations of DHEA, DHEAS, 17α-hydroxyprogesterone, Δ4-androstenedione, testosterone and their ratios in children, adolescents and young adults. Clin Chim Acta. 2014;437:6–13. doi: 10.1016/j.cca.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Keefe CC, Goldman MM, Zhang K, Clarke N, Reitz RE, Welt CK. Simultaneous measurement of thirteen steroid hormones in women with polycystic ovary syndrome and control women using liquid chromatography-tandem mass spectrometry. PLoS One. 2014;9:e93805. doi: 10.1371/journal.pone.0093805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greaves RF, Pitkin J, Ho CS, Baglin J, Hunt RW, Zacharin MR. Hormone modeling in preterm neonates: establishment of pituitary and steroid hormone reference intervals. J Clin Endocrinol Metab. 2015;100:1097–103. doi: 10.1210/jc.2014-3681. [DOI] [PubMed] [Google Scholar]
- 50.Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Performance enhancement in the measurement of 5 endogenous steroids by LC-MS/MS combined with differential ion mobility spectrometry. Clin Chim Acta. 2015;438:330–6. doi: 10.1016/j.cca.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 51.Greaves RF, Jevalikar G, Hewitt JK, Zacharin MR. A guide to understanding the steroid pathway: new insights and diagnostic implications. Clin Biochem. 2014;47:5–15. doi: 10.1016/j.clinbiochem.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 52.Greaves RF. A guide to harmonisation and standardisation of measurands determined by liquid chromatography - tandem mass spectrometry in routine clinical biochemistry. Clin Biochem Rev. 2012;33:123–32. [PMC free article] [PubMed] [Google Scholar]
- 53.Carter GD, Jones JC. Use of a common standard improves the performance of liquid chromatography–tandem mass spectrometry methods for serum 25-hydroxyvitamin-D. Ann Clin Biochem. 2009;46:79–81. doi: 10.1258/acb.2008.008135. and 46:434 (Erratum). [DOI] [PubMed] [Google Scholar]
- 54.Greaves RF, Woollard GA, Hoad KE, Walmsley TA, Johnson LA, Briscoe S, et al. Laboratory medicine best practice guideline: vitamins a, e and the carotenoids in blood. Clin Biochem Rev. 2014;35:81–113. [PMC free article] [PubMed] [Google Scholar]
- 55.Owen LJ, MacDonald PR, Keevil BG. Is calibration the cause of variation in liquid chromatography tandem mass spectrometry testosterone measurement? Ann Clin Biochem. 2013;50:368–70. doi: 10.1177/0004563212474563. [DOI] [PubMed] [Google Scholar]
- 56.Ooi LS, Panesar NS, Masarei JR. Within- and between-subject variation in, and associations between, serum concentrations and urinary excretion of testosterone and estradiol in Chinese men. Clin Chim Acta. 1995;236:87–92. doi: 10.1016/0009-8981(95)06036-0. [DOI] [PubMed] [Google Scholar]
- 57.Ricós C, Arbós MA. Objetivos de calidad para las determinaciones de hormonas. Endocrinologia. 1990;37:230–3. [Google Scholar]
- 58.Ricós C, Arbós MA. Quality goals for hormone testing. Ann Clin Biochem. 1990;27:353–8. doi: 10.1177/000456329002700412. [DOI] [PubMed] [Google Scholar]
- 59.Valero-Politi J, Fuentes-Arderiu X. Within- and between-subject biological variations of follitropin, lutropin, testosterone, and sex-hormone-binding globulin in men. Clin Chem. 1993;39:1723–5. [PubMed] [Google Scholar]
- 60.Ahokoski O, Virtanen A, Huupponen R, Scheinin H, Salminen E, Kairisto V, et al. Biological day-to-day variation and daytime changes of testosterone, follitropin, lutropin and oestradiol-17beta in healthy men. Clin Chem Lab Med. 1998;36:485–91. doi: 10.1515/CCLM.1998.081. [DOI] [PubMed] [Google Scholar]
- 61.Andersson AM, Carlsen E, Petersen JH, Skakkebaek NE. Variation in levels of serum inhibin B, testosterone, estradiol, luteinizing hormone, follicle-stimulating hormone, and sex hormone-binding globulin in monthly samples from healthy men during a 17-month period: possible effects of seasons. J Clin Endocrinol Metab. 2003;88:932–7. doi: 10.1210/jc.2002-020838. [DOI] [PubMed] [Google Scholar]
- 62.Ricós C, Juvany R, Jiménez CV, Perich C, Minchinela J, Hernández A, et al. Procedure for studying commutability validated by biological variation. Clin Chim Acta. 1997;268:73–83. doi: 10.1016/s0009-8981(97)00172-1. [DOI] [PubMed] [Google Scholar]
- 63.Maes M, Mommen K, Hendrickx D, Peeters D, D’Hondt P, Ranjan R, et al. Components of biological variation, including seasonality, in blood concentrations of TSH, TT3, FT4, PRL, cortisol and testosterone in healthy volunteers. Clin Endocrinol (Oxf) 1997;46:587–98. doi: 10.1046/j.1365-2265.1997.1881002.x. [DOI] [PubMed] [Google Scholar]
- 64.Yun Y-M, Botelho JC, Chandler DW, Katayev A, Roberts WL, Stanczyk FZ, et al. Performance criteria for testosterone measurements based on biological variation in adult males: recommendations from the Partnership for the Accurate Testing of Hormones. Clin Chem. 2012;58:1703–10. doi: 10.1373/clinchem.2012.186569. [DOI] [PubMed] [Google Scholar]
- 65.Owen LJ, Keevil BG. Testosterone measurement by liquid chromatography tandem mass spectrometry: the importance of internal standard choice. Ann Clin Biochem. 2012;49:600–2. doi: 10.1258/acb.2012.012037. [DOI] [PubMed] [Google Scholar]
- 66.Clinical and Laboratory Standards Institute. Mass Spectrometry in the Clinical Laboratory: General Principles and Guidance. Approved Guideline. Wayne PA: CLSI; 2007. CLSI document C50-A (ISBN 1-562380648-4). [Google Scholar]
- 67.Clinical and Laboratory Standards Institute. Characterization and Qualification of Commutable Reference Materials for Laboratory Medicine; Approved Guideline. Wayne PA: CLSI; 2010. CLSI document C53-A. [Google Scholar]
- 68.Miller WG, Myers GL, Rej R. Why commutability matters. Clin Chem. 2006;52:553–4. doi: 10.1373/clinchem.2005.063511. [DOI] [PubMed] [Google Scholar]
- 69.Fasce CF, Jr, Rej R, Copeland WH, Vanderlinde RE. A discussion of enzyme reference materials: applications and specifications. Clin Chem. 1973;19:5–9. [PubMed] [Google Scholar]
- 70.Hawkins RC, Johnson RN. The significance of significant figures. Clin Chem. 1990;36:824. [PubMed] [Google Scholar]
- 71.Badrick T, Wilson SR, Dimeski G, Hickman PE. Objective determination of appropriate reporting intervals. Ann Clin Biochem. 2004;41:385–90. doi: 10.1258/0004563041731583. [DOI] [PubMed] [Google Scholar]