

Abstract
Acute kidney injury (AKI) is a clinical syndrome that complicates the course and worsens the outcome in a significant number of hospitalised patients. Recent advances in clinical and basic research will help with a more accurate definition of this syndrome and in the elucidation of its pathogenesis. With this knowledge we will be able to conduct more accurate epidemiologic studies in an effort to gain a better understanding of the impact of this syndrome. AKI is a syndrome that rarely has a sole and distinct pathophysiology. Recent evidence, in both basic science and clinical research, is beginning to change our view for AKI from a single organ failure syndrome to a syndrome where the kidney plays an active role in the progress of multi-organ dysfunction. Accurate and prompt recognition of AKI and better understanding of the pathophysiologic mechanisms underlying the various clinical phenotypes are of great importance to research for effective therapeutic interventions. In this review we provide the most recent updates in the definition, epidemiology and pathophysiology of AKI.
Introduction
The concept of Acute Renal Failure (ARF)1 has undergone significant re-examination in recent years. Traditionally, emphasis was given to the most severe acute reduction in kidney function, as manifested by severe azotaemia and often by oliguria or anuria. However, recent evidence suggests that even relatively mild injury or impairment of kidney function manifested by small changes in serum creatinine (sCr) and/or urine output (UO), is a predictor of serious clinical consequences.2–5
Acute Kidney Injury (AKI) is the term that has recently replaced the term ARF. AKI is defined as an abrupt (within hours) decrease in kidney function, which encompasses both injury (structural damage) and impairment (loss of function). It is a syndrome that rarely has a sole and distinct pathophysiology. Many patients with AKI have a mixed aetiology where the presence of sepsis, ischaemia and nephrotoxicity often co-exist and complicate recognition and treatment. Furthermore the syndrome is quite common among patients without critical illness and it is essential that health care professionals, particularly those without specialisation in renal disorders, detect it easily.
Classification of AKI includes pre-renal AKI, acute post-renal obstructive nephropathy and intrinsic acute kidney diseases. Of these, only ‘intrinsic’ AKI represents true kidney disease, while pre-renal and post-renal AKI are the consequence of extra-renal diseases leading to the decreased glomerular filtration rate (GFR). If these pre- and/or post-renal conditions persist, they will eventually evolve to renal cellular damage and hence intrinsic renal disease.
The current diagnostic approach of AKI is based on an acute decrease of GFR, as reflected by an acute rise in sCr levels and/or a decline in UO over a given time interval.6–8 Recently several biomarkers have been proposed for the diagnosis of AKI and these are in various stages of development and validation.9–12 Nevertheless, it is not clear, if a single or multiple biomarker approach is necessary to diagnose the complicated and multifactorial aspects of AKI.13–16
However, in addition to the analytical difficulties associated with each specific biomarker, there is also an issue concerning the appropriate reference point, and more specifically about using sCr as the standard, for the clinical evaluation of these biomarkers. It is known that sCr is insensitive to acute changes of renal function and levels can vary widely with age, gender, muscle mass, diet, medications and hydration status. Moreover it is not a direct marker of tubular damage, but rather a marker of GFR, and substantial increases in sCr can be observed in renal hypo-perfusion even when the kidneys are structurally intact, resulting in pre-renal azotaemia. For these reasons sCr is considered an ‘imperfect gold standard’ for the diagnosis of AKI.17 Another issue with sCr is that in most clinical situations its true baseline value is not known, which makes the evaluation of patients very difficult.18–20 Moreover, given the phenotypic variability of AKI (different clinical phenotypes with distinct underlying pathophysiologies), it is not clear whether different approaches are necessary for diagnosis and monitoring of the clinical course and therapy.
In this review we will discuss the epidemiology and the definition of AKI. We will also discuss the clinical phenotypes, their pathophysiology and the link between AKI and remote organ dysfunction.
History
The first description of ARF, then termed ischuria renalis, was by William Heberden in 1802.21 At the beginning of the twentieth century, ARF, then named Acute Bright’s disease, was described in William Osler’s Textbook for Medicine (1909), to be “as a consequence of toxic agents, pregnancy, burns, trauma or operations on the kidneys”. During the First World War the syndrome was named war nephritis,22 and was reported in several publications. The syndrome was forgotten until the Second World War, when Bywaters and Beall published their classical paper on crush syndrome.23 Acute tubular necrosis (ATN) was the term that was used to describe this clinical entity, because of histological evidence for patchy necrosis of renal tubules at autopsy. For many years in clinical practice, the terms ATN and ARF were used interchangeably. However, it is Homer W. Smith who is credited for the introduction of the term acute renal failure, in a chapter on Acute renal failure related to traumatic injuries in his 1951 textbook The kidney-structure and Function in Health and Disease. Until recently, a precise biochemical definition for ARF was missing. As a consequence there was no consensus on the diagnostic criteria, resulting in multiple different definitions. A 2002 survey revealed at least 35 definitions in the scientific literature.24
Epidemiology
The lack of standard definition of the syndrome had a great impact in the reported incidence and clinical significance of AKI and its true impact is not well known. The incidence varies, depending on the definition used, patient population and geographical area studied.25–27
Large differences are observed in the incidence and the causes of AKI between developing and developed countries. A recent review described the similarities and differences in incidence, cause, pathophysiology, and public health implications of AKI in developed and developing regions of the world.28
In urban areas of developing countries, main causes of AKI are hospital acquired (renal ischaemia, sepsis and nephrotoxic drugs) while in rural areas it is more commonly a consequence of community acquired disease (diarrhoea, dehydration, infectious diseases, animal venoms etc.). Under-reporting of AKI especially in developing countries is also a major problem that relates with the true knowledge of its impact in many parts of the world.29
In developed countries the prevalence of AKI is increasing. In hospital inpatients it is estimated to occur up to 15% and is more common in critically ill patients, in whom its prevalence is estimated to be up to 60%.28,30–34 On the other hand community AKI is usually uncommon although a recent study estimated its incidence at 4.3% among all hospital admissions.35 However even this incidence remains an underestimate of the true impact of community acquired AKI due to non-referral of patients to hospitals.
Although several studies have focused on special populations (the elderly and children), large epidemiologic studies with children are missing and the incidence of paediatric AKI is inadequately described. The reason is that most paediatric epidemiologic studies were limited to a single centre with small number of patients and focused on critical care populations or on children requiring dialysis.36–40 In a recent large scale epidemiologic study, the incidence of AKI in hospitalised children in the US was found to occur in 3.9 per 1000 admissions.36 The majority of AKI cases in children are secondary to volume responsive mechanisms (e.g. diarrhoea, renal hypoperfusion after surgery) and secondary to sepsis.41 Other conditions such as uraemic haemolytic syndrome and glomerulonephritis have been shown to be of increased frequency in different parts of the world with varied outcomes usually due to late referral of children to hospitals.
Multiple studies have shown that AKI in the elderly (usually defined as older than 65 years) is increasingly common and that there is an age-dependent relationship between AKI and older age.42,43 This has been attributed in part to anatomic and physiologic changes in the ageing kidney and in part to various comorbidities - i.e. hypertension, cardiovascular disease, chronic kidney disease (CKD) - that may require procedures and/or medications that act as kidney stressors and alter renal haemodynamics or are nephrotoxic.44–46
Several studies have also shown that AKI is associated with short and long term adverse outcomes. These have been reviewed recently.47–49 Ricci et al., in a systematic review of 24 studies that involved more than 71000 patients where the RIFLE criteria (see Table 1 for terminology and definition) were used for AKI definition and staging found that the mortality rate was 18.9% in the ‘risk’ class, 36.1% in ‘injury’ class and 46.5% in ‘failure’ class. In non-AKI patients the mortality was 6.9%. Among AKI patients the relative risk for death (with respect to non-AKI patients) was 2.40 for ‘risk’ class, 4.15 for ‘injury’ class and 6.15 for ‘failure class’.50 In intensive care unit (ICU) populations, observational studies have shown that 4–5% of all critically ill patients develop severe AKI requiring renal replacement therapy (RRT) with the mortality rate often exceeding 60%.31,51,52
Table 1.
RIFLE criteria for classification and staging AKI and the modifications proposed by the AKIN network - modified from references (6 and 7)
| RIFLE criteria for classification/staging AKI | AKIN criteria for classification/staging AKI | ||||
|---|---|---|---|---|---|
| Stage | GFR criteria | Urine output criteria | Stage | Serum Creatinine criteria | Urine output criteria |
| Risk | 1.5fold increase in sCr or >25% decrease in GFR | UO < 0.5mL/kg/h for 6h | Stage 1 | Absolute increase in sCr ≥ 0.3 mg/dL (≥26.5 μmol/L) or ≥ 1.5 to 2.0 fold from baseline | UO < 0.5mL/kg/h for 6h |
| Injury | 2.0fold increase in sCr or >50% decrease in GFR | UO < 0.5mL/kg/h for 12h | Stage 2 | Increase in sCR> 2.0 to 3.0 fold from baseline | UO < 0.5mL/kg/h for 12h |
| Failure | 3.0fold increase in sCr or >75% decrease in GFR or sCr>4.0 mg/dL with an acute increase of 0.5 mg/dL | UO < 0.3mL/kg/h for 24h or anuria for 12 h | Stage 3 | Increase in sCr > 3fold from baseline or increase of sCr to ≥4.0 mg/dL (≥ 354 μmol/L) with an acute increase of at least 0.5 mg/dL (44 μmol/L) | UO < 0.3mL/kg/h for 24h or anuria for 12h |
| Loss | Complete loss of kidney function for > 4 weeks | ||||
| ESKD | End stage kidney disease for > 3 months | ||||
ESKD=end stage kidney disease, AKI=acute kidney injury, GFR=glomerular filtration rate, sCr= serum creatinine, UO=urinary output
Although mortality and development of CKD are reported with increased rates in these studies their incidence is quite variable. The factors that associate with long-term prognosis (especially with CKD progression) are poorly understood. Pre-AKI baseline renal function must be known and post-AKI recovery must be clearly defined in order to determine such outcomes. The first is often missing or is guessed in many studies and the second lacks a standard definition.47,53,54
Terminology and definitions
The term Acute Kidney Injury (AKI) was used for the first time by William MacNider in 1918 in a situation of acute mercury poisoning, but became the preferred term in 2004 when ARF was redefined with the now widely accepted consensus criteria known as RIFLE (an acronym of the Risk-Injury-Failure-Loss-End stage kidney disease).6,55
Principal tools to detect AKI were consecutive measurements of sCr, serum urea (sUr), urinalysis, and measurements of UO. Urine indices such as fractional excretion of sodium (FeNa) and urea (FeUr) were also used to differentiate transient from persistent AKI. Agreement in diagnostic criteria for AKI came later from multiple consensus groups. First was the Acute Dialysis Quality Initiative (ADQI) group. In 2002 they developed a system for diagnosis and classification of acute impairment of kidney function through a broad consensus of experts, resulting in the RIFLE criteria. The characteristics of this diagnostic system are summarised in Table 1. With this system three severity grades are defined (Risk, Injury and Failure) and two outcome classes (Loss and End-Stage Renal Disease (ESRD)). The severity criteria of AKI are defined on the basis of the changes in sCr or UO where the worst of each criterion is used. The outcome criteria are defined by the duration of impairment of kidney function.6
The importance of RIFLE criteria is that they move beyond ARF. The term “acute kidney injury/impairment” has been proposed to encompass the “entire spectrum of the syndrome from minor changes in markers of renal function to requirement for renal replacement therapy (RRT)”. Therefore the concept of AKI, as defined by RIFLE, creates a new paradigm. AKI encompasses ATN and ARF as well as other, less severe conditions. It includes patients without actual damage to the kidney but with functional impairment relative to physiologic demand. Including such patients in the classification of AKI is conceptually attractive because these are precisely the patients that may benefit from early intervention. The RIFLE criteria have also been modified for use in the paediatric setting.56 Nevertheless the RIFLE definition is not free of ambiguities. Pickering et al57 showed that there was a mismatch between increases in sCr concentration and decreases in GFR (estimated with MDRD or Cockroft-Gault formulae) in the descriptions of Risk and Failure severity grades. A 1.5-fold increase in sCr corresponds to a one-third decrease (not 25%) in GFR and a three-fold increase corresponds to a two-third decrease in GFR (not 75%). If the GFR is not directly measured but estimated by a formula then results might be also different depending on the formula used. With the MDRD formula a 1.5-fold increase in sCr corresponds to a 37% decrease in GFR, and a three-fold increase in sCr to a 72% decrease in GFR.57
In 2007, the Acute Kidney Injury Network (AKIN) group proposed a modified version of the RIFLE criteria, which aimed to improve the sensitivity of AKI diagnostic criteria.7 There were several changes: an absolute increase in sCr of at least 0.3 mg/dL (26.5 μmol/L) was added to stage 1; the GFR criterion was removed; patients starting RRT were classified as stage 3, irrespectively of sCr values; and outcome classes were removed. The characteristics of this system are summarised in Table 1.
Only one criterion (sCr or UO) has to be fulfilled in order to qualify for a stage. Time becomes more important for AKI diagnosis in the AKIN definition: changes between two sCr values within a 48-hour period are required, while one week was proposed by the ADQI group in the original RIFLE criteria. Severity of AKI in AKIN is staged over the course of 7 days by the fold-change in sCr from baseline.
The latest classification of AKI proposed by the Acute Kidney Injury Working Group of KDIGO (Kidney Disease: Improving Global Outcomes), is based on the previous two classifications, and had the aim of unifying the definition of AKI.8 By the KDIGO definition, AKI is diagnosed by an absolute increase in sCr, at least 0.3 mg/dL (26.5 μmol/L) within 48 hours or by a 50% increase in sCr from baseline within 7 days, or a urine volume of less than 0.5 mL/kg/h for at least 6 hours (Table 2).
Table 2.
AKI definition and staging according to KDIGO criteria - modified from reference (8)
| AKI is defined as any of the following: | ||
|
| ||
| 1 | Increase in sCr ≥0.3 mg/dL (≥26.5 μmol/L) within 48 hours; or | |
| 2 | Increase in sCr ≥1.5 times baseline, which is known or presumed to have occurred within the prior 7 days; or | |
| 3 | Urine volume <0.5 mL/kg/h for 6 hours. | |
|
| ||
| AKI is staged for severity according to the following criteria | ||
|
| ||
| Stage 1 | 1.5–1.9 times baseline OR ≥0.3 mg/dL (≥26.5 μmol/L) absolute increase in sCr | Urine volume <0.5 mL/kg/h for 6–12 hours |
| Stage 2 | sCr ≥2.0–2.9 times baseline sCr ≥3.0 times from baseline OR | Urine volume <0.5 mL/kg/h for ≥12 hours |
| Stage 3 | Increase in sCr to ≥4.0 mg/dL(≥353.6 μmol/L) OR Initiation of renal replacement therapy OR, In patients <18 years, decrease in eGFR to <35 mL/min per 1.73 m2 | Urine volume <0.3 mL/kg/h for ≥24 hours OR Anuria for ≥12 hours |
sCr=serum creatinine, eGFR= estimated glomerular filtration rate
A patient’s progress can be staged over the entire time frame encompassed by an episode of AKI. An increase in sCr up to 3 times from baseline, or a sCr of more than 4.0 mg/dL (354 μmol/L) or initiation of RRT, are all classified as stage 3. KDIGO removes the 0.5 mg/dL increase for sCr >4 mg/dL to diagnose stage 3. KDIGO explicitly states that a rolling baseline can be used over 48-hour and 7-day periods for diagnosis of AKI, while in RIFLE or AKIN it is not clear how this is handled. Changes were also made to severity stage 3 to enable incorporation of paediatric population into both definition and staging.
Aetiology of AKI
There are numerous potential causes of AKI, mainly related to a focal mismatch between oxygen and nutrient delivery (because of impaired microcirculation) to the nephrons and increased energy demands (due to cellular stress).58 For many years the diagnosis and management of AKI was based on the concept of classification to three main categories: pre-renal, intrinsic and post-renal (Figure 1).59–61
Figure 1.
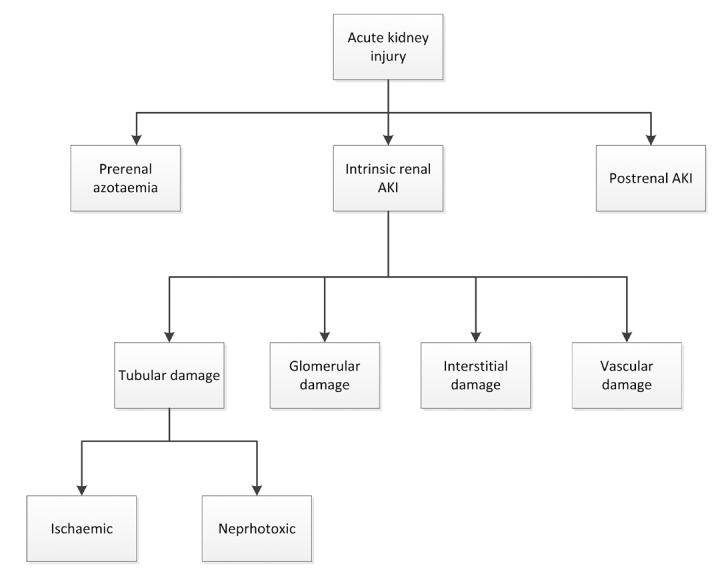
Aetiologies of acute kidney injury
In Pre-renal AKI, renal hypoperfusion leads to a decreased GFR (without damage to the renal parenchyma), as an adaptive response to various extra-renal insults.62 It is known that maintaining a normal GFR is dependent on adequate renal perfusion. The kidneys receive up to 25% of cardiac output and thus any failure of the systematic circulating blood volume or isolated failure of the intra-renal circulation can have a profound impact on renal perfusion (Table 3).
Table 3.
Causes of acute kidney injury.
| Category | Abnormality | Possible causes |
|---|---|---|
| Prerenal | Hypovolaemia | Haemorrhage Volume depletion Renal fluid loss (over-diuresis) Third space (burns, peritonitis, muscle trauma) |
| Impaired cardiac function | Congestive heart failure Acute myocardial infarction Massive pulmonary embolism |
|
| Systemic vasodilatation | Anti-hypertensive medications Gram negative bacteraemia Cirrhosis Anaphylaxis |
|
| Increased vascular resistance | Anaesthesia Surgery Hepatorenal syndrome NSAID medications Drugs that cause renal vasoconstriction (i.e. cyclosporine) |
|
| Instrinsic | Tubular | Renal ischaemia (shock, complications of surgery, haemorrhage, trauma, bacteraemia, pancreatitis, pregnancy) Nephrotoxic drugs (antibiotics, antineoplastic drugs, contrast media, organic solvents, anaesthetic drugs, heavy metals) Endogenous toxins (myoglobin, haemoglobin, uric acid) |
| Glomerular | Acute post-infectious glomerulonephritis Lupus nephritis IgA glomerulonephritis Infective endocarditis Goodpasture syndrome Wegener disease |
|
| Interstitium | Infections (bacterial, viral) Medications (antibiotics, diuretics, NSAIDs, and many more drugs) |
|
| Vascular | Large vessels (bilateral renal artery stenosis, bilateral renal vein thrombosis) Small vessels (vasculitis, malignant hypertension, atherosclerotic or thrombotic emboli, haemolytic uraemic syndrome, thrombotic thrombocytopenic purpura) |
|
| Postrenal | Extrarenal obstruction | Prostate hypertrophy Improperly placed catheter Bladder, prostate or cervical cancer Retroperitoneal fibrosis |
| Intrarenal obstruction | Nephrolithiasis Blood clots Papillary necrosis |
NSAID=non-steroid anti-inflammatory drug
Post-renal AKI occurs after acute obstruction of the urinary flow, which increases intra-tubular pressure and thus decreases GFR.63 In addition, acute urinary tract obstruction can lead to impaired renal blood flow and inflammatory processes that also contribute to diminished GFR.64 Post-renal AKI can develop if the obstruction is located at any level within the urinary collection system (from the renal tubule to urethra). In case the obstruction is above the bladder it must involve both kidneys (or one kidney in the case of a patient with a single functioning kidney) to produce significant renal failure.65 However, a patient with pre-existing renal insufficiency may develop AKI with obstruction of only one kidney. Urinary obstruction may present as anuria or intermittent urine flow (such as polyuria alternating with oliguria) but may also present as nocturia or nonoliguric AKI (Table 3). Timely reversion of pre-renal or post-renal causes usually results in prompt recovery of function, but late correction can lead to kidney damage.
Intrinsic renal aetiologies of AKI can be challenging to evaluate because of the wide variety of injuries that can occur to the kidney. Generally, four structures of the kidney are involved including tubules, glomeruli, the interstitium, and intra-renal blood vessels. (Figure 1 and Table 3)
Acute tubular necrosis (ATN) is the term used to designate AKI resulting from damage to the tubules. It is the most common type of intrinsic kidney injury. AKI from glomerular damage occurs in severe cases of acute glomerulonephritis (GN). AKI from vascular damage occurs because injury to intra-renal vessels decreases renal perfusion and diminishes GFR and finally acute interstitial nephritis occurs due to an allergic reaction to a variety medications or an infection.
The different clinical phenotypes of AKI and their pathophysiology
Essentially AKI is a term used to describe the clinical syndrome that occurs when renal function is acutely decreased to a point that the body accumulates waste products and becomes unable to maintain electrolyte, acid-base and water balance.58
The pathophysiology of AKI is multifactorial and complex. The most common cause of AKI is ischaemia, which can occur for a number of reasons (Table 3). Physiological adaptations, in response to the reduction in blood flow can compensate to a certain degree, but when delivery of oxygen and metabolic substrates becomes inadequate, the resulting cellular injury leads to organ dysfunction. The kidney is highly susceptible to injury related to ischaemia, resulting in vasoconstriction, endothelial injury, and activation inflammatory processes.66 This susceptibility can be explained in part from structural associations between renal tubules and blood vessels in the outer medulla of the kidney, with ischaemia compromising blood flow to critical nephron structures present therein. Following the reduction in effective kidney perfusion, the epithelial cells are unable to maintain adequate intracellular ATP for essential processes. This ATP-depletion leads to cell injury and if it is severe enough can lead to cell death by necrosis or apoptosis. During an ischaemic insult all segments of the nephrons can be affected but proximal tubular cells are the most commonly injured. In addition, the nephron’s natural function is to filter, concentrate and reabsorb many substances from tubular lumen, and the concentration of these substances may reach toxic levels for the surrounding epithelial cells. A detailed description of the sequence of events and the cellular changes during ischaemic AKI can be found elsewhere.67–69
AKI is also very common in the setting of sepsis. In sepsis the circulation is hyperdynamic and blood flow is altered, albeit not necessarily in the ischaemic range, and GFR drops rapidly.70 The pathophysiology of septic-AKI is very complex and involves inflammation, oxidative stress microvascular dysfunction and amplification of injury via secretion of cytokines by tubular cells.71 The traditional classification of AKI into pre-renal, intrinsic-renal and post-renal has recently been challenged since histological diagnosis is performed very rarely and distinction between pre-renal azotaemia and tubular damage cannot be confirmed and only hypothesised retrospectively. Our knowledge is mainly obtained from animal studies where the ischaemia-reperfusion model has been extensively studied. Other models (toxic injury, septic model) are less studied.72 However, these latter models are quite extreme and are not representative of the clinical manifestations of AKI in humans, where renal blood flow never fully stops (except in certain surgical procedures i.e. abdominal aortic aneurysm repair) but less severe forms of low blood flow followed by reperfusion generally occur. Controversy also exists regarding the extent of damage as well as the cell types affected by this damage (proximal vs distal tubular cells).73 The animals used in the studies are usually young and healthy but most patients developing AKI are old and with significant comorbidities (diabetes, CKD, hypertension). Moreover in experimental animals AKI is mono-causal while in humans is often of multiple coexisting aetiologies. A further analysis of pathophysiologic mechanisms is beyond the scope of this review. The reader can refer to several excellent reviews analysing pathophysiologic mechanisms in AKI.44,58,63,66,68,69,71,74–77
Special clinical scenarios
Rhabdomyolysis
Rhabdomyolysis is a syndrome that is characterised by the breakdown and necrosis of damaged skeletal muscle and subsequent release of its contents (i.e. myoglobin, sarcoplasmic proteins) into extracellular fluid and circulation.78–80 These products may be filtered through the glomeruli, leading to AKI via different mechanisms, such as intratubular obstruction secondary to protein precipitation, renal vasoconstriction, inflammation and tubular damage associated with reactive oxygen species production. Rhabdomyolysis usually develops in the setting of one or more of the following situations: disruption of the substrates and/or oxygen for metabolism (i.e. ischaemia, hypoxia, crush injuries), excessive metabolic demand (i.e. strenuous exercise), impaired cellular energy production (i.e. hereditary enzymatic disorders, toxins), and/or increased intracellular calcium influx.81,82
The clinical presentation of this multifactorial and multicausal syndrome varies from an asymptomatic but detectable elevations of CK and myoglobin in blood to a life threatening condition with fulminant AKI. The ability to predict rhabdomyolysis induced AKI is critical since it is one of the leading causes of AKI. Rhabdomyolysis contributes to 5–25% of all AKI cases and 10–50% of patients with some degree of rhabdomyolysis develop AKI.82
Drug-induced AKI
Medications frequently show toxic effects on the kidney as glomerular, interstitial and tubular cells encounter significant concentrations of medications and their metabolites, which can induce changes in kidney function and structure. Renal tubular cells are particularly vulnerable to the toxic effects of drugs because of their role in concentrating and reabsorbing glomerular filtrate, which exposes them to high levels of circulating toxins. Renal toxicity can be a result of haemodynamic changes, direct injury to cells and tissue, inflammatory tissue injury and obstruction of renal excretion. The true incidence of drug-induced nephrotoxicity is difficult to determine. Subtle renal damage (i.e. acid-base abnormalities, disorders of water balance, electrolyte imbalances) and mild urinary sediment abnormalities associated with commonly used medications are frequently unrecognised and the detection is often delayed until an overt change in renal function is apparent, usually by an increase in sCr. Three recent reviews explore in detail the mechanisms underlying renal injury related to the use of most common drugs used in clinical practice.83–85
Contrast Induced Acute Kidney Injury (CI-AKI)
Contrast induced AKI (CI-AKI) previously known as contrast induced nephropathy (CIN) is a syndrome in which acute renal dysfunction is diagnosed following intravascular administration of contrast agents. Contrast agents are used widely for diagnostic and therapeutic purposes. Their nephrotoxic potential was first suggested at least 50 years ago and today are considered one of the most common causes of AKI among hospitalised patients.86,87 The risk of CIN has long been assumed to be proportional to the degree of preexisting renal dysfunction and it is associated with extended length of hospital stay, accelerated onset of end stage renal disease, need for dialysis, increased mortality and increased costs.86,88–90 Although in the past many different definitions were used to define CI-AKI, the new KDIGO definition of AKI applies to CI-AKI and will help us to use a common language in research and in clinical diagnosis of this syndrome. The pathophysiology of CI-AKI is not very well defined. Animal models of CI-AKI suggest several potential mechanisms of nephrotoxicity, including renal ischaemia, vasoconstriction, formation of reactive oxygen species and direct tubular toxicity, which lead to decreased renal perfusion.91–94 However, the physiologic relevance of these models may be limited since multiple renal insults are required to express the desired phenotype and such injury is not typically seen in human patients. The causal association between contrast media and nephrotoxicity has been established from several studies. However the non-existence of a uniform definition and poorly designed studies may have led to overestimation of the frequency and severity of CI-AKI.95–98
Acute kidney injury and extra-renal organ dysfunction
Recent evidence in both basic science and clinical research are beginning to change our view for AKI from a single organ failure syndrome, to a syndrome where the kidney plays an active role in the evolution of multi-organ dysfunction. Recent clinical evidence suggests that AKI is not only an indicator for severity of illness, but also leads to earlier onset of multi-organ dysfunction with significant effects on mortality. Animal models of renal injury have been used extensively in order to elucidate the mechanism of remote organ dysfunction after AKI despite their limitations due to interspecies differences. These studies have shown a direct effect of AKI on distant organs.99–102 These animal studies include models of ischaemiareperfusion injury and sepsis, mainly lipopolysaccharide endotoxin induced sepsis due to its reproducibility in creating distant organ failure.103 AKI is not an isolated event and it results in remote organ dysfunction to the lungs, heart, liver, intestines and brain through a pro-inflammatory mechanism that involves neutrophil cell migration, cytokine expression and increased oxidative stress (Figure 2). Three recent excellent reviews explore the mechanisms and the long-term consequences of AKI other organ systems.104–106
Figure 2.
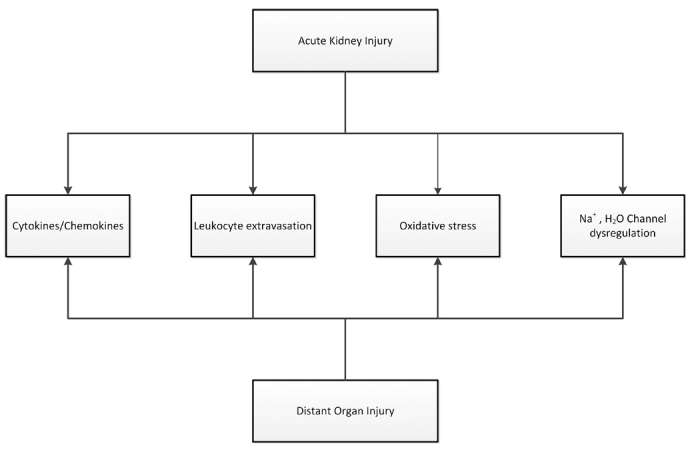
Proposed mechanism of distal organ injury. AKI leads to distant organ injury through a combination of pro-inflammatory and oxidative stress-mediated mechanisms. Serum and distal organ cytokine levels (IL1, IL6, IL10 and TNFa) increase in conjunction with Leukocyte trafficking (neutrophil, lymphocyte and macrophage) and increased oxidative stress (superoxide dismutase, malondialdehyde, and glutathione depletion. In addition, sodium and water channel dysregulation in the lungs aggravate pulmonary edema. - adapted from ref (105).
Kidney-lung crosstalk in the critically ill patient
The kidney and the lung are the two most commonly involved organs in multi-organ failure. Acute lung injury (ALI) and AKI are common complications of sepsis and the development of either increases mortality.107,108 Currently there is growing interest in the potential cross-talk that exists between these organs when injured, with one organ causing or contributing to injury to the other. Animal studies have shown that AKI can cause ALI and vice versa. The mechanism of AKI associated lung injury remains incompletely understood. Several studies have shown the involvement of pro-inflammatory and proapoptotic factors (leukocyte trafficking, cytokines activation of caspases, oxidative stress and uraemic toxins). AKI leads to lung injury and inflammation and ALI in turn facilitates and exacerbates kidney dysfunction via metabolic and biochemical derangements (Figure 3).109
Figure 3.
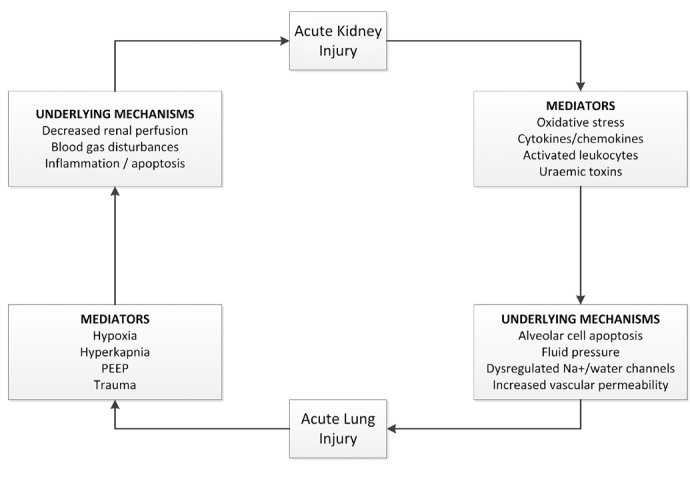
Kidney-lung interactions. AKI induces pathologic effects on the lung via cellular and soluble mediators. ALI in turn facilitates and exacerbates Kidney injury via metabolic and biochemical derangements. - adapted from ref (108).
Heart-kidney crosstalk: the cardiorenal syndrome
Kidney and cardiac disease are not only common but often coexist. Both acute and chronic cardiac disease can contribute directly to acute and/or chronic worsening of renal function and vice versa.110 The term cardiorenal syndrome (CRS) is often used to describe this condition and represents an important model for the exploration of the pathophysiology of cardiac and renal dysfunction. Recently a consensus definition/classification scheme has been proposed for the CRS.111–113 According to this definition, five subtypes of the CRS exist. Each subtype’s etymology reflects the primary and secondary pathology, cardiac and renal as well as dysfunction secondary to systemic disease. (Table 4)
Table 4.
Cardiorenal syndrome classification system – adapted from reference (111)
| Classification | Abbreviation | Characteristic | Primary Event | Secondary Event | Criteria for primary/secondary Events |
|---|---|---|---|---|---|
| Acute cardio-renal syndrome | CRS type1 | Abrupt worsening of cardiac function leading to AKI | AHF, ACS cardiogenic shock | AKI | ESC,AHA,ACC/RIFLE-AKIN |
| Chronic cardio-renal syndrome | CRS type2 | Chronic worsening of cardiac function leading to progressive and permanent chronic kidney disease | CHD | CKD | ESC,AHA,ACC/ KDOQI |
| Acute reno-cardiac syndrome | CRS type3 | AKI causing acute cardiac dysfunction | AKI | AHF, ACS arrhythmias shock | RIFLE-AKIN/ ESC,AHA,ACC |
| Chronic reno-cardiac syndrome | CRS type4 | CKD leading to impairment of cardiac function and/or increased risk of adverse cardiovascular events | CKD | CHD, AHF ACS | KDOQI/ESC,AHA,ACC |
| Secondary cardio-renal syndrome | CRS type5 | Systemic disorders causing both cardiac and renal dysfunction (i.e. septic shock, vasculitis) | Systemic disease (i.e. sepsis) | AKI, CKD AHF, CHD ACS | Disease specific criteria/RIFLE-AKIN, ESC, AHA, ACC,KDOQI |
AKI= acute kidney injury, CKD=chronic kidney disease, ACS= acute coronary syndrome, AHF=acute heart failure, CHD=chronic heart disease,
Kidney-liver interactions: Hepatorenal syndrome
Here it is important to distinguish hepatic dysfunction as a result of AKI as distinct from the well-recognised hepatorenal syndrome (HRS). Liver injury often correlates with severity of kidney injury. Ischaemic AKI induces oxidative stress and promotes inflammation apoptosis and tissue damage to hepatocytes.109,114 A recent review examines in detail the experimental evidence for hepatic dysfunction as a consequence of AKI.115
On the other hand the concept of HRS is very well recognised; it is a reversible functional renal impairment that occurs in patients with advanced liver cirrhosis or in patients with fulminant hepatic failure.116–120 It is characterised by a marked decrease in GFR and renal blood flow in the absence of other causes of renal injury. HRS is not uncommon and occurs in approximately 40% of patients with advanced cirrhosis.121 Two forms of HRS have been described.122,123
Type 1, is characterised by a rapid and progressive impairment of renal function which is defined by a two-fold increase of sCr to a level >220 μmol/L (>2.5 mg/dL) in a period of less than two weeks. Type 2, is a less severe form of HRS, characterised by a stable or slowly progressive impairment of renal function over weeks or months and with a sCr>133 and up to 226 μmol/L (or >1.5 and up to 2.5 mg/dL).
The diagnostic criteria require cirrhosis with ascites, a sCr>133μmol/L (1.5 mg/dL), no improvement in sCr after >2 day upon of withdrawal of diuretics and volume expansion with albumin, absence of shock, no current treatment with nephrotoxic drugs, absence of parenchymal injury as indicated by proteinuria haematuria and/or abnormal ultrasonography.122
These diagnostic criteria present several shortcomings. The sCr should be interpreted with caution in patients with cirrhosis. Reduced endogenous creatinine production which is related to decreased hepatic synthesis and decreased muscle mass from malnutrition, medication-related increases in tubular secretion of creatinine, fluctuations in sCr levels in patients with cirrhosis and large volume ascites and laboratory method dependencies in interference from bilirubin are some of the causes of low sCr values in these patients. Therefore sCr based measurements pose the risk to overestimate renal function and underestimate the severity of renal damage.124–126
Moreover the single cut-off value of a serum creatinine level of 133 μmol/L (1.5 mg/dL) may limit treatment to patients with the most severe degree of renal dysfunction. The changes that lead to the development of HRS are not an ‘all-or-none’ phenomenon, but evolve progressively with the natural history of cirrhosis. It is unclear whether patients who have milder degrees of renal dysfunction will also experience adverse outcomes.
As AKI has not been formally defined in patients with cirrhosis, the ADQI and the International Ascites Club (IAC) formed a Working Group in March 2010 and proposed a revised definition of renal dysfunction (both acute and chronic) in patients with cirrhosis.127 With this definition Type 1 HRS can be regarded as a specific form of AKI, and Type 2 as a specific form of CKD and the concept of ‘acute-on-chronic’ was introduced (Table 5).
Table 5.
Proposed diagnostic criteria of kidney dysfunction in cirrhosis - adapted from reference (126)
| Diagnosis | Definition |
|---|---|
| Acute kidney injury | Rise in serum creatinine of ≥ 50% from baseline or a rise of serum creatinine by ≥ 26.5 μmol/L (≥ 0.3 mg/dL) in <48 h HRS type 1 is a specific form of acute kidney injury |
| Chronic kidney disease | Glomerular filtration rate of <60 ml/min for >3 months calculated using MDRD6 formula HRS type 2 is a specific form of chronic kidney disease |
| Acute-on-chronic kidney disease | Rise in serum creatinine of ≥ 50% from baseline or a rise of serum creatinine by ≥ 26.5 μmol/L (≥ 0.3 mg/dL) in <48 h in a patient with cirrhosis whose glomerular filtration rate is <60 ml/min for >3 months calculated using MDRD6 formula |
• Both the acute deterioration in renal function and the background chronic renal dysfunction can be functional or structural in nature.
• MDRD6=Modification of Diet in Renal Disease formula calculated using six variables of serum creatinine, age, gender, albumin, blood urea nitrogen and whether or not the patient is African-American
Conclusion
AKI is an important clinical syndrome associated with poor clinical outcomes for hospitalised patients. Considerable advances have been made in refining the definition of this syndrome and in the elucidation of the underlying pathophysiologic mechanisms of the different clinical phenotypes. It is obvious that all clinical phenotypes of AKI cannot fit into a single pathophysiologic pathway. AKI facilitates organ cross-talk and distant organ injury. These innovations will aid in the design of epidemiologic studies and randomised trials of preventive and therapeutic interventions.
Footnotes
Competing Interests: None declared.
References
- 1.Eknoyan G. Emergence of the concept of acute renal failure. Am J Nephrol. 2002;22:225–30. doi: 10.1159/000063766. [DOI] [PubMed] [Google Scholar]
- 2.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–70. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 3.Uchino S, Bellomo R, Goldsmith D, Bates S, Ronco C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit Care Med. 2006;34:1913–7. doi: 10.1097/01.CCM.0000224227.70642.4F. [DOI] [PubMed] [Google Scholar]
- 4.Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA. 1996;275:1489–94. [PubMed] [Google Scholar]
- 5.Hoste EA, Clermont G, Kersten A, Venkataraman R, Angus DC, De Bacquer D, et al. RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care. 2006;10:R73. doi: 10.1186/cc4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–12. doi: 10.1186/cc2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidney Disease Improving Global Outcomes (KDIGO) Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1–138. [Google Scholar]
- 9.Vanmassenhove J, Vanholder R, Nagler E, Van Biesen W. Urinary and serum biomarkers for the diagnosis of acute kidney injury: an in-depth review of the literature. Nephrol Dial Transplant. 2013;28:254–73. doi: 10.1093/ndt/gfs380. [DOI] [PubMed] [Google Scholar]
- 10.Siew ED, Ware LB, Ikizler TA. Biological markers of acute kidney injury. J Am Soc Nephrol. 2011;22:810–20. doi: 10.1681/ASN.2010080796. [DOI] [PubMed] [Google Scholar]
- 11.Coca SG, Yalavarthy R, Concato J, Parikh CR. Biomarkers for the diagnosis and risk stratification of acute kidney injury: a systematic review. Kidney Int. 2008;73:1008–16. doi: 10.1038/sj.ki.5002729. [DOI] [PubMed] [Google Scholar]
- 12.Soni SS, Pophale R, Ronco C. New biomarkers for acute renal injury. Clin Chem Lab Med. 2011;49:1257–63. doi: 10.1515/CCLM.2011.664. [DOI] [PubMed] [Google Scholar]
- 13.Basu RK, Wong HR, Krawczeski CD, Wheeler DS, Manning PB, Chawla LS, et al. Combining functional and tubular damage biomarkers improves diagnostic precision for acute kidney injury after cardiac surgery. J Am Coll Cardiol. 2014;64:2753–62. doi: 10.1016/j.jacc.2014.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katagiri D, Doi K, Matsubara T, Negishi K, Hamasaki Y, Nakamura K, et al. New biomarker panel of plasma neutrophil gelatinase-associated lipocalin and endotoxin activity assay for detecting sepsis in acute kidney injury. J Crit Care. 2013;28:564–70. doi: 10.1016/j.jcrc.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Katagiri D, Doi K, Honda K, Negishi K, Fujita T, Hisagi M, et al. Combination of two urinary biomarkers predicts acute kidney injury after adult cardiac surgery. Ann Thorac Surg. 2012;93:577–83. doi: 10.1016/j.athoracsur.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 16.Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int. 2012;82:537–47. doi: 10.1038/ki.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waikar SS, Betensky RA, Emerson SC, Bonventre JV. Imperfect gold standards for kidney injury biomarker evaluation. J Am Soc Nephrol. 2012;23:13–21. doi: 10.1681/ASN.2010111124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaião S, Cruz DN. Baseline creatinine to define acute kidney injury: is there any consensus? Nephrol Dial Transplant. 2010;25:3812–4. doi: 10.1093/ndt/gfq454. [DOI] [PubMed] [Google Scholar]
- 19.Pickering JW, Endre ZH. Baseline creatinine: where to from here? Nephrol Dial Transplant. 2011;26:2056. doi: 10.1093/ndt/gfr099. author reply 2056–7. [DOI] [PubMed] [Google Scholar]
- 20.Pickering JW, Endre ZH. Back-calculating baseline creatinine with MDRD misclassifies acute kidney injury in the intensive care unit. Clin J Am Soc Nephrol. 2010;5:1165–73. doi: 10.2215/CJN.08531109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eknoyan G, Agodoa L. On improving outcomes and quality of dialysis care, and more. Am J Kidney Dis. 2002;39:889–91. doi: 10.1053/ajkd.2002.32720. [DOI] [PubMed] [Google Scholar]
- 22.Davies FC, Weldon RP. A contribution to the study of “war nephritis”. Lancet. 1917;190:118–20. [Google Scholar]
- 23.Bywaters EG, Beall D. Crush injuries with impairment of renal function. Br Med J. 1941;1:427–32. doi: 10.1136/bmj.1.4185.427. D BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kellum JA, Levin N, Bouman C, Lameire N. Developing a consensus classification system for acute renal failure. Curr Opin Crit Care. 2002;8:509–14. doi: 10.1097/00075198-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Li PK, Burdmann EA, Mehta RL. World Kidney Day 2013: acute kidney injury-global health alert. Am J Kidney Dis. 2013;61:359–63. doi: 10.1053/j.ajkd.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Zappitelli M. Epidemiology and diagnosis of acute kidney injury. Semin Nephrol. 2008;28:436–46. doi: 10.1016/j.semnephrol.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Zappitelli M, Parikh CR, Akcan-Arikan A, Washburn KK, Moffett BS, Goldstein SL. Ascertainment and epidemiology of acute kidney injury varies with definition interpretation. Clin J Am Soc Nephrol. 2008;3:948–54. doi: 10.2215/CJN.05431207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382:170–9. doi: 10.1016/S0140-6736(13)60647-9. [DOI] [PubMed] [Google Scholar]
- 29.Prakash J, Singh TB, Ghosh B, Malhotra V, Rathore SS, Vohra R, et al. Changing epidemiology of community-acquired acute kidney injury in developing countries: analysis of 2405 cases in 26 years from eastern India. Clin Kidney J. 2013;6:150–5. doi: 10.1093/ckj/sfs178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Case J, Khan S, Khalid R, Khan A. Epidemiology of acute kidney injury in the intensive care unit. Crit Care Res Pract. 2013;2013:479730. doi: 10.1155/2013/479730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–8. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 32.Liaño F, Pascual J, Madrid Acute Renal Failure Study Group Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Kidney Int. 1996;50:811–8. doi: 10.1038/ki.1996.380. [DOI] [PubMed] [Google Scholar]
- 33.Lameire N, Van Biesen W, Vanholder R. The changing epidemiology of acute renal failure. Nat Clin Pract Nephrol. 2006;2:364–77. doi: 10.1038/ncpneph0218. [DOI] [PubMed] [Google Scholar]
- 34.Lameire N, Van Biesen W, Vanholder R. The rise of prevalence and the fall of mortality of patients with acute renal failure: what the analysis of two databases does and does not tell us. J Am Soc Nephrol. 2006;17:923–5. doi: 10.1681/ASN.2006020152. [DOI] [PubMed] [Google Scholar]
- 35.Wonnacott A, Meran S, Amphlett B, Talabani B, Phillips A. Epidemiology and outcomes in community-acquired versus hospital-acquired AKI. Clin J Am Soc Nephrol. 2014;9:1007–14. doi: 10.2215/CJN.07920713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sutherland SM, Ji J, Sheikhi FH, Widen E, Tian L, Alexander SR, et al. AKI in hospitalized children: epidemiology and clinical associations in a national cohort. Clin J Am Soc Nephrol. 2013;8:1661–9. doi: 10.2215/CJN.00270113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldstein SL. Acute kidney injury in children and its potential consequences in adulthood. Blood Purif. 2012;33:131–7. doi: 10.1159/000334143. [DOI] [PubMed] [Google Scholar]
- 38.Ball EF, Kara T. Epidemiology and outcome of acute kidney injury in New Zealand children. J Paediatr Child Health. 2008;44:642–6. doi: 10.1111/j.1440-1754.2008.01373.x. [DOI] [PubMed] [Google Scholar]
- 39.Mehta P, Sinha A, Sami A, Hari P, Kalaivani M, Gulati A, et al. Incidence of acute kidney injury in hospitalized children. Indian Pediatr. 2012;49:537–42. doi: 10.1007/s13312-012-0121-6. [DOI] [PubMed] [Google Scholar]
- 40.McDonald SP, Craig JC, Australian and New Zealand Paediatric Nephrology Association Long-term survival of children with end-stage renal disease. N Engl J Med. 2004;350:2654–62. doi: 10.1056/NEJMoa031643. [DOI] [PubMed] [Google Scholar]
- 41.Cerdá J, Lameire N, Eggers P, Pannu N, Uchino S, Wang H, et al. Epidemiology of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:881–6. doi: 10.2215/CJN.04961107. [DOI] [PubMed] [Google Scholar]
- 42.Coca SG. Acute kidney injury in elderly persons. Am J Kidney Dis. 2010;56:122–31. doi: 10.1053/j.ajkd.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdel-Kader K, Palevsky PM. Acute kidney injury in the elderly. Clin Geriatr Med. 2009;25:331–58. doi: 10.1016/j.cger.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Bonventre JV, Parrish AR. The aging kidney: increased susceptibility to nephrotoxicity. Int J Mol Sci. 2014;15:15358–76. doi: 10.3390/ijms150915358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chronopoulos A, Cruz DN, Ronco C. Hospital-acquired acute kidney injury in the elderly. Nat Rev Nephrol. 2010;6:141–9. doi: 10.1038/nrneph.2009.234. [DOI] [PubMed] [Google Scholar]
- 46.Chronopoulos A, Rosner MH, Cruz DN, Ronco C. Acute kidney injury in the elderly: a review. Contrib Nephrol. 2010;165:315–21. doi: 10.1159/000313772. [DOI] [PubMed] [Google Scholar]
- 47.Sawhney S, Mitchell M, Marks A, Fluck N, Black C. Long-term prognosis after acute kidney injury (AKI): what is the role of baseline kidney function and recovery? A systematic review. BMJ Open. 2015;5:e006497. doi: 10.1136/bmjopen-2014-006497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;53:961–73. doi: 10.1053/j.ajkd.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bagshaw SM. Short- and long-term survival after acute kidney injury. Nephrol Dial Transplant. 2008;23:2126–8. doi: 10.1093/ndt/gfn300. [DOI] [PubMed] [Google Scholar]
- 50.Ricci Z, Cruz D, Ronco C. The RIFLE criteria and mortality in acute kidney injury: A systematic review. Kidney Int. 2008;73:538–46. doi: 10.1038/sj.ki.5002743. [DOI] [PubMed] [Google Scholar]
- 51.Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. 2005;9:R700–9. doi: 10.1186/cc3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metnitz PG, Krenn CG, Steltzer H, Lang T, Ploder J, Lenz K, et al. Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Crit Care Med. 2002;30:2051–8. doi: 10.1097/00003246-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 53.Swaminathan M, Stafford-Smith M. Changing terminology in renal research: the impact of consensus. Am J Kidney Dis. 2012;59:584–5. doi: 10.1053/j.ajkd.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 54.Macedo E, Bouchard J, Mehta RL. Renal recovery following acute kidney injury. Curr Opin Crit Care. 2008;14:660–5. doi: 10.1097/MCC.0b013e328317ee6e. [DOI] [PubMed] [Google Scholar]
- 55.Kellum JA, Ronco C. Controversies in acute kidney injury: the 2011 Brussels Roundtable. Crit Care. 2011;15:155. doi: 10.1186/cc10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akcan-Arikan A, Zappitelli M, Loftis LL, Washburn KK, Jefferson LS, Goldstein SL. Modified RIFLE criteria in critically ill children with acute kidney injury. Kidney Int. 2007;71:1028–35. doi: 10.1038/sj.ki.5002231. [DOI] [PubMed] [Google Scholar]
- 57.Pickering JW, Endre ZH. GFR shot by RIFLE: errors in staging acute kidney injury. Lancet. 2009;373:1318–9. doi: 10.1016/S0140-6736(09)60751-0. [DOI] [PubMed] [Google Scholar]
- 58.Tögel F, Westenfelder C. Recent advances in the understanding of acute kidney injury. F1000Prime Rep. 2014;6:83. doi: 10.12703/P6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–30. doi: 10.1016/S0140-6736(05)17831-3. [DOI] [PubMed] [Google Scholar]
- 60.Hilton R. Acute renal failure. BMJ. 2006;333:786–90. doi: 10.1136/bmj.38975.657639.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahman M, Shad F, Smith MC. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. 2012;86:631–9. [PubMed] [Google Scholar]
- 62.Blantz RC. Pathophysiology of pre-renal azotemia. Kidney Int. 1998;53:512–23. doi: 10.1046/j.1523-1755.2003_t01-1-00784.x. [DOI] [PubMed] [Google Scholar]
- 63.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2:1303–53. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hegarty NJ, Young LS, Kirwan CN, O’Neill AJ, Bouchier-Hayes DM, Sweeney P, et al. Nitric oxide in unilateral ureteral obstruction: effect on regional renal blood flow. Kidney Int. 2001;59:1059–65. doi: 10.1046/j.1523-1755.2001.0590031059.x. [DOI] [PubMed] [Google Scholar]
- 65.Dager W, Hallilovic J. Acute Kidney Injury. In: DiPiro JT, Talbert RL, Yee GC, et al., editors. Pharmacotherapy: A Pathophysiologic Approach. 8th Edition. New York: McGraw-Hill; 2011. p. 746. [Google Scholar]
- 66.Bonventre JV. Pathophysiology of acute kidney injury: roles of potential inhibitors of inflammation. Contrib Nephrol. 2007;156:39–46. doi: 10.1159/000102069. [DOI] [PubMed] [Google Scholar]
- 67.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 68.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–21. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonventre JV. Pathophysiology of AKI: injury and normal and abnormal repair. Contrib Nephrol. 2010;165:9–17. doi: 10.1159/000313738. [DOI] [PubMed] [Google Scholar]
- 70.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 71.Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J, et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41:3–11. doi: 10.1097/SHK.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh AP, Junemann A, Muthuraman A, Jaggi AS, Singh N, Grover K, et al. Animal models of acute renal failure. Pharmacol Rep. 2012;64:31–44. doi: 10.1016/s1734-1140(12)70728-4. [DOI] [PubMed] [Google Scholar]
- 73.Heyman SN, Rosenberger C, Rosen S. Experimental ischemia-reperfusion: biases and myths-the proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int. 2010;77:9–16. doi: 10.1038/ki.2009.347. [DOI] [PubMed] [Google Scholar]
- 74.Bonventre JV. Pathophysiology of ischemic acute renal failure. Inflammation, lung-kidney cross-talk, and biomarkers. Contrib Nephrol. 2004;144:19–30. [PubMed] [Google Scholar]
- 75.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 76.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–5. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 77.Wong GT, Irwin MG. Contrast-induced nephropathy. Br J Anaesth. 2007;99:474–83. doi: 10.1093/bja/aem237. [DOI] [PubMed] [Google Scholar]
- 78.Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25:332–47. doi: 10.1002/mus.10053. [DOI] [PubMed] [Google Scholar]
- 79.Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol. 2000;11:1553–61. doi: 10.1681/ASN.V1181553. [DOI] [PubMed] [Google Scholar]
- 80.Zhang MH. Rhabdomyolosis and its pathogenesis. World J Emerg Med. 2012;3:11–5. doi: 10.5847/wjem.j.issn.1920-8642.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vanholder R, Sever MS, Erek E, Lameire N. Acute renal failure related to the crush syndrome: towards an era of seismo-nephrology? Nephrol Dial Transplant. 2000;15:1517–21. doi: 10.1093/ndt/15.10.1517. [DOI] [PubMed] [Google Scholar]
- 82.Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: Rhabdomyolysis — an overview for clinicians. Crit Care. 2005;9:158–69. doi: 10.1186/cc2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choudhury D, Ahmed Z. Drug-associated renal dysfunction and injury. Nat Clin Pract Nephrol. 2006;2:80–91. doi: 10.1038/ncpneph0076. [DOI] [PubMed] [Google Scholar]
- 84.Naughton CA. Drug-induced nephrotoxicity. Am Fam Physician. 2008;78:743–50. [PubMed] [Google Scholar]
- 85.Schetz M, Dasta J, Goldstein S, Golper T. Drug-induced acute kidney injury. Curr Opin Crit Care. 2005;11:555–65. doi: 10.1097/01.ccx.0000184300.68383.95. [DOI] [PubMed] [Google Scholar]
- 86.McCullough PA. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419–28. doi: 10.1016/j.jacc.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 87.Mehran R, Nikolsky E. Contrast-induced nephropathy: definition, epidemiology, and patients at risk. Kidney Int Suppl. 2006;100:S11–5. doi: 10.1038/sj.ki.5000368. [DOI] [PubMed] [Google Scholar]
- 88.Weisbord SD, Chen H, Stone RA, Kip KE, Fine MJ, Saul MI, et al. Associations of increases in serum creatinine with mortality and length of hospital stay after coronary angiography. J Am Soc Nephrol. 2006;17:2871–7. doi: 10.1681/ASN.2006030301. [DOI] [PubMed] [Google Scholar]
- 89.Mitchell AM, Jones AE, Tumlin JA, Kline JA. Incidence of contrast-induced nephropathy after contrast-enhanced computed tomography in the outpatient setting. Clin J Am Soc Nephrol. 2010;5:4–9. doi: 10.2215/CJN.05200709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.James MT, Samuel SM, Manning MA, Tonelli M, Ghali WA, Faris P, et al. Contrast-induced acute kidney injury and risk of adverse clinical outcomes after coronary angiography: a systematic review and meta-analysis. Circ Cardiovasc Interv. 2013;6:37–43. doi: 10.1161/CIRCINTERVENTIONS.112.974493. [DOI] [PubMed] [Google Scholar]
- 91.Persson PB, Tepel M. Contrast medium-induced nephropathy: the pathophysiology. Kidney Int Suppl. 2006;100:S8–10. doi: 10.1038/sj.ki.5000367. [DOI] [PubMed] [Google Scholar]
- 92.Seeliger E, Sendeski M, Rihal CS, Persson PB. Contrast-induced kidney injury: mechanisms, risk factors, and prevention. Eur Heart J. 2012;33:2007–15. doi: 10.1093/eurheartj/ehr494. [DOI] [PubMed] [Google Scholar]
- 93.Jorgensen AL. Contrast-induced nephropathy: pathophysiology and preventive strategies. Crit Care Nurse. 2013;33:37–46. doi: 10.4037/ccn2013680. [DOI] [PubMed] [Google Scholar]
- 94.Perrin T, Descombes E, Cook S. Contrast-induced nephropathy in invasive cardiology. Swiss Med Wkly. 2012;142:w13608. doi: 10.4414/smw.2012.13608. [DOI] [PubMed] [Google Scholar]
- 95.McDonald JS, McDonald RJ, Comin J, Williamson EE, Katzberg RW, Murad MH, et al. Frequency of acute kidney injury following intravenous contrast medium administration: a systematic review and meta-analysis. Radiology. 2013;267:119–28. doi: 10.1148/radiol.12121460. [DOI] [PubMed] [Google Scholar]
- 96.Newhouse JH, Kho D, Rao QA, Starren J. Frequency of serum creatinine changes in the absence of iodinated contrast material: implications for studies of contrast nephrotoxicity. AJR Am J Roentgenol. 2008;191:376–82. doi: 10.2214/AJR.07.3280. [DOI] [PubMed] [Google Scholar]
- 97.Newhouse JH, RoyChoudhury A. Quantitating contrast medium-induced nephropathy: controlling the controls. Radiology. 2013;267:4–8. doi: 10.1148/radiol.13122876. [DOI] [PubMed] [Google Scholar]
- 98.McDonald RJ, McDonald JS, Bida JP, Carter RE, Fleming CJ, Misra S, et al. Intravenous contrast material-induced nephropathy: causal or coincident phenomenon? Radiology. 2013;267:106–18. doi: 10.1148/radiol.12121823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003;14:1549–58. doi: 10.1097/01.asn.0000064946.94590.46. [DOI] [PubMed] [Google Scholar]
- 100.Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H. Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int. 1999;55:2362–7. doi: 10.1046/j.1523-1755.1999.00460.x. [DOI] [PubMed] [Google Scholar]
- 101.Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, et al. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol. 2007;293:F30–40. doi: 10.1152/ajprenal.00023.2007. [DOI] [PubMed] [Google Scholar]
- 102.Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol. 2007;18:155–64. doi: 10.1681/ASN.2006050494. [DOI] [PubMed] [Google Scholar]
- 103.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868–78. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.White LE, Hassoun HT. Inflammatory mechanisms of organ crosstalk during ischemic acute kidney injury. Int J Nephrol. 2012;2012:505197. doi: 10.4061/2012/505197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shiao CC, Wu PC, Huang TM, Lai TS, Yang WS, Wu CH, et al. National Taiwan University Hospital Study Group on Acute Renal Failure (NSARF) and the Taiwan Consortium for Acute Renal Diseases (CAKs) Long-term remote organ consequences following acute kidney injury. Crit Care. 2015;19:438. doi: 10.1186/s13054-015-1149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yap SC, Lee HT. Acute kidney injury and extrarenal organ dysfunction: new concepts and experimental evidence. Anesthesiology. 2012;116:1139–48. doi: 10.1097/ALN.0b013e31824f951b. [DOI] [PubMed] [Google Scholar]
- 107.Paladino JD, Hotchkiss JR, Rabb H. Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease? Microvasc Res. 2009;77:8–12. doi: 10.1016/j.mvr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ko GJ, Rabb H, Hassoun HT. Kidney-lung crosstalk in the critically ill patient. Blood Purif. 2009;28:75–83. doi: 10.1159/000218087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White LE, Chaudhary R, Moore LJ, Moore FA, Hassoun HT. Surgical sepsis and organ crosstalk: the role of the kidney. J Surg Res. 2011;167:306–15. doi: 10.1016/j.jss.2010.11.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Virzì G, Day S, de Cal M, Vescovo G, Ronco C. Heart-kidney crosstalk and role of humoral signaling in critical illness. Crit Care. 2014;18:201. doi: 10.1186/cc13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–39. doi: 10.1016/j.jacc.2008.07.051. [DOI] [PubMed] [Google Scholar]
- 112.Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Acute Dialysis Quality Initiative (ADQI) consensus group Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31:703–11. doi: 10.1093/eurheartj/ehp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ronco C, McCullough PA, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Acute Dialysis Quality Initiative (ADQI) consensus group Cardiorenal syndromes: an executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI) Contrib Nephrol. 2010;165:54–67. doi: 10.1159/000313745. [DOI] [PubMed] [Google Scholar]
- 114.Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, et al. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int. 2009;75:783–92. doi: 10.1038/ki.2008.683. [DOI] [PubMed] [Google Scholar]
- 115.Lane K, Dixon JJ, MacPhee IA, Philips BJ. Renohepatic crosstalk: does acute kidney injury cause liver dysfunction? Nephrol Dial Transplant. 2013;28:1634–47. doi: 10.1093/ndt/gft091. [DOI] [PubMed] [Google Scholar]
- 116.Wadei HM, Mai ML, Ahsan N, Gonwa TA. Hepatorenal syndrome: pathophysiology and management. Clin J Am Soc Nephrol. 2006;1:1066–79. doi: 10.2215/CJN.01340406. [DOI] [PubMed] [Google Scholar]
- 117.Wadei HM. Hepatorenal syndrome: a critical update. Semin Respir Crit Care Med. 2012;33:55–69. doi: 10.1055/s-0032-1301735. [DOI] [PubMed] [Google Scholar]
- 118.Low G, Alexander GJ, Lomas DJ. Hepatorenal syndrome: aetiology, diagnosis, and treatment. Gastroenterol Res Pract. 2015;2015:207012. doi: 10.1155/2015/207012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ng CK, Chan MH, Tai MH, Lam CW. Hepatorenal syndrome. Clin Biochem Rev. 2007;28:11–7. [PMC free article] [PubMed] [Google Scholar]
- 120.Møller S, Krag A, Bendtsen F. Kidney injury in cirrhosis: pathophysiological and therapeutic aspects of hepatorenal syndromes. Liver Int. 2014;34:1153–63. doi: 10.1111/liv.12549. [DOI] [PubMed] [Google Scholar]
- 121.Ginès P, Guevara M, Arroyo V, Rodés J. Hepatorenal syndrome. Lancet. 2003;362:1819–27. doi: 10.1016/S0140-6736(03)14903-3. [DOI] [PubMed] [Google Scholar]
- 122.Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310–8. doi: 10.1136/gut.2006.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arroyo V, Ginès P, Gerbes AL, Dudley FJ, Gentilini P, Laffi G, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23:164–76. doi: 10.1002/hep.510230122. [DOI] [PubMed] [Google Scholar]
- 124.Caregaro L, Menon F, Angeli P, Amodio P, Merkel C, Bortoluzzi A, et al. Limitations of serum creatinine level and creatinine clearance as filtration markers in cirrhosis. Arch Intern Med. 1994;154:201–5. [PubMed] [Google Scholar]
- 125.Orlando R, Floreani M, Padrini R, Palatini P. Evaluation of measured and calculated creatinine clearances as glomerular filtration markers in different stages of liver cirrhosis. Clin Nephrol. 1999;51:341–7. [PubMed] [Google Scholar]
- 126.Sherman DS, Fish DN, Teitelbaum I. Assessing renal function in cirrhotic patients: problems and pitfalls. Am J Kidney Dis. 2003;41:269–78. doi: 10.1053/ajkd.2003.50035. [DOI] [PubMed] [Google Scholar]
- 127.Wong F, Nadim MK, Kellum JA, Salerno F, Bellomo R, Gerbes A, et al. Working Party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut. 2011;60:702–9. doi: 10.1136/gut.2010.236133. [DOI] [PubMed] [Google Scholar]