

Abstract
Human platelets display a unique dual receptor system for responding to its primary endogenous activator, α-thrombin. Because of the lack of efficacious antagonists, the field has relied on synthetic peptides and pepducins to describe protease-activated receptor PAR1 and PAR4 signaling. The precise contributions of each receptor have not been established in the context of thrombin. We took advantage of newly discovered PAR antagonists to contrast the contribution of PAR1 and PAR4 to thrombin-mediated activation of the platelet fibrin receptor (GPIIbIIIa). PAR1 is required for platelet activation at low but not high concentrations of thrombin, and maximal platelet activation at high concentrations of thrombin requires PAR4. As the concentration of thrombin is increased, PAR1 signaling is quickly overcome by PAR4 signaling, leaving a narrow window of low thrombin concentrations that exclusively engage PAR1. PAR4 antagonism reduces the maximum thrombin response by over 50%. Thus, although the PAR1 response still active at higher concentrations of thrombin, this response is superseded by PAR4. Truncation of a known PAR4 antagonist and identification of the minimum pharmacophore converted the mechanism of inhibition from noncompetitive to competitive, such that the antagonist could be outcompeted by increasing doses of the ligand. Fragments retained efficacy against both soluble and tethered ligands with lower cLogP values and an increased free fraction in plasma. These reversible, competitive compounds represent a route toward potentially safer PAR4 antagonists for clinical utility and the development of tools such as radioligands and positron emission tomography tracers that are not currently available to the field for this target.
Introduction
Acute coronary syndrome is the leading cause of death and morbidity in the Western world (Grech and Ramsdale, 2003), with up to one-third of patients experiencing secondary events, including myocardial infarction and unstable angina within 6 months (Collinson et al., 2000). The importance of platelet activation in thrombus formation is reflected by the efficacy of antiplatelet reagents in preventing recurrent ischemic events. The advent of dual antiplatelet therapy (aspirin plus clopidogrel) resulted in substantial reductions in cardiovascular events (Bowry et al., 2008); however, an increased risk of bleeding has been reported since the first aspirin/clopidogrel combination therapy clinical trials (Harker et al., 1999; Yusuf et al., 2001; Sabatine et al., 2005). Other P2Y purinoceptor 12 antagonists (ticagrelor, prasugrel) have emerged, showing an incremental reduction in the risk of thrombosis but with a concomitant increase in thrombolysis in myocardial infarction major and fatal bleeding events (Wiviott et al., 2007; Montalescot et al., 2009; Wallentin et al., 2009). The most challenging aspect of developing new antiplatelet reagents is balancing efficacy with safety.
Thrombin is the terminal enzyme of the coagulation cascade at the center of both thrombosis and hemostasis. Thrombin is generated at the site of vascular insult, cleaving fibrinogen for crosslinking and activating vascular cells through protease-activated receptors (PARs). Platelets express a dual receptor system for responding to thrombin: PAR1 and PAR4 (Kahn et al., 1999; Coughlin, 2000). Thrombin activates PARs through cleavage of the extracellular domain of the receptor, revealing an encrypted tethered ligand (TL) that binds intramolecularly to activate the receptor (Vu et al., 1991). PARs can also be activated artificially with a synthetic soluble “activating peptide” (AP) corresponding in sequence to the naturally derived TL (Vu et al., 1991; Xu et al., 1998). PAR1 contains a “hirudin-like domain” with high affinity for thrombin and, consequently, is activated by relatively low concentrations of thrombin (Vu et al., 1991). PAR4 lacks this domain (Xu et al., 1998) and requires more than a full log-order higher concentration of thrombin for activation (Kahn et al., 1999). PAR4, the low-affinity receptor, is engaged after PAR1 in a sequential manner at a 20- to 70-fold slower rate (Covic et al., 2000). A lack of effective small molecule tools has prevented a detailed investigation of the relative roles of PAR1 and PAR4 in thrombin-stimulated human platelet activation.
Thrombin receptor antagonists (TRAs) have been eagerly anticipated in cardiovascular medicine; vorapaxar is a PAR1-specific TRA that underwent two phase III clinical trials: TRA•CER (TRA*CER Executive and Steering Committees, 2009) and TRAo2P (Morrow et al., 2009, 2012; Scirica et al., 2012; Tricoci et al., 2012). After a safety review, the TRA•CER trial was halted early, and the TRAo2P secondary prevention trial was partially discontinued due to an alarming increase in bleeding (Tricoci et al., 2012; Morrow et al., 2013; Bohula et al., 2015). Vorapaxar was approved with a black box warning against “use of zontivity (vorapaxar) in patients with a history of stroke, transient ischemic attack, or intracranial hemorrhage or active pathological bleeding,” (www.fda.gov) greatly limiting the scope of its clinical utility. Efforts to target the alternate PAR on platelets (PAR4) were initiated. In 2013, Bristol-Myers Squibb published a patent describing a series of efficacious and bioavailable PAR4 antagonists (Lawrence et al., 2013). We synthesized the lead from this patent as a tool reagent, BMS-3 (Fig. 1) (internally assigned as VU0652925), which is an analog of BMS986120 (4-(4-(((6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)benzofuran-4-yl)oxy)methyl)-5-methylthiazol-2-yl)morpholine), the recently disclosed clinical candidate. In conjunction with an analog of vorapaxar (structure provided in Fig. 1) (SCH602539) that was kindly provided by Merck (Kenilworth, NJ), we interrogated the relative contribution of PAR1 and PAR4 to thrombin-mediated platelet activation.
Fig. 1.

Specificity and potency of PAR tool compounds. (A) Chemical structures of PAR1 (SCH602539) and PAR4 (VU0652925) antagonists. (B) The effect of PAR1 and PAR4 antagonists on PAR1-AP–induced aggregation. Washed human platelets were preincubated for 20 minutes with 1 μM SCH602539 or 316 nM VU0652925 before initiating aggregation with 20 μM PAR1-AP. Representative tracings of three independent experiments are shown. (C) The effect of PAR1 and PAR4 antagonists on PAR4-AP–induced aggregation. Platelets were pretreated with antagonist as in (B). Aggregation was initiated with 200 μM PAR4-AP. Representative tracings of three independent experiments are shown. (D) The effect of SCH602539 on PAR1-AP– and PAR4-AP–induced platelet activation. GPIIbIIIa inside-out activation (PAC1) and P-selectin expression (P-Sel) were measured by flow cytometry with PAC1 and CD62p antibodies. Platelets were treated with increasing concentrations of SCH602539 for 20 minutes prior to activation with 20 μM PAR1-AP or 200 μM PAR4-AP. Data are normalized to vehicle control. Means ± S.E.M. are shown (n = 3). (E) The effect of VU0652925 on PAR1-AP– and PAR4-AP–induced platelet activation. Platelets were treated as in (D). Data are normalized to vehicle control. Means ± S.E.M. are shown (n = 3). DMSO, dimethylsulfoxide; P-Sel, P-selectin.
We chose inside-out activation of the platelet integrin complex GPIIbIIIa as a primary readout. Activation of GPIIbIIIa allows interaction with divalent fibrinogen or multivalent von Willebrand factor, linking platelets and leading to aggregation (Savage et al., 2001). GPIIbIIIa-dependent aggregation is central to thrombosis, as evidenced by the effectiveness of GPIIbIIIa inhibitors (Coller et al., 1983; Bhatt and Topol, 2000). Both tools are selective for their respective receptors and are effective against the TL. We demonstrated that antagonism of PAR1 can be completely overcome by increasing concentrations of thrombin. PAR4, on the other hand, is responsible for the majority of platelet response to higher concentrations of thrombin. PAR4 antagonists reduce GPIIbIIIa activation by over 50%. Because of concerns with the safety of a noncompetitive antagonist that does not allow full recovery of the platelet thrombin response, we deconstructed the PAR4 antagonist, identified a minimum pharmacophore, and converted the compound’s mechanism of inhibition to a classic competitive modality such that it could be outcompeted by reasonable concentrations of thrombin.
Materials and Methods
Materials.
Activating peptides for PAR1 (PAR1-AP; SFLLRN) and PAR4 (PAR4-AP; AYPGKF) were purchased from GL Biochem (Shanghai, China). α-Thrombin and γ-thrombin were purchased from Enzyme Research Laboratories (South Bend, IN). Fluorescein isothiocyanate (FITC)–conjugated PAC1 and phycoerythin (PE)-conjugated P-selectin were purchased from Becton Dickinson (Franklin Lakes, NJ).
Blood Collection and Platelet Isolation.
Human platelets were obtained from healthy volunteers. The Vanderbilt University Internal Review Board approved these studies. Informed consent was obtained from all individuals prior to the blood draw. Blood was collected into sodium citrate anticoagulant (final concentration, 0.32%) through a 19-gauge needle. Platelet-rich plasma was collected after centrifugation at 1100 rpm (15 minutes, at room temperature). Acid citrate dextrose was added and incubated for 10 minutes (room temperature) before centrifugation at 2400 rpm for 10 minutes to isolate platelets. Platelets were washed and equilibrated with Tyrode’s buffer (15 mM HEPES, 0.33 mM NaH2PO4, pH 7.4, 138 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 5.5 mM dextrose) with 0.1% bovine serum albumin. Platelets were collected, counted on a Z1 Coulter Particle Counter (Beckman Coulter, Brea, CA), and diluted in Tyrode’s buffer with 0.1% bovine serum albumin to the indicated concentrations.
Platelet Aggregation.
Platelets were diluted to 2.0 × 108/ml and aliquoted into glass cuvettes. Antagonists were allowed to equilibrate with platelets for 20 minutes prior to stimulation with the indicated agonists. Aggregations were recorded for 10 minutes on a model 700 Optical Lumi Aggregometer (Chrono-Log, Havertown, PA).
Flow Cytometry.
For detection of PAC1 (GPIIbIIIa activation) and CD62p (P-selectin expression) binding, washed platelets at 1.5 × 107 cells/ml were preincubated with PE-conjugated CD62P and FITC-conjugated PAC1 for 20 minutes before stimulation with the appropriate agonist for 10 minutes. Samples were fixed with 1% paraformaldehyde in phosphate-buffered saline for 20 minutes before dilution with phosphate-buffered saline. Data were collected on a BD LSRII 5 laser (Becton Dickinson) and analyzed with FlowJo software (FlowJo LLC, Ashland, OR). Mean fluorescence intensity (geometric) of PE and FITC was determined from 30,000 events after compensation correction. Data were normalized to vehicle controls. EC50 values were gleaned from nonlinear regression analysis (four-parameter, variable slope) performed with GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). For Schild analysis, dose ratios (DRs) were constructed from PAR4-AP EC50 values with and without antagonist conducted on the same day with the same donor.
Plasma Protein Binding.
The protein binding of each compound was determined in plasma via equilibrium dialysis using RED plates (Thermo Fisher Scientific, Rochester, NY). Plasma was added to the 96-well plate containing test compound and mixed thoroughly for a final concentration of 5 µM. Subsequently, an aliquot of the plasma-compound mixture was transferred to the cis chamber (red) of the RED plate, with a phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated for 4 hours at 37°C with shaking. At completion, aliquots from each chamber were diluted 1:1 with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile containing internal standard (50 ng/ml carbamazepine) (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 relative centrifugal force, 10 minutes) and supernatants transferred and diluted 1:1 (supernatant/water) into a new 96-well plate, which was then sealed in preparation for liquid chromatography (LC)–tandem mass spectrometry (MS/MS) analysis. Each compound was assayed in triplicate within the same 96-well plate. The unbound fraction was determined using the following equation:
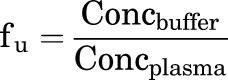 |
where fu is the unbound fraction, Concbuffer is the concentration of compound in buffer and Concplasma is the concentration of compound in plasma.
Intrinsic Clearance.
Human or rat hepatic microsomes (0.5 mg/ml) and 1 µM test compound were incubated in 100 mM potassium phosphate buffer (pH 7.4) with 3 mM MgCl2 at 37°C with constant shaking. After a 5-minute preincubation, the reaction was initiated by addition of NADPH (1 mM). At selected time intervals (0, 3, 7, 15, 25, and 45 minutes), aliquots were taken and subsequently placed into a 96-well plate containing cold acetonitrile with internal standard (50 ng/ml carbamazepine). Plates were then centrifuged at 3000 relative centrifugal force (4°C) for 10 minutes, and the supernatant was transferred to a separate 96-well plate and diluted 1:1 with water for LC-MS/MS analysis. The in vitro half-life (t1/2, in minutes; eq. 1), intrinsic clearance (CLint, in milliliters per minute per kilogram; eq. 2), and subsequent predicted hepatic clearance (CLhep, in milliliters per minute per kilogram; eq. 3) were determined using the following equations:
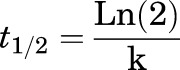 |
(1) |
where k represents the slope from linear regression analysis of the natural log percent remaining of test compound as a function of incubation time.
 |
(2) |
where a indicates scale-up factors of 20 (human) or 45 (rat).
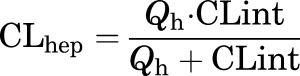 |
(3) |
where Qh (hepatic blood flow, in milliliters per minute per kilogram) is 21 (human) or 70 (rat).
LC-MS/MS Bioanalysis of Samples from Plasma Protein Binding and Intrinsic Clearance Assays.
Samples were analyzed on a TSQ Quantum Ultra triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA) via electrospray ionization with two Accela pumps (Thermo Electron) and a CTC PAL autosampler (Leap Technologies, Carrboro, NC). Analytes were separated by gradient elution on a dual-column system with two Hypersil Gold (2.1 × 30 mm, 1.9 µm; Thermo Fisher Scientific) columns thermostated at 40°C. High-performance LC mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile. The gradient started at 10% B after a 0.2-minute hold and was linearly increased to 95% B over 0.8 minutes, held at 95% B for 0.2 minutes, and returned to 10% B in 0.1 minutes. The total run time was 1.3 minutes and the high-performance LC flow rate was 0.8 ml/min. While pump 1 ran the gradient method, pump 2 equilibrated the alternate column isocratically at 10% B. Compound optimization and data collection and processing were performed using Thermo QuickQuan (version 2.3; Thermo Electron) and Xcalibur (version 2.0.7 SP1; Thermo Fisher Scientific) software.
Results
Specificity and Potency of PAR Tool Compounds.
We synthesized a PAR4 antagonist (VU0652925) recently presented in a Bristol-Myers Squibb patent (Fig. 1A) (Lawrence et al., 2013) and Merck kindly provided an analog of vorapaxar (SCH602539) to study PAR1. Using soluble peptides corresponding in sequence to the TL of each receptor, we profiled the specificity of each compound in human platelets. SCH602539 had no effect on PAR4-AP–induced aggregation at concentrations effective against PAR1-AP. Similarly, VU0652925 had no effect on PAR1-AP–induced aggregation at concentrations effective against PAR4-AP (Fig. 1, B and C). Flow cytometry analysis of GPIIbIIIa activation (PAC1) and P-selectin expression was consistent with aggregation demonstrating no major off-target effects at concentrations as high as 1 μM for each compound (Fig. 1, D and E). Both were fully effective against their respective APs and displayed comparable potency against both GPIIbIIIa activation and P-selectin secretion. Against PAR1-AP, SCH602539 had a PAC1 IC50 (-pIC50 ± S.E.M.) of 26.5 nM (7.58 ± 0.05) and P-selectin IC50 of 36.5 nM (7.44 ± 0.05). Against PAR4-AP, VU0652925 had a PAC1 IC50 of 43.0 pM (10.4 ± 0.04) and a P-selectin IC50 of 39.2 pM (10.41 ± 0.04).
Inhibition of PAR1 and PAR4 Abolish Thrombin-Mediated Signaling on Human Platelets.
To dissect the roles of PAR1 and PAR4 in the context of thrombin-mediated platelet activation and accurately determine the relative contribution of each, tool compounds must display full efficacy against the TL. Incubation with SCH602539 or VU0652925 alone had only partial effects on thrombin-mediated activation (Fig. 2A). SCH602539 had no effect on 10 nM thrombin but reduced 2 nM thrombin-mediated platelet activation by 27.3% ± 4.73%. VU0652925, on the other hand, reduced activation by up to 74.4% ± 8.72% in the context of 2 nM thrombin and 64.2% ± 7.40% in the context of 10 nM thrombin. When combined at the most effective concentrations (316 nM VU0652925 and 1 μM SCH602539), thrombin-mediated platelet activation (Fig. 2B) and aggregation (Fig. 2C) was abolished. The fact that there was no residual activation with combined PAR1 and PAR4 antagonism indicates that the antagonists are capable of abolishing TL-mediated activation of their respective receptor.
Fig. 2.

Inhibition of PAR1 and PAR4 abolish thrombin-mediated signaling on human platelets. (A) The effect of PAR1 and PAR4 antagonists individually on thrombin-mediated platelet activation. Platelets were treated with increasing doses of SCH602539 or VU0652925 for 20 minutes prior to activation with 2 or 10 nM α-thrombin. GPIIbIIIa activation was monitored by PAC1 binding. Means ± S.E.M. are shown (n = 4). (B) The effect of PAR1 and PAR4 antagonists, combined, on platelet activation by thrombin. Platelets were treated for 20 minutes with 3.16 μM SCH62539 or 1 μM VU652965 prior to activation with 2 or 10 nM α-thrombin. Means ± S.E.M. are shown (n = 3). Two-sided t-test was used. Software used was GraphPad Prism (GraphPad Software, La Jolla, CA). *, P < 0.05; ***, P < 0.005. (C) The effect of PAR1 and PAR4 antagonists on platelet aggregation induced by thrombin. Platelets were pretreated for 20 minutes with antagonist prior to activation with 10 nM α-thrombin. Representative tracings of three independent experiments are shown. DMSO, dimethylsulfoxide.
Relative Contributions of PAR1 and PAR4 to Thrombin-Mediated Platelet Signaling.
Because of the lack of tool compounds with appropriate attributes, the precise contributions of PAR1 and PAR4 to platelet signaling have not yet been established in the context of its endogenous ligand thrombin. When exactly is PAR4 engaged as the concentration of thrombin increases? How much of the response at high concentrations of thrombin can be attributed to PAR4? To date, these questions about a relevant pharmacological target remain only vaguely defined. To address this, thrombin concentration response curves (CRCs) were challenged with increasing doses of SCH602539 and VU0652925 individually. Displacement of the thrombin activation curve by each antagonist eventually saturated, representing the point at which the response was exclusively driven by the other PAR (Fig. 3, A and B). However, the nature of displacement was very different. Increasing doses of SCH602539 induced a parallel rightward shift, whereas increasing doses of VU0652925 induced a depression of the maximal response. The parallel rightward shift with SCH602539 resembles the shift in agonist potency typically observed with competitive antagonists. As more agonist is added, the antagonist is outcompeted and the cellular response is restored; however, the displacement with SCH602539 saturates. Consistently, in the presence of a fixed concentration of VU0652925, increasing concentrations of SCH602539 lead to full suppression of the thrombin response. Thus, within the timeframe of the assay dictated by the kinetics of GPIIbIIIa activation on the human platelet, SCH602539 exhibits the characteristics of a noncompetitive antagonist (Fig. 3C) (Becker et al., 2009; Chintala et al., 2010). Since we demonstrated that SCH602539 is capable of abolishing PAR1-mediated GPIIbIIIa activation, the full restoration of the response with increasing concentrations of thrombin can only be attributed to engagement of PAR4. Importantly, these data indicate that PAR1 exclusively contributes to GPIIbIIIa activation at only a very narrow window of thrombin concentrations. VU0652925, on the other hand, induces a progressive and saturable depression of the maximum thrombin response with no rightward shift. The residual response in the presence of VU0652925 (at high concentrations of thrombin) is likely PAR1 mediated. When platelets were preincubated with SCH6052965, increasing concentrations of VU0652925 fully suppressed the thrombin CRC, indicating that VU0652925 is also exhibiting noncompetitive characteristics in our assay. Thus, the residual response at high concentrations of thrombin in the presence of saturating doses of VU0652925 is PAR1 mediated. It is difficult to definitively define the mechanism of action with functional assays alone. It is possible that these compounds are reversible but have an extremely slow off rate. However, we were careful to conduct our assays in human tissue with a physiologically relevant readout, allowing us to contrast biologic activities of these antagonists and make suggestions about their physiologic implications based on the functional consequences. Importantly, these data indicate that although PAR4 requires slightly higher concentrations of thrombin to be engaged, it is responsible for the majority of thrombin-mediated GPIIbIIIa activation on the human platelet.
Fig. 3.

Relative contributions of PAR1 and PAR4 to thrombin-mediated platelet signaling. (A) The effect of increasing doses of SCH602539 on α-thrombin–induced GPIIbIIIa activation (PAC1) CRCs. CRCs were constructed with PAC1 binding data measured by flow cytometry. (B) The effect of increasing doses of VU0652925 on α-thrombin PAC1 CRCs. (C) The effect of increasing doses of SCH602539 on PAR1 isolated thrombin-mediated platelet activation. Platelets were pretreated with a fixed concentration of 316 nM VU0652925 and the indicated concentrations of SCH602539 before activation with increasing concentrations of α-thrombin. (D) The effect of increasing concentrations of VU0652925 on PAR4 isolated thrombin-mediated platelet activation. Platelets were pretreated with a fixed concentration of 1 μM SCH602539 and the indicated concentrations of VU0652925 before activation with increasing concentrations of α-thrombin. Means are shown (n = 3). DMSO, dimethylsulfoxide.
Identification of the Minimum Pharmacophore.
As shown in example 1 in Fig. 4 and Table 1, BMS-3 (VU0652925) is a large molecular weight compound with implicit plasma binding and toxicology concerns. Given its large size and noncompetitive nature in our assay, we began an effort to identify the minimum pharmacophore within VU0652925 that retains specificity, activity against AP, and TL-mediated activation. Activity against PAR4-AP, γ-thrombin, and PAR1-AP was monitored as compounds representing progressive truncations of VU0652925 were synthesized. Figure 4 illustrates the route taken to arrive at the minimum pharmacophore, a 2-methoxyimidazo[2,1-b][1,3,4]thiadiazole ring on an otherwise unsubstituted benzofuran core (example 6) and Table 1 shows the biologic activity. Although potency suffered with the deletion of the 2-phenylthiazole and methoxy moieties from the benzofuran core, specificity and activity against AP and TL were retained.
Fig. 4.

Structures of VU0652925 analogs.
TABLE 1.
Identification of the minimum pharmacophore: biologic activities and molecular weights of VU0652925 analogs
Platelet activation was induced by 200 μM PAR4-AP, 100 nM γ-thrombin, or 20 μM PAR1-AP. Data are presented as the means of at least three independent experiments. Experiments were conducted at 10 μM antagonist for the percent maximum values.
| Example | VU Number | PAR4-AP |
γ-Thrombin |
PAR1-AP |
MW | ||
|---|---|---|---|---|---|---|---|
| Max PAC-1 | IC50 PAC-1 | Max PAC-1 | IC50 PAC-1 | Max PAC-1 | |||
| % | pM | % | nM | % | |||
| 1 | VU0652925 | 0 | 43 | 0 | 229b | 94 | 460.53 |
| 2 | VU0661247 | 0 | 210 | 0 | 3.03 | 51.5 | 407.44 |
| 3 | VU0661245 | 0 | 472 | 0 | 8.42 | 92.6 | 331.35 |
| 4 | VU0807074 | 0.19 | 1.68a | 21.1 | 16.2 | 105 | 301.32 |
| 5 | VU0807081 | 0.27 | 176 | 0 | 7.18 | 102 | 301.32 |
| 6 | VU0806526 | 0.20 | 1.69a | 6.99 | 58.8 | 111 | 271.04 |
MW, molecular weight.
Values are given in nanomoles.
Values are given in picomoles.
Schild Analysis and Identification of Competitive PAR4 Antagonists.
We performed Schild analysis to determine the mechanism of action of each compound in the series (Fig. 5). Schild analysis of VU0652925 with AP suggests a noncompetitive mode of action, consistent with the α-thrombin data (Fig. 3D). Increasing concentrations of VU0652925 led to full depression of the PAR4-AP CRC and log-log plots of DR-1 versus [VU0652925] yielded a slope of 2.86 ± 0.53, which is clearly inconsistent with a competitive mode of inhibition. Replacement of the 2-phenylthiazole group (example 2, VU0661247) leads to an apparent switch in modality. Increasing concentrations of antagonist failed to induce significant depression of the maximum response, suggesting that the antagonist was fully reversible within the time frame of GPIIbIIIa activation. However, the DR-1 versus [antagonist] plot yielded a slope slightly greater than 1 (m = 1.23 ± 0.11). Removal of all benzofuran substituents (example 6, VU0806526) resulted in a slope of 1.02 ± 0.02, consistent with a classic competitive mode of inhibition.
Fig. 5.

Schild analysis and identification of competitive PAR4 antagonists. Progressive fold-shift experiments and accompanying Schild analysis with VU0652925 fragments. A) VU0652925, B) VU0661247, C) VU0661245, D) VU0807074, E) VU0807081, F) VU0806526. Platelet activation was monitored by PAC1 binding. Platelets were pretreated with increasing concentrations of each antagonist for 20 minutes prior to activation with increasing concentrations of PAR4-AP. Each curve was constructed from at least three independent experiments. DRs were calculated from the EC50s of each individual experiment (vehicleEC50/VU#EC50) and plotted against the administered concentration of antagonist. A) Shown on the right are the means ± S.E.M. of log DR-1 (n = 3). In the graph insert, m is the slope from linear regression. DMSO, dimethylsulfoxide.
Smaller PAR4 Antagonists Display Better Drug Metabolism and Pharmacokinetic Dispositions.
Initial characterization of VU0652925 revealed a compound with an undetectable free fraction and relatively high cLogP values (>5), indicative of potentially poor bioavailability. Therefore, drug metabolism and pharmacokinetic parameters such as plasma protein binding and clearance (CLhep, CLint) were also followed during modification of VU0652925. Replacement of the 2-phenylthiazole moiety with a methoxy group (example 3) resulted in a detectable free fraction, and the successive truncation of the compound down to example 6 resulted in increasing concentrations of unbound compound in plasma and more favorable cLogP values (Table 2). Clearance rates increased concomitantly as the unbound fraction increased and cLogP values decreased.
TABLE 2.
In vitro drug metabolism and pharmacokinetic parameters of VU0652925 fragments
Values were determined as described in the Materials and Methods.
| Example |
MW |
PPB fu | CLInt | CLhep | CLogP |
|||
|---|---|---|---|---|---|---|---|---|
| h |
r |
h |
r |
h |
r |
|||
| ml/min per kg | ||||||||
| 1 (VU0652925) | 460.53 | <0.001 | <0.001 | 2.17 | 2.14 | 1.91 | 16.4 | 5.44 |
| 2 (VU0661247) | 407.44 | 0.001 | 0.003 | 17.7 | 37.9 | 9.62 | 24.6 | 5.09 |
| 3 (VU0661245) | 331.35 | 0.008 | 0.031 | 20.7 | 61.5 | 10.4 | 32.7 | 3.32 |
| 4 (VU0807074) | 301.32 | 0.007 | 0.03 | 133 | 336 | 18.1 | 57.9 | 3.23 |
| 5 (VU0807081) | 301.32 | 0.015 | 0.022 | 65.3 | 210 | 15.9 | 52.5 | 3.23 |
| 6 (VU0806526) | 271.04 | 0.01 | 0.047 | 89.4 | 285 | 17 | 56.2 | 3.31 |
cLogP, octanol/water partition coefficient; fu, unbound fraction; MW, molecular weight; PPB, plasma protein binding.
Novel PAR4 Antagonists Are Effective against the TL and Completely Reversible.
Schild analysis with the BMS-3 fragments indicates that they are reversible within the time constraints of AP-mediated GPIIbIIIa activation on human platelets. However, for this to be a tractable strategy toward novel PAR4 antagonists, compounds should be effective against the TL but reversible as the concentration of thrombin increases. Therefore, we compared Schild analysis with VU0652925 and one of our lead compounds developed from the minimum pharmacophore identified here (VU0661224). Platelets were activated with increasing concentrations of γ-thrombin, a product of α-thrombin cleavage that does not interact with or activate PAR1. In the context of γ-thrombin, VU0652925 is able to suppress GPIIbIIIa activation even at extremely high concentrations. By contrast, although it is effective at lower concentrations of γ-thrombin, antagonism of TL-mediated PAR4 activation with VU661224 is reversed at high concentrations. Clearly these antagonists display distinct pharmacodynamics. It remains to be seen whether this will translate to distinct outcomes in vivo.
Discussion
With a PAR4 cleavage-blocking antibody and a small molecule PAR1 antagonist, Kahn et al. (1999) initially noted the synergy between PAR1 and PAR4 in the induction of platelet aggregation. As we also observed (Fig. 2), PAR1 is able to substitute for PAR4 and vice versa to induce ex vivo platelet aggregation in the context of relatively high concentrations of thrombin. Covic et al. (2000) subsequently described the biphasic kinetics of PAR1 and PAR4 activation, noting that the slow signal from PAR4 (20- to 70-fold slower than PAR1) is responsible for the majority of the Ca2+ response to thrombin on human platelets. However, the lack of improved pharmacological tools has prevented any further detail of the relationship between PAR1 and PAR4 in the context of thrombin from being revealed. Our data with SCH602539 and VU0652925 are in agreement with these historical data. PAR1 is able to substitute for PAR4 and vice versa to induce aggregation; however, detailed investigation of the individual contributions of PAR1 and PAR4 to GPIIbIIIa activation suggests that the receptors are not redundant. Using noncompetitive antagonists, we determined that over one-half of the GPIIbIIIa activation response at higher concentrations of thrombin can be attributed to PAR4 and cannot be substituted by PAR1 activity. The physiologic relevance of such high concentrations of thrombin has not been established because it is currently unfeasible to accurately measure local concentrations of circulating thrombin. However, results presented in the Bristol-Myers Squibb patent (Lawrence et al., 2013) around the efficacy of VU0652925 in reducing thrombus volume in a cynomolgus electrolytic carotid artery injury model of thrombosis with a PAR4 antagonist, and their movement into phase II clinical trials, speak to the relevance of these higher concentrations of thrombin and the efficacy of inhibiting their action on platelets. Precedent for the strong and superseding PAR4 response comes from work conducted by our group and others (Vretenbrant et al., 2007; Fälker et al., 2011; Duvernay et al., 2013).
In our primary assay, we observed noncompetitive modes of inhibition for both SCH602539 and VU0652925. Vorapaxar (the orally bioavailable analog of SCH602539) is reportedly a reversible compound but with an extremely slow off rate. The terminal half-life is 126–269 hours and antiplatelet effects can be expected for 4 weeks beyond discontinuation of dosing; therefore, the compound is described by Merck as “essentially irreversible” (Becker et al., 2009; Chintala et al., 2010). SCH602539 likely has a similarly slow off rate, which contributes to the noncompetitive mode of pharmacology that we observed. SCH602539 may not have adequate time to dissociate within the window of platelet activation. The kinetics of platelet activation, once stimulated by thrombin, are expected to be similar ex vivo and in vivo, making the alteration of thrombin-induced GPIIbIIIa activation by SCH602539 that we observed in our ex vivo assay physiologically relevant. VU0652925 may have a similarly extremely slow off rate rendering the compound “essentially irreversible” within the kinetics of thrombin-induced GPIIbIIIa activation. This would explain the mode of inhibition we observed.
The unanticipated results of the vorapaxar clinical trials indicating significant bleeding risk heed caution in designing new TRAs. The nature of the dual receptor system, as illustrated by these results, suggests distinct safety implications for PAR1 and PAR4 antagonists. The effects of a PAR1 antagonist, no matter whether it is competitive or noncompetitive, can be overcome by engagement of PAR4. There are no other lower-affinity thrombin receptors on the platelet, and since PAR4 mediates the majority of thrombin-induced GPIIbIIIa activation, a noncompetitive PAR4 antagonist will permanently depress the thrombin response. Given the role of thrombin-mediated platelet activation in hemostasis, it may be important that the effects of PAR4 antagonists are surmountable so that they can be effective against thrombosis but overcome in the context of life-threatening bleeding (i.e., surgery or trauma). A competitive PAR4 antagonist should inhibit platelet activation at low concentrations of thrombin but should eventually allow reversal of the antagonism and full rescue of the thrombin-mediated platelet response. This would not necessarily render the PAR4 antagonists safer than vorapaxar. However, unless it is competitive and reversible, a PAR4 antagonist may present an even greater risk than vorapaxar. Given the bleeding risk noted in the vorapaxar clinical trials with administration of the antagonist in addition to the standard of care, it is important to compare multiple pharmacological modes of PAR4 inhibition to determine the safest and most efficacious strategy.
A reversible, competitive PAR4 antagonist has not only lower safety concerns but also advantages in terms of its utility as a tool compound. A radiolabeled reversible, competitive antagonist would be capable of defining the binding site of the TL, which has remained elusive until now. A competitive antagonist that interacts with the TL binding pocket would also be critical for determining whether a novel ligand is an allosteric modulator. Finally, reversibility is a requisite for developing positron emission tomography tracers. PAR4 expression has been demonstrated to be dynamic and reflective of various pathologic conditions (Rohatgi et al., 2003; Henrich-Noack et al., 2006; Dabek et al., 2009; Zhang et al., 2014a,b; Yu et al., 2015) and, therefore, has the potential for developing into a biomarker.
The last example presented in this article (example 6, VU0806526) represents a route forward to developing additional specific PAR4 antagonists that can be fully outcompeted by higher concentrations of thrombin. A companion article published elsewhere will describe the development of a series of competitive, reversible PAR4 antagonists around the minimum pharmacophore identified here. That work will present the compound highlighted in Fig. 6B (VU0661224) showing efficacy against the TL but full reversibility. Future efforts will focus on engineering potency into these novel PAR4 antagonists for clinical and basic science research so that we may better understand this important pharmacological target.
Fig. 6.

Novel PAR4 antagonists are effective against the TL and completely reversible. Progressive fold-shift experiments with VU0652925 (A) and novel PAR4 antagonist VU0661224 (B). Platelet activation was monitored by PAC1 binding. Platelets were pretreated with increasing concentrations of each antagonist for 20 minutes prior to activation with increasing concentrations of PAR4-AP. Each curve was constructed from at least three independent experiments. DMSO, dimethylsulfoxide.
Acknowledgments
The authors thank Merck for kindly providing sufficient quantities of the PAR1 antagonist SCH602539 for isolation and study of thrombin-mediated PAR4 activation.
Abbreviations
- AP
activating peptide
- BMS986120
(4-(4-(((6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)benzofuran-4-yl)oxy)methyl)-5-methylthiazol-2-yl)morpholine)
- CRC
concentration response curve
- DR
dose ratio
- FITC
fluorescein isothiocyanate
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- PAR
protease-activated receptor
- TL
tethered ligand
- TRA
thrombin receptor antagonist
Authorship Contributions
Participated in research design: Duvernay, Stauffer, Lindsley, Hamm.
Conducted experiments: Duvernay, Temple, Maeng, Blobaum.
Performed data analysis: Duvernay, Temple, Maeng, Blobaum.
Wrote or contributed to the writing of the manuscript: Duvernay, Lindsley, Hamm.
Footnotes
This research was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants R01NS082198 and R01NS081669].
References
- Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, Ziada KM, Berman G, Strony J, Joseph D, et al. TRA-PCI Investigators (2009) Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet 373:919–928. [DOI] [PubMed] [Google Scholar]
- Bhatt DL, Topol EJ. (2000) Current role of platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes. JAMA 284:1549–1558. [DOI] [PubMed] [Google Scholar]
- Bohula EA, Aylward PE, Bonaca MP, Corbalan RL, Kiss RG, Murphy SA, Scirica BM, White H, Braunwald E, Morrow DA. (2015) Efficacy and safety of vorapaxar with and without a thienopyridine for secondary prevention in patients with previous myocardial infarction and no history of stroke or transient ischemic attack: results from TRA 2°P-TIMI 50. Circulation 132:1871–1879. [DOI] [PubMed] [Google Scholar]
- Bowry AD, Brookhart MA, Choudhry NK. (2008) Meta-analysis of the efficacy and safety of clopidogrel plus aspirin as compared to antiplatelet monotherapy for the prevention of vascular events. Am J Cardiol 101:960–966. [DOI] [PubMed] [Google Scholar]
- Chintala M, Strony J, Yang B, Kurowski S, Li Q. (2010) SCH 602539, a protease-activated receptor-1 antagonist, inhibits thrombosis alone and in combination with cangrelor in a Folts model of arterial thrombosis in cynomolgus monkeys. Arterioscler Thromb Vasc Biol 30:2143–2149. [DOI] [PubMed] [Google Scholar]
- Coller BS, Peerschke EI, Scudder LE, Sullivan CA. (1983) A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest 72:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson J, Flather MD, Fox KA, Findlay I, Rodrigues E, Dooley P, Ludman P, Adgey J, Bowker TJ, Mattu R. (2000) Clinical outcomes, risk stratification and practice patterns of unstable angina and myocardial infarction without ST elevation: Prospective Registry of Acute Ischaemic Syndromes in the UK (PRAIS-UK). Eur Heart J 21:1450–1457. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. (2000) Thrombin signalling and protease-activated receptors. Nature 407:258–264. [DOI] [PubMed] [Google Scholar]
- Covic L, Gresser AL, Kuliopulos A. (2000) Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39:5458–5467. [DOI] [PubMed] [Google Scholar]
- Dabek M, Ferrier L, Roka R, Gecse K, Annahazi A, Moreau J, Escourrou J, Cartier C, Chaumaz G, Leveque M, et al. (2009) Luminal cathepsin G and protease-activated receptor 4: a duet involved in alterations of the colonic epithelial barrier in ulcerative colitis. Am J Pathol 175:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernay M, Young S, Gailani D, Schoenecker J, Hamm HE. (2013) Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol Pharmacol 83:781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fälker K, Haglund L, Gunnarsson P, Nylander M, Lindahl TL, Grenegård M. (2011) Protease-activated receptor 1 (PAR1) signalling desensitization is counteracted via PAR4 signalling in human platelets. Biochem J 436:469–480. [DOI] [PubMed] [Google Scholar]
- Grech ED, Ramsdale DR. (2003) Acute coronary syndrome: unstable angina and non-ST segment elevation myocardial infarction. BMJ 326:1259–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker LA, Boissel JP, Pilgrim AJ, Gent M. (1999) Comparative safety and tolerability of clopidogrel and aspirin: results from CAPRIE. CAPRIE Steering Committee and Investigators. Clopidogrel versus aspirin in patients at risk of ischaemic events. Drug Saf 21:325–335. [DOI] [PubMed] [Google Scholar]
- Henrich-Noack P, Riek-Burchardt M, Baldauf K, Reiser G, Reymann KG. (2006) Focal ischemia induces expression of protease-activated receptor1 (PAR1) and PAR3 on microglia and enhances PAR4 labeling in the penumbra. Brain Res 1070:232–241. [DOI] [PubMed] [Google Scholar]
- Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. (1999) Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest 103:879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MR, Miller MM, Seiffert DA, Posy SL, Wong PC, Banville J, Ruediger EH, Deon DH, Martel A, Tremblay F, et al. (2013) inventors, Bristol-Myers Squibb Company, assignee. Imidazothiadiazole derivatives as protease activated receptor 4 (PAR4) inhibitors for treating platelet aggregation. Patent WO2013163244 A1. 2013 Oct 31.
- Montalescot G, Wiviott SD, Braunwald E, Murphy SA, Gibson CM, McCabe CH, Antman EM, TRITON-TIMI 38 investigators (2009) Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet 373:723–731. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Alberts MJ, Mohr JP, Ameriso SF, Bonaca MP, Goto S, Hankey GJ, Murphy SA, Scirica BM, Braunwald E, Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events–TIMI 50 Steering Committee and Investigators (2013) Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke 44:691–698. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. TRA 2P–TIMI 50 Steering Committee and Investigators (2012) Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 366:1404–1413. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Scirica BM, Fox KA, Berman G, Strony J, Veltri E, Bonaca MP, Fish P, McCabe CH, Braunwald E, TRA 2(o)P-TIMI 50 Investigators (2009) Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2 degrees P)-TIMI 50 trial. Am Heart J 158:335.e3–341.e3. [DOI] [PubMed] [Google Scholar]
- Rohatgi T, Sedehizade F, Sabel BA, Reiser G. (2003) Protease-activated receptor subtype expression in developing eye and adult retina of the rat after optic nerve crush. J Neurosci Res 73:246–254. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Cannon CP, Gibson CM, López-Sendón JL, Montalescot G, Theroux P, Claeys MJ, Cools F, Hill KA, Skene AM, et al. CLARITY-TIMI 28 Investigators (2005) Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 352:1179–1189. [DOI] [PubMed] [Google Scholar]
- Savage B, Cattaneo M, Ruggeri ZM. (2001) Mechanisms of platelet aggregation. Curr Opin Hematol 8:270–276. [DOI] [PubMed] [Google Scholar]
- Scirica BM, Bonaca MP, Braunwald E, De Ferrari GM, Isaza D, Lewis BS, Mehrhof F, Merlini PA, Murphy SA, Sabatine MS, et al. TRA 2°P-TIMI 50 Steering Committee Investigators (2012) Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2°P-TIMI 50 trial. Lancet 380:1317–1324. [DOI] [PubMed] [Google Scholar]
- TRA*CER Executive and Steering Committees (2009) The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA*CER) trial: study design and rationale. Am Heart J 158:327.e4–334.e4. [DOI] [PubMed] [Google Scholar]
- Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. TRACER Investigators (2012) Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 366:20–33. [DOI] [PubMed] [Google Scholar]
- Vretenbrant K, Ramström S, Bjerke M, Lindahl TL. (2007) Platelet activation via PAR4 is involved in the initiation of thrombin generation and in clot elasticity development. Thromb Haemost 97:417–424. [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64:1057–1068. [DOI] [PubMed] [Google Scholar]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, et al. PLATO Investigators (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361:1045–1057. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, et al. TRITON-TIMI 38 Investigators (2007) Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 357:2001–2015. [DOI] [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. (1998) Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA 95:6642–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Jiang P, Xiang Y, Zhang Y, Zhu Z, Zhang C, Lee S, Lee W, Zhang Y. (2015) Increased expression of protease-activated receptor 4 and Trefoil factor 2 in human colorectal cancer. PLoS One 10:e0122678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK, Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators (2001) Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 345:494–502. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. (2014a) F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ Health Perspect 122:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yang R, Burwinkel B, Breitling LP, Holleczek B, Schöttker B, Brenner H. (2014b) F2RL3 methylation in blood DNA is a strong predictor of mortality. Int J Epidemiol 43:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]