

Abstract
The intestinal microbiota exerts a marked influence in the mammalian host, both during homeostasis and disease. However, until very recently, there has been relatively little focus on the potential effect of commensal microorganisms on viral infection of the intestinal tract. In this Progress article, I review the recent advances that elucidate the mechanisms by which enteric viruses use commensal bacteria to enhance viral infectivity. These mechanisms segregate into two general categories: the direct facilitation of viral infection, including bacterial stabilization of viral particles and the facilitation of viral attachment to host target cells; and the indirect skewing of the antiviral immune response in a manner that promotes viral infection. Finally, I discuss the implications of these interactions for the development of vaccines and novel therapeutic approaches.
The human body is colonized by an immense population of microorganisms that is collectively referred to as the microbiota. The intestinal lumen contains a particularly rich microbial community numbering in the trillions and comprised of more than 1,000 different bacterial species as well as a diverse array of viruses, fungi and archaea1,2. This incredibly complex microbial community is regulated by environmental, dietary and host genetic factors3,4. In recent years there has been an expansion of research aimed at defining the influence of the intestinal microbiota on homeostatic and disease states in mammalian hosts. A wealth of evidence indicates that intestinal microorganisms exist in a complex symbiotic relationship that benefits their human host. For example, intestinal microorganisms degrade dietary substances that would otherwise be indigestible, which yields end products that act as energy sources for cellular metabolism, modulators of immune responses and regulators of gut motility5,6. Intestinal microorganisms also have a crucial role in immune development and functionality, as highlighted by the inability of germ-free mice (BOX 1) to develop mature lymphoid structures within the gastrointestinal tract and their severely compromised ability to mount immune responses, which includes a reduction in the levels of secretory immunoglobulin A (IgA) and intestinal T cells, compared with wild-type mice7–9. Finally, commensal bacteria in the gut can protect the mammalian host from infection by pathogenic microorganisms. For example, bacterial species of the microbiota can induce intestinal epithelial cells to secrete antimicrobial proteins, such as angiogenin and the C-type lectin RegIIIγ10,11. Overall, the intestinal microbiota affects host physiology, shapes the mucosal immune system and provides protection from pathogenic bacteria.
Box 1. Experimental approaches to elucidate the role of commensal bacteria during infection with enteric viruses.
Two general strategies have been used to assess the effect of the intestinal microbiota on infection with enteric viruses: the infection of germ-free mice, which are naturally devoid of commensal microbiota; and the administration of a cocktail of oral antibiotics to deplete mice of their commensal microbiota before infection with virus. Although mice used in research are typically devoid of many known pathogens and are thus referred to as specific-pathogen free, germ-free mice are raised in sterile isolators and are considered to be free of all microorganisms, including commensal microorganisms that reside in the intestinal lumen. The benefit of the germ-free state is that it facilitates the investigation of the effect of commensal microorganisms on homeostatic and disease states in the host. However, germ-free mice have several abnormalities in intestinal morphology and mucosal immune development that should be considered when using them to address specific research questions.
A common antibiotic-depletion method is to orally gavage mice with a cocktail of broad-spectrum antibiotics, for example, vancomycin, ampicillin, metronidazole and neomycin, once per day for five days (see the figure). After the fifth day of gavage, antibiotics are then added to the drinking water. Efficient microbial depletion is confirmed by plating fresh faecal samples after the fifth day of gavage and examining bacterial growth under anaerobic conditions followed by aerobic conditions. A control group of mice treated with saline, instead of antibiotics, should be included in all steps, which represents microbially colonized mice (also referred to as control mice throughout the article). The major benefits of the antibiotic-depletion strategy are that the mice do not require specialized housing in sterile isolators, such as germ-free mice do, and they develop normal mucosal immune responses. However, although antibiotic depletion can substantially reduce the microbial load in the intestine, it does not remove 100% of the commensal microorganisms, so one must consider the effect of residual antibiotic-resistant microorganisms on experimental outcomes. Therefore, both approaches to study the role of commensal microorganisms on infection with enteric viruses — germ-free mice and antibiotic depletion — have advantages and disadvantages.

Various viruses, including rotaviruses, noroviruses and astroviruses, infect the gastrointestinal tract and are responsible for an immense disease burden worldwide, causing severe childhood diarrhoea and outbreaks of gastroenteritis. These three virus families are each comprised of non-enveloped RNA viruses that are highly infectious and transmitted through the faecal–oral route. Rotavirus infections are a principal cause of paediatric diarrhoea. They have been recently estimated to cause approximately 200,000 deaths annually in children under 5 years of age worldwide, a reduction from 453,000 deaths in 2008 probably owing to the introduction of effective rotavirus vaccines12,13. With the large-scale implementation of effective rotavirus vaccines, noroviruses are now recognized as the leading cause of severe childhood diarrhoea in developed countries14,15 and they are also considered the most common cause of foodborne disease16. Noroviruses can infect all age groups and are associated with approximately 18% of the total acute cases of gastroenteritis worldwide17. Astroviruses have been reported to account for approximately 2–9% of paediatric cases of gastroenteritis worldwide, although this is undoubtedly a substantial underestimate considering the recent discovery of numerous novel astroviruses that are quite divergent from the previously recognized classic human astroviruses18. There are also enteric viruses that replicate in the intestinal tract but are generally asymptomatic (for example, reovirus), and those that can cause severe disease after dissemination to peripheral tissues (for example, poliovirus). Certain retroviruses, including mouse mammary tumour virus (MMTV), can be transmitted orally through milk from an infected mother to her pups in which they infect the gastro-intestinal tract19, and are therefore also considered enteric in nature.
Because all enteric viruses encounter the dense population of microorganisms residing in the gut lumen, it is likely that there are substantial interactions between commensal bacteria and enteric viruses that could have beneficial or inhibitory outcomes for viral infections at this site. Indeed, several studies have now revealed that the intestinal microbiota has significant and varied roles in enhancing infection with enteric viruses. In this Progress article, I will briefly describe the fundamental observations supporting the bacterial promotion of infection with enteric viruses and subsequently describe in detail the mechanisms underlying these observations. Finally, I discuss how this information could lead to novel treatment and prevention strategies.
The microbiota enhances infection
A role for commensal bacteria in influencing enteric viral infections was first demonstrated in a set of landmark studies, published in 2011, that used poliovirus, reovirus and MMTV20,21. By administering a cocktail of oral antibiotics to mice (BOX 1), the depletion of commensal bacteria was shown to result in substantial attenuation of poliovirus infection compared with infection of microbially colonized mice (mice with a normal microbiota) (BOX 1), as indicated by reduced mortality and reduced faecal shedding of the virus in the antibiotic-treated mice, which reflect a reduction in the level of intestinal virus replication20. Reconstitution of intestinal microorganisms into antibiotic-treated mice was sufficient to restore poliovirus pathogenesis. Moreover, intraperitoneal poliovirus infection occurred efficiently irrespective of the status of the intestinal microbiota. These findings highlight the unique role that intestinal bacteria have in promoting poliovirus infection in the gut. Similarly, oral reovirus infection of interferon (IFN)-deficient mice, which are susceptible to symptomatic infection, that had been treated with antibiotics failed to cause intestinal pathology, whereas reovirus infection of microbially colonized IFN-deficient mice caused faecal abnormalities and tissue pathology. Consistent with this attenuation of infection, intestinal titres of reovirus were substantially reduced in antibiotic-treated IFN-deficient mice, compared with control animals20. In addition, the transmission of MMTV from an infected mother to her pups was prevented by antibiotic treatment of the mother, germ-free mothers that were infected with MMTV failed to transmit the virus to their offspring, and reconstitution of germ-free mothers with a defined gut microbiota fully restored viral transmission21.
Since these initial reports of the microbiota-driven enhancement of infection with enteric viruses, similar observations have been made for infection with rotaviruses and noroviruses. For example, rotavirus-infected antibiotic-treated mice have reduced levels of viral antigen in faeces, reduced levels of viral genomes in intestinal tissue and delayed viral shedding compared with controls22. Furthermore, the incidence and duration of diarrhoea caused by rotavirus infection in suckling mice was reduced by antibiotic treatment22. Similarly, three independent reports have demonstrated that the microbiota can promote murine norovirus infection. In the first study, antibiotic-treated mice showed a reduction in acute virus titres in the distal ileum, mesenteric lymph nodes and colon compared with control mice, which reflects a decrease in viral replication in vivo following antibiotic treatment23. In the second report, germ-free mice showed a reduction in the amount of infectious virus shed in the faeces compared with colonized hosts24. In the third report, a murine norovirus failed to establish persistent infection in antibiotic-treated mice, a phenotype that could be fully rescued by faecal transplantation from colonized (but not from antibiotic-treated) mice25. As observed with poliovirus, systemic murine norovirus infection was not dependent on the intestinal microbiota25.
Collectively, these studies demonstrate that a diverse set of enteric viruses require the intestinal microbiota for efficient infection of the gastrointestinal tract following oral inoculation. The remainder of this Progress article will focus on the mechanisms by which bacteria enhance viral infection.
Direct mechanisms
The intestinal microbiota can directly facilitate enteric virus replication by several mechanisms, including by stabilizing virions, which potentially enhances viral transmissibility, and by promoting virus attachment to host cells.
Virion stabilization
The mechanism by which bacteria enhance infection with enteric viruses has been demonstrated for poliovirus. Poliovirus isolated from the intestinal lumen of microbially colonized mice 2 h post-infection, before the production of progeny virions, had higher infectivity than virus isolated from the lumens of antibiotic-treated or germ-free mice, and tissue-culture-derived virus incubated with faeces from microbially colonized mice or with individual bacteria was more viable than virus incubated with faeces from antibiotic-treated or germ-free mice20. Importantly, the enhancement of viral infectivity did not require live bacteria20. In fact, it was shown that poliovirus directly binds to the bacterial outer-membrane component lipopolysaccharide (LPS), and that LPS and other N-acetylglucosamine-containing bacterial surface polysaccharides that were longer than six monosaccharides were sufficient for stimulatory activity20,26 (FIG. 1a). Virus incubated with bacteria or with bacterial surface polysaccharides showed an increase in thermostability and resistance to inactivation by dilute chlorine bleach, a phenotype that correlated with the delayed release of viral genomes26 (FIG. 1a). Collectively, these data suggest that viral binding to bacterial surface polysaccharides promotes virion stability and prevents premature conformational changes that result in genome extrusion into target cells on receptor binding. Importantly, viral genome uncoating is an event required for the successful infection of target cells; therefore, there must be a fine-tuned balance between enhanced virion stability and the ability to undergo the conformational changes that are required for uncoating.
Figure 1. Enteric virus interactions with bacterial surface glycans can facilitate viral stability and binding to target cells.

a | The binding of lipopolysaccharide (LPS), or other bacterial polysaccharides, to poliovirus leads to an increase in viral thermostability and resistance to inactivation by dilute chlorine bleach, which has been linked with the delayed release of viral genomes. This increase in viral stability could potentiate viral transmissibility. b | Poliovirus associated with LPS binds to the known poliovirus receptor (PVR) more efficiently on the surface of target cells. Several lines of evidence show that LPS enhancement is conferred by facilitating viral binding to the known PVR: pre-treatment of permissive cells with PVR-specific antibody inhibits viral binding in both the presence and absence of LPS; virus bound to LPS does not gain competency to infect non-PVR-expressing cells; and virus incubated with LPS has increased binding to soluble PVR compared with virus alone. c | Human norovirus infection of B cells is facilitated by commensal bacteria that express the appropriate histo-blood group antigen (HBGA). The first indication that commensal bacteria stimulate human norovirus infection of B cells was provided by the observation that the filtration of virus-positive stool to remove commensal bacteria reduced viral infectivity. Providing direct evidence, the supplementation of filtered stool with HBGA-expressing bacteria fully restores infectivity. E. cloacae, Enterobacter cloacae.
A specific residue in a surface-exposed loop of the poliovirus VP1 capsid protein (a threonine at position 99) was shown to be crucial for bacterial polysaccharide-mediated stabilization of the virion26, as the introduction of a lysine at this position (creating the T99K mutant) rendered poliovirus insensitive to LPS-mediated stabilization at temperatures below 40°C. However, this T99K mutant virus was stabilized by LPS at temperatures above 40°C, and the deletion of residue 99 did not alter LPS stabilization compared with wild-type virus. Based on these results, the authors of this study suggested that residue 99 influences LPS binding at physiological temperatures although it is not directly involved in binding. Highlighting the physiological relevance of the LPS-mediated stabilization of virions, when mice were co-infected in a 1:1 ratio with wild-type and T99K mutant polioviruses, substantially more wild-type virus was retained in the faeces of mice 96h after shedding. These findings support the notion that interactions with commensal bacteria can promote the transmission of enteric viruses by enhancing their environmental stability.
Promoting virus attachment
In addition to enhancing virion stability, bacterial surface polysaccharides, such as LPS, promote poliovirus attachment to the surface of target cells26 (FIG. 1b). This activity involves the direct facilitation of viral binding to the poliovirus receptor (PVR), which is supported by the observations that the pre-treatment of permissive cells with PVR-specific antibody inhibited viral attachment irrespective of the presence of LPS, the incubation of poliovirus with LPS did not promote viral infection of non-PVR expressing cells, and virus incubated with LPS bound more efficiently to purified PVR than virus that had not been incubated with LPS26 (FIG. 1b). Several lines of evidence suggest that the stimulatory effects of LPS on poliovirus stability and on cell attachment represent distinct mechanisms. First, the enhancement of viral binding to the poliovirus receptor required 20-fold less LPS than virion stabilization26. Second, the VP1 T99K mutant poliovirus (described above) was not impaired in the LPS-mediated enhancement of cell binding despite the fact that it was impaired in the LPS-mediated enhancement of virion stabilization.
Bacteria-mediated enhancement of norovirus infection also functions, at least in part, by stimulating viral attachment to target cells. Specifically, B cells were recently shown to be permissive to human and murine norovirus infections, which represents a major breakthrough in norovirus research considering the long history of human norovirus uncultivability23. Infection of human B cells by a human norovirus required a commensal bacterial cofactor, as first identified by a substantial reduction in viral infectivity after filtration of the virus-positive stool inoculum through a 0.2 μm membrane (FIG. 1c). Supporting the notion that the filterable cofactor was indeed a microorganism, this phenotype could be fully rescued by pre-incubation of the filtered inoculum with a single type of heat-killed bacteria (FIG. 1c). In contrast to studies of poliovirus, LPS was not capable of rescuing this phenotype. Instead, bacterial expression of an appropriate histo-blood group antigen (HBGA) correlated with the ability of norovirus to infect B cells. Human noroviruses have long been known to require specific HBGA glycans for infectivity27 and recent work has identified that commensal bacteria expressing these glycans are competent for binding to human norovirus particles in a virus-strain-specific manner28. Additional studies identified that Enterobacter cloacae that express the H-type HBGA could enhance human norovirus infection of B cells, whereas an H-type-negative Escherichia coli strain could not23,29. Furthermore, pre-incubation of filtered human norovirus inoculum with the H-type HBGA alone was sufficient to stimulate viral attachment to the surface of B cells. The host receptor, or receptors, used by human noroviruses has not been defined so it remains unknown as to whether bacterial HBGA stimulates viral attachment to a host receptor. However, these data are entirely consistent with a mechanism similar to the LPS-mediated stimulation of poliovirus attachment to its host receptor.
In summary, enteric viruses can interact with bacterial surface glycans, which results in enhanced virion stability and enhanced binding to the surface of target host cells. Therefore, it may be possible to develop novel antiviral strategies that target these virus–bacteria interactions. Considering the physical separation between most commensal microorganisms and intestinal epithelial cells, an intriguing question remains as to how a virus–bacterium complex comes into contact with target cells such that a bacterial glycan can stimulate viral infection (BOX 2).
Box 2. How do commensal bacteria stimulate viral attachment to host cells?
A thick mucus layer and other host defence strategies maintain a physical separation between the epithelium and most commensal microorganisms10,52. Therefore, it is unclear how bacterial glycans directly facilitate viral binding to the surface of target cells in the intestinal tract23,26. There are several possible explanations that will be important to examine experimentally in future studies. First, the interactions between enteric viruses and bacteria that interact directly with enterocytes, for example, segmented filamentous bacteria53,54, could preferentially stimulate viral infections. Second, viruses bound to bacterial surface glycans that are not physically associated with intact bacteria could preferentially enhance viral infections. For example, histo-blood group antigens (HBGAs) that are expressed by commensal bacteria were demonstrated to primarily localize to the extracellular polymeric substance28, which suggests that they may be secreted into the gut lumen. Enteric viruses could also bind to bacterial glycans contained in the secreted outer membrane vesicles (OMVs) of commensal microorganisms. Consistent with this idea, OMVs express bacterial glycans, such as lipopolysaccharide (LPS), and they can interact directly with enterocytes55,56. Third, virus–bacteria–host cell interactions may occur preferentially at sites of reduced host defences, specifically targeting microfold cells (M cells) overlying gut-associated lymphoid tissue, including Peyer’s patches. Because M cells are specialized in sampling luminal contents, they do not secrete mucus or contain microvilli on their apical surface57. In fact, many enteric pathogens, including poliovirus, reovirus, MMTV and norovirus, have evolved to exploit this host vulnerability58–62.
An additional complexity pertaining to norovirus infection is the established tropism of the virus for immune cells that underlie the intestinal epithelium23,47, which led to the inference that norovirus–bacteria complexes transit across the non-permissive epithelium where bacterial HBGA can stimulate the viral infection of underlying immune cells. Indeed, noroviruses can be internalized by enterocytes in the absence of apparent viral replication63–65. Moreover, norovirus applied into the apical supernatant of confluent epithelial cells can be released into the basal chamber and target immune cells23,59. As observed in direct B cells infections, B cell infection in this co-culture system also required a bacterial cofactor23,29, although it has not been elucidated whether the entire bacterium or only its stimulatory glycan co-transcytoses across the epithelial barrier. It should be noted that OMVs can directly interact with intestinal immune cells; therefore, it will be interesting to determine whether HBGAs localize to OMVs55.
Indirect mechanisms
The intestinal microbiota can indirectly enhance infection with enteric viruses by skewing the antiviral immune response. For example, the microbiota can induce a tolerogenic microenvironment that promotes viral evasion of the host immune system, it can suppress the production of antiviral antibodies or it can alter virus-induced IFN signalling.
Inducing a tolerogenic microenvironment
In the steady-state, intestinal epithelial cells can sense commensal bacteria through several innate immune receptors, which results in their secretion of cytokines that regulate immune responses in the gut, leading to the establishment of a tolerogenic microenvironment30,31. Moreover, regulatory T cells (Treg cells) that are specific to commensal bacterial antigens are highly prevalent in the intestine where they maintain immunological tolerance to the tremendous numbers and different species of non-pathogenic microorganisms that comprise the microbiota. However, the establishment of a tolerogenic microenvironment during the immune recognition of commensal bacteria by intestinal epithelial cells and Treg cells could, in theory, influence antiviral immune responses. For example, once Treg cells are stimulated by their specific antigen, they can suppress other cell types in an antigen-nonspecific manner using both cell contact-dependent (for example, the engagement of co-stimulatory receptors on antigen presenting cells) and contact-independent (for example, the secretion of immunoregulatory cytokines) mechanisms32–35. Therefore, it is possible that the immune recognition of commensal bacteria that are complexed with enteric viruses results in bystander suppression of antiviral immune responses.
Supporting this concept, the establishment of persistent MMTV infection in pups that were exposed to provirus-containing maternal milk required the LPS receptor Toll-like receptor 4 (TLR4) and the anti-inflammatory cytokine interleukin-10 (IL-10)36. Notably, MMTV seems to directly bind to LPS, as virus isolated from pups that ingested MMTV-laden maternal milk was associated with LPS, and virus and LPS co-fractionated in ultracentrifugation density gradients21. Furthermore, recent studies have provided mechanistic insight into this virus–LPS interaction, revealing that MMTV incorporates LPS-binding host proteins, such as CD14, MD2 (also known as LY96) and TLR4, into its envelope, and that virions isolated from mice deficient in LPS binding proteins were unable to bind to LPS37. Together with the observation that this MMTV–LPS interaction was required for virally induced IL-10 secretion from cultured splenocytes21, a model develops whereby MMTV bound to bacterial LPS activates TLR4 signalling and ultimately leads to the secretion of immunoregulatory cytokines, such as IL-10 (FIG. 2). Previous studies have identified that dendritic cells and macrophages were activated through TLR4 in response to infection with MMTV, whereas B cells were the main source of IL-10 (REF. 36). An additional series of in vitro and in vivo experiments further examined the chain of events in which the engagement of TLR4 by LPS drives the production of IL-6, which then induces IL-10 secretion; in this immunosuppressive microenvironment, MMTV is able to establish a persistent infection (FIG. 2). Intriguingly, LPS bound to MMTV was more potent at inducing the secretion of IL-6 from splenocytes than virus-free LPS, suggesting that viral binding can augment the immunostimulatory nature of LPS37. Providing evidence for the immunological tolerance to MMTV antigens recognized in this microenvironment, mice that were exposed to MMTV-laden milk as pups failed to produce detectable antiviral antibodies when immunized with viral antigens as adults, although they were fully competent to produce antibodies to a non-viral antigen21. Highlighting the intestinal specificity of this phenomenon, pups that were exposed to MMTV intraperitoneally were not tolerant to MMTV antigens later in life21. Therefore, these data suggest that MMTV–microbiota interactions promote tolerance to viral antigens and facilitate the establishment of a persistent viral infection.
Figure 2. Commensal bacteria can induce a tolerogenic microenvironment that facilitates the establishment of MMTV persistence.
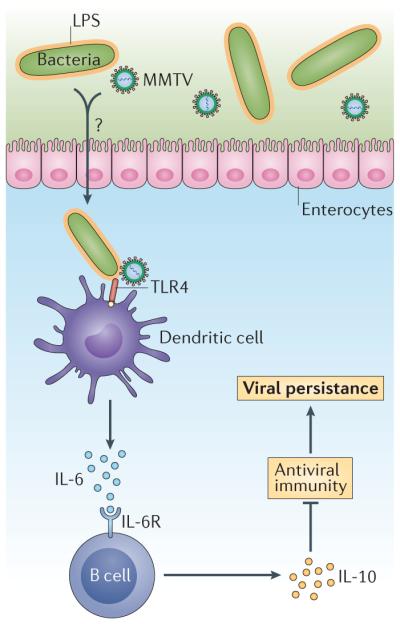
Available evidence pertaining to the establishment of mouse mammary tumour virus (MMTV) persistence supports a model whereby MMTV-bound lipopolysaccharide (LPS) activates Toll-like receptor (TLR) signalling on dendritic cells, or macrophages, inducing the secretion of interleukin-6 (IL-6), which acts on B cells to stimulate their secretion of IL-10. In this immunosuppressive microenvironment, MMTV can establish persistence and immunological tolerance is established to MMTV antigens. Several key observations support this model: the establishment of MMTV persistence requires the LPS receptor TLR4 and the immunosuppressive cytokine IL-10; MMTV binds to LPS and this interaction is required for the induction of IL-10; and mice exposed to MMTV-laden milk as pups are tolerant to MMTV antigens later in life. IL-6R, interleukin-6 receptor.
Indirect evidence suggests that the immune recognition of commensal bacteria may also result in bystander suppression during norovirus infection. Although infections with norovirus are only modestly inflammatory in immunocompetent hosts38–44, infection of mice deficient in the production of IL-10 (IL-10−/− mice) with murine norovirus induces significant intestinal inflammation45. Moreover, this phenotype was microbiota-dependent as no inflammation was observed in germ-free IL-10-deficient mice that were infected with a murine norovirus, and colonization of germ-free IL-10-deficient mice with a defined microbiota was sufficient to restore virally induced inflammation45. Future studies will be required to assess the effect of this inflammatory response on the control of acute viral infection and the development of adaptive immune responses to the virus.
Suppressing antibody production
Despite a reduction in rotavirus infectivity in the absence of intestinal microorganisms, the antiviral antibody response was substantially higher in antibiotic-treated mice compared with microbially colonized controls22. This was true for faecal IgA, serum IgA and serum IgG levels 9–11 weeks post-infection, although antibody levels prior to 9 weeks post-infection were comparable in the two groups. Consistent with these findings, the serum antiviral antibody response was higher in germ-free mice than in conventionally housed mice. Therefore, these data suggest that commensal bacteria suppress the maintenance of an antiviral antibody response. This idea is also supported by an increase in the number of antibody secreting cells in the intestinal lamina propria and Peyer’s patches of antibiotic-treated mice compared with control mice 7 weeks, but not 2 weeks, post-infection22.
Importantly, antibiotic-treated mice that were infected with a murine norovirus had reduced antiviral serum IgG levels 35 days post-infection compared with microbially colonized mice25, which contrasts to the rotavirus studies that showed enhanced antiviral immunity under bacteria-depleted conditions. Therefore, it will be important to carry out a kinetic examination of this response as well as to examine the mucosal antibody response that is probably key to providing protection from secondary noroviruschallenges.
Suppressing IFN signalling
To test the hypothesis that the bacteria-mediated enhancement of persistent murine norovirus infection was related to the skewing of the antiviral immune response, various mouse strains that were deficient in specific immune components were treated with antibiotics and subsequently infected with murine norovirus25. This strategy identified that type I and type II IFN responses were dispensable for the bacterial regulation of viral persistence, as were the pattern recognition receptors TLR2, TLR4 and melanoma differentiation-associated protein 5 (MDA5; also known as IFIH1), adaptive immune responses and the autophagy pathway. By contrast, mice lacking the IFNλ receptor (also known as the type III IFN receptor), signal transducer and activator of transcription 1 (STAT1; which is a key signalling molecule upon the engagement of IFNλ with its cognate receptor) or interferon-regulatory factor 3 (IRF3; which is required for expression of IFNλ), were all persistently infected with a murine norovirus irrespective of the presence of commensal microorganisms. These results support a model whereby commensal bacteria suppress the production of a key antiviral cytokine — IFNλ — upon their interaction with norovirus (FIG. 3). Moreover, these data are consistent with the recent demonstration of a crucial role for type III IFN responses in preventing persistent murine norovirus infection in the colon46. Importantly, this latter study revealed that IFNλ acted on non-haematopoietic cells to prevent the establishment of persistent murine norovirus infection, although available evidence indicates that noroviruses have a specific tropism for immune cells23,46–49. Therefore, IFNλ is likely to inhibit the persistence of norovirus infection through an unknown indirect mechanism. Notably, IFNλ also controls rotavirus infection in mice50; thus, it will be interesting to determine whether this response is similarly regulated by the interactions between the enteric virus and commensal bacteria.
Figure 3. Commensal bacteria can suppress the type III interferon response, facilitating the establishment of murine norovirus persistence.

Although the establishment of murine norovirus persistence requires commensal bacteria in immunocompetent mice, the establishment of persistence occurs in a microbiota-independent manner in mice lacking the type III interferon receptor (IFNλR), known as IFNλR-deficient mice. Together with the observations that IFNλ acts on non-haematopoietic cells and that the tropism of murine norovirus is specific to immune cells, a model develops whereby norovirus interactions with commensal bacteria normally suppress the production of antiviral IFNλ (part a). In microorganism-depleted mice, IFNλ is produced and activates the IFNλR in enterocytes or in other bystander cells to indirectly inhibit the establishment of norovirus persistence (part b). In mice lacking functional type III IFN signalling pathways, the establishment of persistence does not require interactions with commensal bacterial because the bystander cells are impaired in their ability to respond to IFNλ (part c).
In summary, by interacting with components of the immune system, the intestinal microbiota can modulate antiviral immune responses, which enhance infection by enteric viruses. Therefore, elucidating the mechanistic details that govern these interactions may also inform the design of novel antiviral strategies.
Outlook
Although the influence of commensal bacteria on infection with enteric viruses was largely unexplored until recent years, it is rapidly becoming clear that the intestinal microbiota has a profound effect on the overall outcome of virus infections at this mucosal site. Considering the density and diversity of microorganisms residing in the gut lumen, this is perhaps not surprising. However, it is crucial to understand these interactions to design effective therapeutic and preventive strategies. For all enteric viruses that have been studied thus far in microbially depleted environments, including poliovirus, reovirus, rotavirus, an enteric retrovirus and norovirus, the intestinal microbiota has been demonstrated to have a stimulatory role in viral infection. As the mechanisms for this enhancement begin to be identified, it is clear that enteric viruses have evolved diverse strategies to exploit the microorganisms they encounter in the gut lumen. However, these mechanisms segregate into two general categories: direct facilitation of viral infections, with two subcategories of virion stabilization and the promotion of viral binding to the surface of target cells; and the indirect skewing of the antiviral immune response in a manner that is conducive to viral infection.
Understanding the molecular requirements for virus–bacteria interactions should illuminate novel approaches to combat enteric viruses. For example, based on the bacterial enhancement of poliovirus thermostability and bleach resistance, it may be possible to design strategies to inactivate enteric viruses in environmental reservoirs by targeting their binding to microorganisms. Antiviral drugs that disrupt virus–bacteria interactions may be particularly useful in controlling the transmission of certain enteric viruses during large outbreaks. Finally, novel vaccination approaches may develop from understanding these interactions. Especially pertinent to this point, a major obstacle in vaccine design may be the bystander suppression that is induced by the co-recognition of viruses and commensal bacteria by the host immune system. However, this knowledge may be used to design vaccines that override the tolerogenic signal provided by the commensal bacterial antigens. Providing proof-of-principle for this idea, it was recently demonstrated that bacterial flagellin, an immunostimulatory bacterial antigen, prevented rotavirus infection in mice51. Although this protection occurred independent of adaptive immune responses, it is interesting to speculate that bacterial adjuvants can be designed that express the appropriate viral ligand and immunostimulatory bacterial antigens for driving robust and lasting protective immunity to enteric viruses.
In this Progress article, although I have focused exclusively on the effect of commensal bacteria on infection by pathogenic enteric viruses, it is important to recognize the immense community of non-pathogenic viruses, archaea, meiofauna and fungi that reside in the gut lumen as well. These undoubtedly also interact with pathogenic viruses that enter the host in this compartment and future studies should investigate whether these additional microbiota–virus interactions also influence the outcome of infection.
Finally, it would be remiss not to acknowledge the idea that antibiotics could be used to treat infection with enteric viruses. Although this argument could logically develop from the studies highlighted in this Progress article, one must recognize that the antibiotic strategy used in these collective experiments was designed to deplete mice of the majority of their commensal microorganisms. Considering the multitude of health benefits provided by our microbiota — for example, protection from pathogenic bacterial infections and the promotion of host metabolic and immune functions — the negative consequences of broad-spectrum antibiotic treatment would far outweigh the benefit of resolving an enteric virus infection. Moreover, it is widely recognized that antibiotic overuse contributes to the emergence of antibiotic-resistant bacteria that can cause life-threatening infections.
In summary, understanding how the intestinal microbiota promotes infection with enteric viruses is a very exciting and developing field. I anticipate that additional details into the molecular and immunological mechanisms that govern the interactions between the microbiota and enteric viruses will continue to be uncovered and will deepen our understanding of the complex relationships between the mammalian host and its commensal microorganisms as well as pathogenic invaders.
Acknowledgements
This work was supported by the US National Institutes of Health (NIH; grant R01AI116892).
Footnotes
Competing interests statement
The author declares no competing interests.
References
- 1.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norman JM, Handley SA, Virgin HW. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology. 2014;146:1459–1469. doi: 10.1053/j.gastro.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sommer F, Bäckhed F. The gut microbiota — masters of host development and physiology. Nat. Rev. Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson JK, et al. Host–gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 6.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 7.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamada N, Núñez G. Regulation of the immune system by the resident intestinal bacteria. Gastroenterology. 2014;146:1477–1488. doi: 10.1053/j.gastro.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl Acad. Sci. USA. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaishnava S, et al. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 12.Tate JE, et al. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect. Dis. 2012;12:136–141. doi: 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- 13.Lanata CF, et al. Global causes of diarrheal disease mortality in children<5 years of age: a systematic review. PLoS ONE. 2013;8:e72788. doi: 10.1371/journal.pone.0072788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payne DC, et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013;368:1121–1130. doi: 10.1056/NEJMsa1206589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koo HL, et al. Noroviruses: the most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vaccination. J. Pediatr. Infect. Dis. Soc. 2013;2:57–60. doi: 10.1093/jpids/pis070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koo HL, Ajami N, Atmar RL, DuPont HL. Noroviruses: the leading cause of foodborne disease worldwide. Discov. Med. 2010;10:61–70. [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed SM, et al. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect. Dis. 2014;14:725–730. doi: 10.1016/S1473-3099(14)70767-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosch A, Pintó RM, Guix S. Human astroviruses. Clin. Microbiol. Rev. 2014;27:1048–1074. doi: 10.1128/CMR.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross SR. Mouse mammary tumor virus molecular biology and oncogenesis. Viruses. 2010;2:2000–2012. doi: 10.3390/v2092000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuss SK, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchiyama R, Chassaing B, Zhang B, Gewirtz AT. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014;210:171–182. doi: 10.1093/infdis/jiu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones MK, et al. Enteric bacteria promote human and murine norovirus infection of B cells. Science. 2014;346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014;516:94–98. doi: 10.1038/nature13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldridge MT, et al. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science. 2015;347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson CM, Jesudhasan PR, Pfeiffer JK. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe. 2014;15:36–46. doi: 10.1016/j.chom.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan M, Jiang X. Norovirus and its histo-blood group antigen receptors: an answer to a historical puzzle. Trends Microbiol. 2005;13:285–293. doi: 10.1016/j.tim.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Miura T, et al. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013;87:9441–9451. doi: 10.1128/JVI.01060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karst SM. Identification of a novel cellular target and a co-factor for norovirus infection – B cells and commensal bacteria. Gut Microbes. 2015;6:266–271. doi: 10.1080/19490976.2015.1052211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 31.Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell. 2013;153:812–827. doi: 10.1016/j.cell.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi T, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 33.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 34.Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int. Immunol. 2009;21:1105–1111. doi: 10.1093/intimm/dxp095. [DOI] [PubMed] [Google Scholar]
- 35.Caridade M, Graca L, Ribeiro RM. Mechanisms underlying CD4+ Treg immune regulation in the adult: from experiments to models. Front. Immunol. 2013;4:378. doi: 10.3389/fimmu.2013.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jude BA, et al. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 2003;4:573–578. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- 37.Wilks J, et al. Mammalian lipopolysaccharide receptors incorporated into the retroviral envelope augment virus transmission. Cell Host Microbe. 2015;18:456–462. doi: 10.1016/j.chom.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blacklow NR, et al. Acute infectious nonbacterial gastroenteritis: etiology and pathogenesis. Ann. Intern. Med. 1972;76:993–1008. doi: 10.7326/0003-4819-76-6-993. [DOI] [PubMed] [Google Scholar]
- 39.Dolin R, Levy AG, Wyatt RG, Thornhill TS, Gardner JD. Viral gastroenteritis induced by the Hawaii agent. Jejunal histopathology and serologic response. Am. J. Med. 1975;59:761–768. doi: 10.1016/0002-9343(75)90461-1. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber DS, Blacklow NR, Trier JS. The mucosal lesion of the proximal small intestine in acute infectious nonbacterial gastroenteritis. N. Engl. J. Med. 1973;288:1318–1323. doi: 10.1056/NEJM197306212882503. [DOI] [PubMed] [Google Scholar]
- 41.Mumphrey SM, et al. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J. Virol. 2007;81:3251–3263. doi: 10.1128/JVI.02096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Souza M, Azevedo MSP, Jung K, Cheetham S, Saif LJ. Pathogenesis and immune responses in gnotobiotic calves after infection with the genogroup II.4-HS66 strain of human norovirus. J. Virol. 2008;82:1777–1786. doi: 10.1128/JVI.01347-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Troeger H, et al. Structural and functional changes of the duodenum in human norovirus infection. Gut. 2009;58:1070–1077. doi: 10.1136/gut.2008.160150. [DOI] [PubMed] [Google Scholar]
- 44.Kahan SM, et al. Comparative murine norovirus studies reveal a lack of correlation between intestinal virus titers and enteric pathology. Virology. 2011;421:202–210. doi: 10.1016/j.virol.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basic M, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel Dis. 2014;20:431–443. doi: 10.1097/01.MIB.0000441346.86827.ed. [DOI] [PubMed] [Google Scholar]
- 46.Nice TJ, et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wobus CE, et al. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004;2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bok K, et al. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc. Natl Acad. Sci. USA. 2011;108:325–330. doi: 10.1073/pnas.1014577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duizer E, et al. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 2004;85:79–87. doi: 10.1099/vir.0.19478-0. [DOI] [PubMed] [Google Scholar]
- 50.Pott J, et al. IFN-λ determines the intestinal epithelial antiviral host defense. Proc. Natl Acad. Sci. USA. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B, et al. Prevention and cure of rotavirus infection via TLR5/NLRC4–mediated production of IL-22 and IL-18. Science. 2014;346:861–865. doi: 10.1126/science.1256999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johansson MEV, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl Acad. Sci. USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blumershine RV, Savage DC. Filamentous microbes indigenous to the murine small bowel: a scanning electron microscopic study of their morphology and attachment to the epithelium. Microb. Ecol. 1977;4:95–103. doi: 10.1007/BF02014280. [DOI] [PubMed] [Google Scholar]
- 54.Klaasen HLBM, Koopman JP, Poelma FGJ, Beynen AC. Intestinal, segmented, filamentous bacteria. FEMS Microbiol. Rev. 1992;8:165–179. doi: 10.1111/j.1574-6968.1992.tb04986.x. [DOI] [PubMed] [Google Scholar]
- 55.Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015;15:375–387. doi: 10.1038/nri3837. [DOI] [PubMed] [Google Scholar]
- 56.Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 2015;13:605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013;6:666–677. doi: 10.1038/mi.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Golovkina TV, Shlomchik M, Hannum L, Chervonsky A. Organogenic role of B lymphocytes in mucosal immunity. Science. 1999;286:1965–1968. doi: 10.1126/science.286.5446.1965. [DOI] [PubMed] [Google Scholar]
- 59.Gonzalez-Hernandez MB, et al. Murine norovirus transcytosis across an in vitro polarized murine intestinal epithelial monolayer is mediated by M-like cells. J. Virol. 2013;87:12685–12693. doi: 10.1128/JVI.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez-Hernandez MB, et al. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J. Virol. 2014;88:6934–6943. doi: 10.1128/JVI.00204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siciński P, et al. Poliovirus type 1 enters the human host through intestinal M cells. Gastroenterology. 1990;98:56–58. doi: 10.1016/0016-5085(90)91290-m. [DOI] [PubMed] [Google Scholar]
- 62.Wolf JL, et al. Intestinal M cells: a pathway for entry of reovirus into the host. Science. 1981;212:471–472. doi: 10.1126/science.6259737. [DOI] [PubMed] [Google Scholar]
- 63.Marionneau S, et al. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology. 2002;122:1967–1977. doi: 10.1053/gast.2002.33661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamura M, Natori K, Kobayashi M, Miyamura T, Takeda N. Interaction of recombinant norwalk virus particles with the 105-kilodalton cellular binding protein, a candidate receptor molecule for virus attachment. J. Virol. 2000;74:11589–11597. doi: 10.1128/jvi.74.24.11589-11597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White LJ, et al. Attachment and entry of recombinant norwalk virus capsids to cultured human and animal cell lines. J. Virol. 1996;70:6589–6597. doi: 10.1128/jvi.70.10.6589-6597.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]