

Abstract
Oxidative stress, mainly contributed by reactive oxygen species (ROS), has been implicated in pathogenesis of several diseases. We review two primary examples; fibrosis and cancer. In fibrosis, ROS promote activation and proliferation of fibroblasts and myofibroblasts, activating TGF-β pathway in an autocrine manner. In cancer, ROS account for its genomic instability, resistance to apoptosis, proliferation, and angiogenesis. Importantly, ROS trigger cancer cell invasion through invadopodia formation as well as extravasation into a distant metastasis site. Use of antioxidant supplements, enzymes, and inhibitors for ROS-generating NADPH oxidases (NOX) is a logical therapeutic intervention for fibrosis and cancer. We review such attempts, progress, and challenges. Lastly, we review how nanoparticles with inherent antioxidant activity can also be a promising therapeutic option, considering their additional feature as a delivery platform for drugs, genes, and imaging agents.
Keywords: ROS, Antioxidant, Fibrosis, Cancer, Metastasis, Nanoparticles
Graphical abstract
Highlights
-
•
Reactive Oxygen Species (ROS) have several important roles in fibrosis and cancer.
-
•
Scavenging ROS with antioxidants can intervene fibrosis and cancer progression.
-
•
Nanoparticles with inherent antioxidant activity have therapeutic implications.
1. Introduction
The regulation of redox homeostasis is crucial for the maintenance of normal cellular growth, metabolism, and survival. Oxidative stress is defined as the imbalance between the production of reactive oxygen species (ROS) and the capability of the cell to elicit an effective antioxidant response. At lower concentrations, ROS are important signaling molecules involved in cellular proliferation, migration, and apoptosis [1], [2]. Several sources of ROS in cells and tissue have been identified, including mitochondrial electron transfer chain [3] and NADPH oxidase (NOX) enzymes [4]. At higher concentrations, these molecules could be useful against pathogens, resulting in increased leukocyte and platelet activation, and increased leukocyte recruitment [5]. While this is true in the context of innate immunity and inflammatory signaling in the immune cells, most ROS are harmful to cells due to the accumulation of irreversible damages to proteins, lipids, and most importantly, to DNA leading to mutations and cell death [6], [7].
ROS and oxidative stress have been implicated in a number of diseases, including fibrosis and cancer [8]. NOX-derived ROS have been identified as the main source of oxidative stress, which promotes key events in the development of fibrotic diseases (such as skin fibrosis [9], idiopathic pulmonary fibrosis [10], liver fibrosis [11], and kidney fibrosis [12]) as well as the initiation and progression of cancer [13]. To date, there is no cure for most of these diseases. Current approaches are limited to attempts on slowing down disease progression in fibrotic diseases (such as pirfenidone for pulmonary fibrosis). For cancer, there are several treatment approaches including, chemotherapy, surgery, radiation, immunotherapy and other novel targeted therapies. Cures can be achieved in some cases (e.g., when tumors are diagnosed early), but resistance and recurrence are common. Chemotherapy and radiation also generate ROS, which, at high levels, are toxic to cancer cells. Nevertheless, sub-lethal ROS generated by these treatments were also reported to promote cancer invasion and metastasis [14]. ROS are thus considered one of the mediators of drug resistance and metastasis in cancer [14], [15], [16].
In recent years, antioxidants have drawn much attention as potential therapeutic interventions due to their ability to fight oxidative stress (and thereby negate its role) in fibrosis and cancer development. The main function of antioxidants is to scavenge or neutralize free radical formation and to inhibit the deleterious downstream effects of ROS. However, most antioxidants, taken orally, have limited absorption profile, which leads to low bioavailability and insufficient concentrations at the target site [17], [18]. To overcome this issue, research has been focused on developing nanoparticles with intrinsic antioxidant properties which can be functionalized to provide localized or targeted therapy. These nanoparticles are mostly made up of inorganic materials such as mesoporous silica, cerium oxide, and fullerene, which exhibit antioxidant activities, protecting cells against oxidative stress when evaluated in vitro and in animal models. The antioxidant capacities of these nanoparticles are thought to be contributed by their redox and catalytic properties, electronic configuration, oxygen vacancy defects, and high-surface-to-volume ratio [19], [20]. Additionally, nanoparticles can be designed to be multi-functional, also serving as delivery platforms for other therapeutics.
2. Overview of reactive oxygen species (ROS)
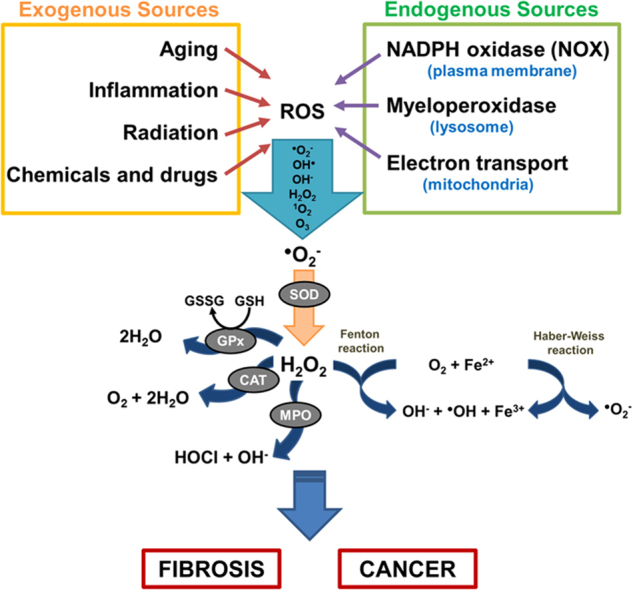
Reactive species are broadly categorized into 4 groups: ROS, reactive nitrogen species (RNS), reactive sulfur species (RSS), and reactive chloride species (RCS) [21]. Among these groups, ROS are found to be most abundantly produced [21]. ROS are generally defined as oxygen-containing small species including superoxide anion radical (O2•−), hydroxyl radical (OH•), hydroxyl ion (OH−), hydrogen peroxide (H2O2), singlet oxygen (1O2), and ozone (O3) [4], [21]. ROS can be generated either by exogenous sources such as UV radiation, toxic chemicals and drugs, physiological changes such as aging or injury/inflammation [22], or by intracellular (endogenous) sources such as NOX enzymes on the plasma membrane [4], myeloperoxidases (MPO) in phagocytes [23], and as by-products of respiratory chain function in mitochondria [3]. As highlighted in Fig. 1, ROS generation is a cascade of reactions initiated by the production of O2•− inside the cells, contributed by endogenous and exogenous cellular sources. Cellular defenses against these ROS molecules involve endogenous antioxidants, such as glutathione peroxidases (GPx), catalases (CAT), and superoxide dismutases (SOD) [24]. Under normal physiological conditions, the formation and elimination of ROS is tightly regulated through the help of the ROS-scavengers/endogenous antioxidants to maintain homeostasis and avoid the harmful effects of oxidative stress [24]. However, the elimination process can become saturated and the increased accumulation of ROS leads to permanent changes and/or damages to the DNA, lipids and proteins with detrimental effects, such as cell death, mutagenesis, carcinogenesis and fibrosis.
Fig. 1.

Sources of ROS and key ROS molecules in signaling. ROS generation is a cascade of reaction initiated by the production of O2•− inside the cells, contributed by endogenous and exogenous cellular sources. Molecular oxygen is reduced to superoxide anion (O2•−) by enzymes such as NOX and nitric oxide synthases (NOS), or as by-products of redox reactions in mitochondrial respirations. O2•−, being cell-impermeant molecule, is then rapidly dismutated to H2O2 either spontaneously or enzymatically by antioxidant enzyme superoxide dismutases (SODs). The intracellular removal of H2O2 can be categorized into three different mechanisms: 1) by the action of catalase (CAT) and glutathione peroxidases (GPx) which reduces H2O2 to water, 2) through conversion of H2O2 into hypochlorous acid (HOCl) and 1O2 by the heme enzyme myeloperoxidase (MPO) the neutrophils, which results in antimicrobial activity, and 3) by Fenton reaction whereby H2O2 is converted to the highly reactive OH• through oxidation of Fe2+ to Fe3+. The OH• produced will then react with H2O2 to form O2•−, which, again, reacts with H2O2 to form OH• and OH−, as a part of Haber-Weiss reaction.
2.1. Roles of ROS in fibrosis
Fibrosis is a complex disease characterized by excessive synthesis and accumulation of extracellular matrices that occur as a result of activation and proliferation of fibroblasts and myofibroblasts. Fibrogenesis can be broadly categorized into four different stages: 1) initiation of tissue injury, 2) inflammation and activation of fibroblasts, 3) extracellular matrix (ECM) synthesis, and 4) deposition of ECM, which eventually leads to organ failure [25]. The causes of fibrosis vary greatly, but common contributing factors include i) physical or chemical injury, ii) autoimmune disease (e.g., systemic sclerosis) [26], iii) virus-induced (e.g., hepatitis C virus-induced liver fibrosis) [27], iv) alcohol-induced (e.g., liver fibrosis) [28], v) hypertension (e.g., hypertensive myocardial fibrosis), or vi) unknown (e.g., idiopathic pulmonary fibrosis) [26], [29], [30]. Notably, nearly 45% of all naturally-occurring deaths in the western world are attributed to some form of fibrotic disease [31].
The release of ROS along with the secretion of chemokines and growth factors (such as platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), connective tissue growth factor (CTGF), interleukin-6 (IL-6), and interleukin-13 (IL-13)) by immune cells during the inflammation phase is known to promote the activation of fibroblast and collagen deposition in fibrosis [32], [33]. Among them, TGF-β is the most potent profibrogenic cytokine, which plays a vital role in regulating important biological processes such as cellular proliferation, extracellular matrix (ECM) production, and epithelial–mesenchymal transition (EMT) [22]. TGF-β mRNA and/or protein expression has been found to be elevated in most fibrotic diseases in patients [34], [35], [36] as well as experimental fibrosis models [37], [38], [39]. As shown in Fig. 2, the presence of ROS could activate TGF-β signaling pathways, which then signal through either SMAD-dependent or SMAD-independent pathways (e.g., phosphatidylinositol-3-kinase (PI3K), c-Jun N-terminal kinases (JNK)) [40]. Increased TGF-β signaling also induces elevated production of NOX4-generated ROS [41], which further stimulates the transcriptional activities of pro-fibrotic genes such as collagen I (COLI), alpha smooth muscle actin (α-SMA), and NOX4. In addition, the presence of NOX4-generated ROS could activate signaling pathways such as JNK and nuclear factor kappa B (NFκB) [42], [43], and trigger DNA oxidation as the initial step in a cascade of events which lead to myofibroblast differentiation and overaccumulation of collagen deposition into ECM, leading to fibrosis.
Fig. 2.

ROS contribute to the induction and persistence of TGF-β-mediated fibrosis. The presence of ROS induces the conversion of latent TGF-β complex to its active form, which binds to its receptor and triggers signaling pathways such as SMAD2/3, PI3K, and JNK. This in turn increases the transcriptional activity of various pro-fibrotic genes, such as NOX4, αSMA, and COL I. Increase in NOX4 expression also results in ROS generation, which leads activation of other ROS-dependent signaling transduction pathways such as, NFκB and JNK. Elevated ROS also causes irreversible DNA damage, through oxidization of its bases. Together, enhanced ROS and activated TGF-β signaling contributes to proliferation and transdifferentiation of fibroblast cells into myofibroblasts, and excessive ECM deposition leading to fibrosis.
2.2. Roles of ROS in cancer
Cancer is the second leading cause of death in the United States and was responsible for 584,872 deaths in 2013 [44]. The number of new cancer cases is estimated to climb to 22 million worldwide within the next two decades [45]. Elevated ROS levels have been detected in most cancer cell lines [46] and have been implicated in malignant progression and resistance to treatment [47]. As highlighted in Fig. 3, ROS play a critical role in various signaling cascades relating to survival, proliferation, resistance to apoptosis, neovascularization, invasion, and extravasation and growth into a distant metastasis site [48], [49]. The roles of ROS in cancer can be described as follows.
Fig. 3.

ROS play multiple roles in cancer progression. ROS generated by multiple sources including chemotherapeutics, radiation, inflammation, and hypoxia conditions contributes to genomic instability of the cancer cells, survival, resistance to apoptosis, proliferation, angiogenesis, invasion (through invadopodia formation), as well as extravasation into a distant metastasis site.
2.2.1. Effects of ROS on redox-mediated cellular mechanisms
ROS are capable of modifying numerous cellular pathways by altering the DNA binding sites of redox-sensitive transcription factors (such as hypoxia-inducible factor-1 alpha (HIF-1α), NFĸB, activator protein-1 (AP-1), and p53), or by oxidizing the cysteine residues on these molecules [43]. At the post-translational level, ROS could also directly oxidize multiple types of amino acids, such as methionine to sulfoxide and cysteine to sulfonic acids [50]. These oxidative modifications on the amino acids will lead to structural and conformational change of the tertiary protein structure, which might cause protein degradation by proteasomes or activation/inhibition of the protein activities. Direct protein carbonylation can also occur through oxidative attack on amino acids involved in catalysis such as lysine, arginine, proline, and threonine, which leads to enzyme inactivation [50].
2.2.2. Role of ROS in genomic instability
ROS generated from either the extracellular/intracellular sources could also lead to DNA damage, which in turn activates a number of stress response genes and DNA repair mechanisms. The redox-sensitive p53 protein is an active transcription factor that is involved in numerous cell processes including cell cycle arrest, senescence, and apoptosis [51]. In the presence of excess ROS, p53 plays a crucial role in preventing the propagation of DNA damage [52]. However, in cancer cells, TP53 (gene which encodes p53) is a commonly mutated gene varying from 10% occurrence in diseases, such as hematopoeitic malignancies, to close to 100% in high-grade serous carcinoma of the ovary [53]. In these cancers, DNA damage will accumulate more readily due to inadequate DNA repair mechanisms, resulting in gene mutation and/or deletion. The genomic instability will further activate a number of oncogenes resulting in abnormal metabolic activity and decreased antioxidant production. All these events will eventually lead to an increase in intracellular ROS production in a positive-feedback manner [54].
2.2.3. Role of ROS in tumor hypoxia
As primary tumors continue to grow, the demand for nutrients and oxygen supply will increase in parallel. However, these demands are not always met in rapidly growing tumors and regions of the tumor will become deprived of oxygen. In order to support tumor growth and proliferation in these hypoxic microenvironments, cancer cells undergo several changes to adapt to this oxygen- and nutrient-deprived state, including genotype selections favouring survival (such as TP53 mutation [55]) and activation of hypoxia inducible factor-1 (HIF-1) transcription factor [56]. The HIF family regulates a broad array of genes in response to oxygen deprivation and has been comprehensively reviewed elsewhere [56], [57]. In hypoxic conditions, the hydroxylation of HIF-1α is inhibited, preventing it from being degraded as in normoxic conditions. The HIF-1α then dimerizes with HIF-1β, which later binds to hypoxia response elements (HREs) on the DNA and stimulates the transcription of its target genes, such as vascular endothelial growth factor (VEGF), N-myc downstream-regulated gene (NDRG), and glucose transporter I [58]. These hypoxia-responsive genes are involved in glucose transport, glycolysis, and angiogenesis, allowing cancer cells to survive in such harsh environment. A hypoxic microenvironment also contributes to ROS formation through the release of superoxide, hydrogen peroxide, and hydroxyl radical from the mitochondrial electron transport chain, and ROS, in turn, also stabilize HIF-1α under both normoxic and hypoxic conditions [59], [60], [61].
2.2.4. Interplay between ROS and TGF-β signaling in cancer
Similar to fibrosis, the cross-talk between ROS and TGF-β signaling in cancer has been well-documented and comprehensively reviewed [62], [63]. TGF-β1 is one of the most potent cytokines known to contribute to immunosuppression of immune cells and promoting angiogenesis and EMT in cancer cells [64].
TGF-β1 induces apoptosis in immune cells by directly suppressing the production of cytolytic factors in T-cells, inhibiting proliferation and differentiation of numerous immune cells, and decreasing the tumor surface immunogenicity through inhibition of major histocompatibility complex class II antigens [65]. Gorelik et al. showed that T-cell specific blockade of TGF-β signaling could enhance anti-tumor immunity by the generation of CD8+-mediated tumor-specific cytotoxic T-cells response [66].
Tumor angiogenesis is vital for tumor growth and can also facilitate the dissemination of tumor cells. TGF-β plays a critical role in promoting angiogenesis. The TGF-β SMAD-dependent signaling pathway has been shown to induce vascular endothelial growth factor (VEGF) expression. In addition, different levels of TGF-β expression show distinct effects on angiogenesis: at low levels, TGF-β upregulates angiogenic factors including VEGF, CTGF, and fibroblast growth factor (FGF), while at high levels, TGF-β stimulates smooth muscle cells recruitment and cell differentiation, while inhibiting endothelial cell growth [67].
TGF-β is a major inducer of EMT and cell migration through a combination of SMAD-dependent and -independent pathways (e.g., p38 MAPK) [68]. The downstream effects of the EMT response include transcriptional reprogramming which promotes inactivation of genes (such as E-cadherin) that encodes for epithelial markers and activation of genes for mesenchymal proteins such as N-cadherin and vimentin [69], [70]. Downregulation of E-cadherin is a common feature in many cancers such as metastatic breast cancer [71] and non-small cell lung cancer (NSCLC) [72]. Studies have shown that forced expression of E-cadherin in cancer cells in vitro could suppress cellular migration and invasiveness [73], while forced expression of N-cadherin in cancer cells caused the opposite effects [74]. The shift in expression from E- to N-cadherin and their distinctive expression patterns reflects the EMT phenotype, which is associated with cancer malignancy and metastasis [69].
In addition, TGF-β has been identified as a major contributor of intracellular ROS production through NOX4 activation. NOX4-derived ROS have been implicated in the EMT phenotype in pancreatic cancer cells [62], increased cell survival in urothelial carcinoma [75], and increased cellular migration and invasiveness in breast cancer [76] and ovarian cells [77], respectively.
2.2.5. Role of ROS in metastases
Metastasis is the main cause of cancer-related mortality, which accounts for 90% of death in cancer patients [78]. The metastatic cascade, as shown in Fig. 3, is a complex process encompassing multiple steps, which lead to cancer cell dissemination, such as: 1) loss of cellular adhesion, 2) increased motility and invasiveness of cancer cells through ECM, 3) intravasation and entry into the circulation, 4) exit into a distant tissue (extravasation), and 5) colonization of a new foreign environment [78]. ROS can activate several pathways involved in metastasis. For example, ROS can activate matrix metalloproteinases (MMPs), which can degrade basement membranes and extracellular matrices, facilitating intravasation and extravasation of cancer cells [79]. Furthermore, ROS generated by the NOX family (to be discussed in the next section) were shown to be crucial for the formation of invadopodia, actin-rich membrane protrusions of cancer cells that facilitate pericellular proteolysis and invasive behavior [80]. Reduction of ROS using antioxidant such as N-acetylcysteine (NAC) or NOX inhibitor, DPI, in cancer cells has the ability to decrease cell viability [81], invasion and invadopodia formation [80], suggesting the role of antioxidant in mitigating metastasis.
3. NOX4 – the main source of ROS in fibrosis and cancer
3.1. NADPH oxidase (NOX) family and ROS
NOX family is comprised of seven members including NOX1-5 and dual oxidase DUOX1-2, which are among the best-characterized intracellular ROS-generating enzymes (as shown in Fig. 4) [4], [82]. All are transmembrane flavoproteins, which generate ROS by transferring an electron to an oxygen molecule, resulting in superoxide anion (O2•−), which is then either spontaneously (by low pH) or catalytically (by SOD) dismutated to H2O2. Most NOXes require additional subunits to be functional, specifically NOX 1-3, bind to the transmembrane protein p22phox, which further recruits cytosolic regulatory subunits, such as organizers (p47phox, p40phox, or NOXO1), activators (p67phox or NOXA1), and small GTPases (Rac1 or Rac2) [83], [84]. NOX4, being the exception, only needs to bind to p22phox and does not require cytosolic subunits for maximal oxidase activity. NOX1-5 are mostly located at the plasma membrane of the cell, with NOX4 being additionally detected in the endoplasmic reticulum (ER), mitochondrial membrane, nuclear membrane, focal adhesions, and invadopodia [84]. Extensive details on the structure and activation of NOX isoforms have been reviewed elsewhere [4], [82], [85].
Fig. 4.

Structure of NADPH oxidase family. (A) NOX1 activity requires p22phox, NOXO1 and NOXA1, and the small GTPase Rac. (B) NOX2 requires p22phox, p47phox, p67phox, and Rac. (C) NOX3 requires p22phox and NOXO1. (D) NOX4 requires p22phox, it is constitutively active without the requirement for other cytosolic subunits. (E) and (F): NOX5, DUOX1, and DUOX2 are activated by Ca2+ and do not appear to require subunits. Reproduced with permission from The American Physiological Society [4].
3.2. High NOX4 expression in fibrosis
NOX4 mRNA expression has been found to be upregulated in both pulmonary fibroblasts isolated from idiopathic pulmonary fibrosis (IPF) patients [86] and skin fibroblasts from scleroderma patients [87], as well as in a number of in vivo fibrosis models, including liver fibrosis [88], pulmonary fibrosis [89], [90], diabetic neuropathy (kidney fibrosis associated with diabetes mellitus) [91].
As mentioned in the previous section (Section 2), TGF-β signaling is the major contributor to fibrogenesis. TGF-β upregulates NOX4 expression through 2 major pathways: the canonical SMAD2/3 [86], [92] and non-canonical PI3K pathways [93], [94]. Inhibition of TGF-β signaling using pharmacological inhibitors for SMAD3 or PI3K abrogated NOX4 expression, suggesting that NOX4 expression is downstream of SMAD [95] and/or PI3K [94] pathway. Suppression of NOX4 activity with a NOX inhibitor diphenyleneiodonium chloride (DPI), siRNA, or the antioxidant N-acetylcysteine (NAC), was shown to decrease the expression of alpha smooth muscle actin (α-SMA) and collagen I (COL I) in fibroblasts collected from pulmonary fibrosis patients and in a bleomycin-induced lung injury mouse model [86], [90].
3.3. High NOX4 expression in cancer
High expression of NOX4 has been detected in several cancer types including gliomas [96], melanoma [97], breast cancer [98], ovarian cancer [98], and pancreatic cancer [62]. In cancer cell lines, elevated levels of NOX4 are associated with PI3K/Akt-regulated cell proliferation and invasion [99], TGF-β/SMAD3-driven EMT and cell migration [95], as well as Tks5-dependent invadopodia formation [80]. Depletion of NOX4 with siRNA treatment significantly reduced tumor growth in the in vivo models of bladder cancer [75], renal cancer [100], and glioblastoma [57]. These results suggest that NOX4 is a potential target for pharmacological intervention for cancer treatment.
4. Strategies to suppress oxidative stress
Antioxidants have been commonly described as substances that can delay, prevent or remove oxidative damage to a target molecule [101]. Given that fibrotic and cancer cells are generally present with higher oxidative stress levels than normal cells, it is believed that patients who suffers from those diseases will benefit from antioxidant supplementation.
4.1. Dietary antioxidant supplements
Dietary antioxidants including vitamin C (ascorbic acid), vitamin E (tocopherol), vitamin A (β-carotene), and selenium have the ability to counteract oxidative damage and can be obtained through food components such as fruits and vegetables.
Vitamin C is a water-soluble and strong antioxidant. Vitamin C exists in two forms: L-ascorbic acid and the oxidized form, dehydro-L-ascorbic acid. It can directly react with hydroxyl and lipid peroxyl radicals to form H2O and lipid hydroperoxides. Vitamin C can also neutralize vitamin E and glutathione radicals, and regenerate these antioxidants [102].
Vitamin E exists in at least 8 different isoforms (α-, β-, γ-, δ-tocopherols, and α-, β-, γ-, δ-tocotrienols), which differ only in the number of methyl groups and in the side chains of its aliphatic tails [103]. Only α-tocopherols isoform is mostly retained in the body due to the preferential transfer of α-tocopherol to the lipid particles by a liver α-tocopherol transfer protein. The main role of vitamin E is to act as a chain-breaking antioxidant, which prevents the propagation of lipid peroxidation [104].
Vitamin A is a fat-soluble vitamin and usually found in the diet as preformed vitamin A from animal products such as meat and fish, and as pro-vitamin A from plant-based products such as fruits and vegetables. β-carotene has the highest provitamin A activity, which is further metabolized to retinoic acid and retinol, the active form of vitamin A. β-carotene can physically quench 1O2 and protect organisms from oxidative damage [105].
Selenium is an essential trace element, which can be acquired from the diet by the consumptions of nuts, meats, and fish. It is co-translationally incorporated into amino acids, such as selenocysteine and selenomethionine [106]. The selenium-containing amino acids act as antioxidants by scavenging free radicals and repairing oxidized selenium species.
A few clinical trials (summarized in Table 1) have been conducted, mainly using the synthetic form of these antioxidants on healthy and at-risk populations [107], [108], [109], [110], [111], [112], [113], [114], [115], [116], [117]. These observational studies were designed to provide evidence on the benefit of antioxidant supplementation for reducing or lowering the risk of patients developing or dying from cancer. However, most of the data were inconclusive, with the majority showing no protection or exhibiting harmful effects in the patient cohort. It is possible that this lack of benefit is due to: 1) the difference in the chemical composition of antioxidants found in food compared to those in supplements, 2) the disease-specificity of certain antioxidants (i.e., some antioxidants are more effective than the others in protecting against certain types of diseases), or 3) due to the low bioavailability, these supplements could not reach sufficient intracellular levels to be effective [118]. Therefore, a more potent antioxidant that can be delivered to a specific diseased tissue and with improved bioavailability is thought to be more beneficial than these antioxidant supplements.
Table 1.
Antioxidant supplements in clinical trials.
| Name of trials | Type of antioxidants | Target population | Length of study | Conclusion of the study | Ref. |
|---|---|---|---|---|---|
| Linxian general population nutrition intervention trial | 15 mg beta-carotene, 30 mg alpha-tocopherol, and 50 μg selenium daily | 29,584 healthy Chinese men and women in North China at increased risk of developing esophageal cancer and gastric cancer were recruited | 5 years | reduction in cancer mortality associated with gastric cancer, but not esophangeal cancer | [107] |
| Alpha-Tocopherol/Beta-Carotene Cancer Prevention Study (ATBC) | alpha-tocopherol (50 mg/day) or beta-carotene (20 mg/day) or both | 29,133 male smokers in Finland | 5–8 years | no overall reduction in the incidence of lung cancer or in mortality in all treatment groups | [112] |
| Carotene and Retinol Efficacy Trial (CARET) | 30 mg of β-carotene plus 25, 000 IU of retinyl palmitate daily | 816 men with substantial occupational exposures to asbestos and 1029 men and women who were either current or former cigarette smokers in United States | 6–12 years | beta-carotene supplementation was associated with increased lung cancer incidence and all caused mortality which persisted up to 6 years after the supplementation was ended | [113] |
| 12 year study showed that beta-carotene had no effect on lung cancer incidence or mortality rate in smokers vs. non-smokers | [116] | ||||
| Physicians' Health Study I (PHS I) | 50 mg β-carotene every other day | 22,071 male physicians between age of 40–84 years in the United States | 12 years | supplementation did not reduce the incidence of prostate cancer or other cancers, including lymphoma, leukemia, melanoma, and cancers of the lung, bladder, pancreas, and colon and rectum | [114] |
| Physicians' Health Study II (PHS II) | 400 IU vitamin E every other day, 500 mg vitamin C every day, 50 mg β-carotene or in combination | 14,642 male physicians older than 50 years old in the United States | 8 years | daily multivitamin use was associated with a reduction in total cancer among 1312 men with a baseline history of cancer, but did not differ significantly from that among 13,329 men initially without cancer | [109] |
| Women's Health Study (WHS) | 50 mg β-carotene every other day, vitamin E supplementation (600 IU every other day), and aspirin (100 mg every other day) | 39,876 women aged 45 years or older | 2 years | no benefit or harm associated with 2 years of beta-carotene supplementation | [115] |
| Selenium and Vitamin E Cancer Prevention Trial (SELECT) | daily supplementation with selenium (200 μg), vitamin E (400 IU), or both | 35,533 men from 427 participating sites in the United States, Canada, and Puerto Rico | 7 years | the use of supplements did not reduce the incidence of prostate or other cancers | [109] |
| 8.5 years | after 1.5 years post supplementation, the follow-up study found 17% increase in prostate cancer incidence among men taking vitamin E alone than among men taking a placebo | [108] |
4.2. Enzyme-related antioxidants
Glutathione (GSH), N-acetylcysteine (NAC), and superoxide dismutase (SOD) are enzyme-related antioxidants that act as the first-line defense against cellular oxidants. The effects of these molecules have also been investigated in several clinical trials (see Table 2), and the results from these studies will be discussed in details below.
Table 2.
Enzyme related antioxidants in clinical trials.
| Drug name | Type | Disease | Sponsor | Phase | Study outcome | Current status | Identifier/Ref. |
|---|---|---|---|---|---|---|---|
| Glutathione (GSH) | GSH | Liver fibrosis | Royal Free Hospital London | N/A | no benefit in oral glutathione in hepatic cirrhosis patients | Completed | [122] |
| N-acetylcysteine (NAC) | Precursor of glutathione (GSH) | Idiopathic Pulmonary Fibrosis (IPF) | Zambon SpA | III | no increased survival and no significant difference between treatment arms at 12 months | Completed | NCT00639496 |
| [125] | |||||||
| Duke University | III | no benefit over placebo | Completed | NCT00650091 | |||
| [128] | |||||||
| Head and neck cancer, lung cancer | The Netherlands Cancer Institute, Amsterdam | N/A | no benefit shown in survival, event-free survival, or second primary tumors-for patients | Completed | [111] | ||
| MnSOD plasmid/liposome | Intraoral MnSOD-plasmid liposome (PL) gene therapy | Radiation-induced esophagitis in Advanced Stage III Non-small cell lung cancer | University of Pittsburgh | I/II | oral administration of MnSOD PL was safe and tolerable | Suspended | NCT0061897[137] |
| (reason unknown) | |||||||
| APN201 | Topical administration of Liposomal Recombinant Human Cu/Zn-Superoxide Dismutase | Radiation-induced Dermatitis in Women With Breast Cancer | Apeiron Biologics | I/II | topical treatment was well tolerated with lower pain score in 36/39 patients and decreased fibrotic size in 50% of the cases | Completed | NCT01513278 |
| [138] |
4.2.1. GSH and NAC
GSH is the main non-protein thiol in cells, which acts as a reducing agent and is essential in regulating cellular redox status. GSH is involved in cell protection against free radicals and many other cellular functions [119]. GSH is also critical for the regeneration of other antioxidants, such as tocopherols and ascorbate [120]. NAC is a cysteine precursor that replenishes the intracellular levels of GSH [121]. A few clinical trials have been conducted with either GSH or NAC as interventions for fibrotic diseases, such as liver fibrosis [122], [123] and lung fibrosis [124], [125], as well as cancers, such as head and neck cancer or lung cancer [111]. However, the data have been largely disappointing, with most of them showing no beneficial effects. This is mostly contributed by the low bioavailability of GSH and NAC. GSH is known to be poorly absorbed when ingested due to the action of the intestinal enzyme, γ-glutamyl transpeptidase, which degrades GSH [126]. High dose of NAC is impossible to achieve in vivo without toxicity concerns. For instance, an estimated 5010 mg/kg loading dose (within the first 60 min) and 2250 mg/kg maintenance dose (for the next 4 h) are needed to reach 10 mM concentration in blood based on the pharmacokinetic data of NAC in human volunteers [127], but NAC is usually prescribed at a much lower dose of only 150 mg/kg loading dose and 50 mg/kg maintenance dose (i.v.) (NAC, Acetadote®, package insert) or 600-mg oral dose (three times daily) for pulmonary fibrosis patients in the PANTHER-IPF trial [128].
SODs are metal-containing proteins that catalyze the conversion of superoxide to hydrogen peroxide. Three isoforms have been identified, namely, cytosolic Cu-Zn SOD (SOD1), mitochondrial MnSOD (SOD2), and extracellular SOD (SOD3). The cytosolic and mitochondrial SODs have been indicated in multiple studies as tumor suppressor genes. Overexpression of MnSOD suppressed the malignancy of human breast cancer cells [129], [130], glioma cells [131], and melanoma cells [132]. In contrast, depletion of MnSOD resulted in increased cell proliferation in vitro and contributed to more aggressive tumor growth in vivo [133]. Likewise, overexpression of Cu-Zn-SOD decreased tumor growth in multiple cancer types [130], [134].
SOD overexpression has been shown to confer protection against radiation and display chemopreventive effects in in vivo cancer models. Preclinical studies in mouse models have shown that intraoral delivery of MnSOD2 plasmid/liposomes (MnSOD-PL) to mice decreased irradiation-induced mucosal ulceration [135] as well as esophagitis [136]. Based on the results from these pre-clinical studies, the chemoprotective effects of MnSOD-PL were investigated in radiation/chemotherapy-induced esophagitis in NSCLC patients (see Table 2). Overall, in the phase I clinical trial study, the response rate for the chemoradiation regime was satisfying at 70% and the treatment was safe and well-tolerated [137]. Unfortunately, the Phase II study was later suspended (reason unknown). In another study [138], topical delivery of Cu-Zn SOD1 (APN 201) was tested on 44 female breast cancer patients for its ability to prevent radiation-induced dermatitis in the patients who undergo radiotherapy after surgery. The topical treatment of Cu-Zn SOD1 reduced pain in the fibrotic region in more than 90% of the cases and decreased the fibrotic size by half in 30% of the patients. In a separate study, topical delivery of liposomal human recombinant Cu-Zn SOD1 (APN 201) was safe and well tolerated in 20 female breast cancer patients who received radiation therapy after breast-preserving surgery [139].
Overall, these studies show some promising results on the chemoprotective effects of the SODs. That said, no clinical trial has been conducted so far to investigate the effectiveness of SODs in combatting cancer growth in humans; however, several SODs mimetics have been shown to be beneficial in the pre-clinical models of prostate [140] and breast cancer [141].
4.3. Inhibiting NOX enzymes
Currently, a few nonspecific NOX inhibitors have been identified (which target more than one NOX isoforms) [88], [142], [143], [144], [145], [146], [147]. Some issues with specificity, potency, and toxicity of these inhibitors have limited the use of these compounds in clinical studies [148]. Gene silencing of NOXes has been carried out with shRNA and siRNA [75], [99], [149] but primarily as a proof-of-concept in in vivo studies without using suitable delivery agents such as liposomes or nanoparticles to safely and efficiently deliver these molecules to the right target. The different types of NOX inhibitors as well as NOX gene therapeutics, and their potential applications from the in-vivo studies are summarized in Table 3.
Table 3.
Pre-clinical applications of NOX inhibitors.
| Type | Name | NOX specificity | In vivoroute | Application | Ref. |
|---|---|---|---|---|---|
| Small molecule | diphenyleneiodonium (DPI) | All types of NOX | i.v. | inhibition of A549 human lung cancer metastasis in mice | [142] |
| i.p. | attenuation of skin fibrosis in bleomycin-induced mouse model | [143] | |||
| Small molecule | Fulvene-5 | NOX1, NOX4 | i.p. | inhibition of hemangioma growth in mice | [144] |
| Small molecule | Celastrol | NOX1, NOX2, NOX4, NOX5 | i.p. | inhibition of B16F10 lung cancer metastasis in vivo | [145], [146] |
| Small molecule | Imipramine blue | NOX4, insufficient characterization data for other NOXes | i.v. | inhibition of HNSCC cancer invasion in mice | [153] |
| inhibition of RT2 glioma invasion in rats | [152] | ||||
| Small molecule | GKT136901 | NOX1, NOX2, NOX4, NOX5 | p.o. | inhibition of tumor growth in B16F0 melanoma and Lewis Lung Carcinoma (LLC1) xenograft in mice | [154] |
| Small molecule | GKT137831 | NOX1, NOX2, NOX4, NOX5 | p.o. | reduction of liver fibrosis in CCL4-induced and BDL mouse models | [88], [147] |
| shRNA | NOX4 | NOX4 | N/A | inhibition of tumor growth in NOX4-shRNA transfected NSCLC cells, A549 and H460 | [99] |
| N/A | inhibition of tumor growth in hepatocarcinoma (Hep3B) xenograft in mice | [149] | |||
| siRNA | NOX4 | NOX4 | single intravesical delivery (with Atelocollagen) | inhibition of tumor growth in orthotopic bladder carcinoma | [75] |
| i.t. | attenuation of pulmonary fibrosis in bleomycin-induced mouse model | [90] |
Note: intravenously (i.v.), intraperitoneally (i.p.), per os (p.o.), or intrathecally (i.t.)
Diphenyleneiodonium (DPI) is a nonspecific inhibitor for NOX enzymes. It inhibits a number of flavoproteins including eNOS, xanthine oxidase, and cholinesterases and the internal calcium pump [150]. Although at least two pre-clinical studies using DPI have shown positive results on lung cancer inhibition [142] as well as skin fibrosis reduction [143], the off-target effects of DPI against other flavoproteins will prevent its translation into clinical use.
Fulvene-5 is a water-soluble small molecule inhibitor for NOX2 and NOX4 enzymes. It has an aromatic structure, which allows electron delocalization. Treatment of Fulvene-5 on bEnd.3 endothelioma cells (hemangioma) in vivo significantly reduced tumor growth and was largely attributed to NOX4 inhibition [144]. Hemangioma is the most common type of benign tumors, which occurs in approximately 5–10% infants [151] .
Celastrol is a compound extracted from Thunder God Vine (Tripterygium wilfordii Hook F.), which could inhibit NOX1, 2, 4, and 5 [145]. Treatment of celastrol on mouse melanoma B16F10 and human lung cancer 95-D cells inhibits cell migration as well as invasion in a dose-dependent manner, without affecting cell viability [146]. The anti-metastatic effect of celastrol was further demonstrated in the B16F10 experimental metastasis model in mice with the reduction in pulmonary nodules.
Imipramine blue is an organic triphenylmethane blue dye that is a derivative for the antidepressant drug, imipramine. It has been shown to target NOX4; however, there was insufficient characterization data provided regarding its selectivity on other NOX isoforms. Treatment with imipramine blue in vivo resulted in tumor inhibition of human glioma cells as well as enhancing treatment efficacy in combination with chemotherapy, doxorubicin [152]. In another study, imipramine blue treatment in head and neck squamous cell carcinoma (HNSCC) tumor in mice inhibited tumor invasion and metastatic colonization in the lungs [153].
GKT136901 and GKT137831 are both small molecule inhibitors developed by a French biotech company, Genkyotex. Both compounds have high specificity to NOX1 and 4 (and less for NOX2 and 5). Both compounds show therapeutic potential in reducing liver fibrosis and tumor growth in mice [88], [147], [154]. GKT137831 is currently a lead compound undergoing Phase II clinical trials in diabetic kidney disease (NCT02010242). The preliminary results from this clinical trial showed that patients receiving treatment for 12 weeks had few adverse effects than placebo. However, the primary efficacy endpoint was not achieved due to negligible change in albuminuria (Genkyotex press release, September 2015).
4.3.1. NOX4 shRNA/siRNA
NOX4 gene silencing using siRNA or shRNA has shown good results in terms of tumor reduction in multiple cancer types including NSCLC, hepatocarcinoma, and bladder cancer [75], [99], [149] as well as reduction in pulmonary fibrosis [90]. However, delivering siRNA into the target cells requires a delivery agent, such as atelocollagen [75], since siRNA is unstable under physiological conditions.
4.4. Nanoparticles with intrinsic antioxidant properties
Nanotechnology has become a main focus of biomedical research in recent years. Several types of inorganic nanoparticles possess intrinsic antioxidant properties by scavenging free radicals and decreasing ROS concentrations. In this section, these inorganic nanoparticles will be discussed and summarized in Table 4.
Table 4.
Nanoparticles with intrinsic antioxidant properties.
| Types of nanoparticles | Mode of actions | In vivoroute | Applications | Ref. |
|---|---|---|---|---|
| Cerium oxide nanoparticle (CeO2) | regenerative capacity of the Ce3+/Ce4+ redox couple | i.p. | inhibition of tumor growth and metastasis in A2780 ovarian cancer in mice | [156], [157] |
| i.v. | reduction of oxidative stress in CCl4-induced liver fibrosis in mice | [158], [159] | ||
| Fullerene | presence of π-electrons over the carbon atoms | i.v. , i.p. | protection from liver injury in CCl4-induced acute hepatoxicity and nephrotoxicity rat models | [162] |
| i.p. | inhibition of tumor growth and metastasis in LLC xenograft mouse models (when co-delivered with doxorubicin) | [163] | ||
| Platinum nanoparticles (PtNP) | catalytic activity due to high ratio of electrons remaining on the particle surface | i.v. | prevention of hepatic injury after hepatic ischemia/reperfusion in mice | [164] |
| Mesoporous silica nanoparticles (MSNP) | free radical scavenger, reduction of NOX4 expression in cells | i.d. | attenuation of dermal fibrosis in bleomycin-induced scleroderma mouse model | [169] |
Note: i.p. (intraperitoneal), i.v. (intravenous), i.d. (intradermal)
4.4.1. Cerium oxide (CeO2)
Cerium is a rare-earth element, which belongs to the lanthanide series in the periodic table. CeO2 can exist in both Ce3+ and Ce4+ valence states, which give it the unique ability as an antioxidant [155]. The ROS-scavenging capability of CeO2 is based on redox cycling between Ce3+ and Ce4+ on the nanoparticle surface. Giri et al. reported that treatment of CeO2 nanoparticles attenuated tumor growth in A2780 ovarian cancer mice and significantly inhibited the metastasis of these cancer cells into the lungs of mice [156]. They also found that angiogenesis in the tumors was reduced in treated mice as indicated by less CD31-positive staining. The same group also reported that surface functionalization of CeO2 nanoparticle with folic acid and its co-delivery with cisplatin further decreased the tumor burden and angiogenesis [157]. In other studies, CeO2 nanoparticle administration to carbon-tetrachloride (CCl4)-induced liver fibrosis mice was shown to inhibit oxidative [158], [159] and endoplasmic reticulum stress signaling pathways as well as reduction in inflammatory cytokines such as TNF-α and IL-1β [158].
4.4.2. Fullerene (C60)
Fullerene is a sphere-like molecule composed of 60 carbon atoms, which are arranged in a hexagonal formation to form a hollow spherical structure. It is a powerful antioxidant due to the delocalization of the π-electrons over the carbon atoms, which can readily react with free radicals [160]. Fullerene is also capable of inactivating hydroxyl radicals via attachment to its double bonds [161]. Due to these attractive antioxidant properties, fullerenes and its derivatives have been studied in numerous biomedical applications including fibrosis and cancer. Prophylactic treatment of fullerene in CCl4-induced hepatic injury rat model prevented damage in the liver and kidney of the rats [162]. Another study has also shown that co-treatment of Doxorubicin chemotherapy drug with fullerene resulted in inhibition of tumor growth and metastasis, and increased survival in LLC tumor-bearing mice [163].
4.4.3. Platinum nanoparticles (PtNPs)
PtNPs are known to possess ROS scavenging ability due to the catalytic activity contributed by its high ratio of electrons to particle surface [164]. PtNPs have demonstrated its therapeutic potential in several pre-clinical applications such as treating aging-related skin disease in SOD1 knockout mice [165], protection against UV-induced apoptosis in HaCaT keratinocytes [166], and prevention of hepatic injury from hepatic ischemia/reperfusion injury in mice [164].
4.4.4. Mesoporous silica nanoparticles (MSNP)
MSNPs have been reported to have reactive oxygen species (ROS), hydroxyl radical, and free radical scavenging capability [167], [168], [169]. ROS scavenging by MSNP also attenuates NOX4 mRNA expression in melanoma cells in vitro [168]. Thus, MSNPs have great potential in the treatment of oxidative-induced pathological conditions such as fibrosis and cancer.
Among all classes of antioxidant nanoparticles discussed herein, MSNP is most widely used for delivery of drugs, genes, and imaging agents. MSNP delivery carriers have many favorable attributes, such as tailorable mesoporous structures, high surface areas, large pore volumes, ease of controlling size, and high scalability [170]. MSNP is soluble in physiological pH to non-toxic silicic acid, which can be cleared by the kidneys [171]. Silica is endogenous in the human body unlike cerium and platinum. Numerous studies have also shown great in vitro [172], [173] and in vivo [174], [175], [176] biocompatibility of silica nanoparticles. These features are desirable since it will prevent long-term toxicity issues of the nanomaterials in vivo. Silica nanoparticles with PET tracers have also recently entered a clinical trial with a favorable safety profile (Cornell dots) [177]. Amorphous silica nanoparticles have been widely used in cosmetics, food and pill additives as anti-caking.
Our group reported successful development of MSNP-based siRNA delivery platform, which is a 50-nm MSNP core coated with cross-linked PEI, PEG, and conjugated with an antibody for targeted systemic delivery for breast cancer treatment [173]. We also showed that the same platform is effective for skin fibrosis treatment by simultaneously utilizing therapeutic siRNAs and inherent antioxidant property of MSNP [169]. Treatment of the polymer-coated MSNP reduced ROS in murine dermal fibroblast cells under TGF-β stimulated conditions. Localized intradermal injection of the material in scleroderma-like (skin fibrosis) mice resulted in reduction of pro-fibrotic gene expressions and skin thickness (Fig. 5) [169]. In brief, we established a skin fibrosis mouse model by repeated intradermal bleomycin injections, resulting in an increase in skin thickness (Fig. 5A–C). The antioxidant MSNP based nanoparticle clearly suppressed the NOX4 expression to the baseline level of mice not receiving bleomycin (Fig. 5E). The anti-fibrotic effect was enhanced by incorporating siRNA against heat shock protein 47 (siHSP47) on the nanoparticles. HSP47 is a collagen-specific molecular chaperone that binds to procollagen molecule and ensure its proper assembly before secretion into the extracellular space [178]. Knocking down HSP47 was confirmed in vivo (Fig. 5D). The HSP47 knockdown coupled with the antioxidant property of MSNP resulted in significant decrease in skin thickness (Fig. 5B–C) and pro-fibrotic markers, α-SMA (Fig. 5F) and COL I (Fig. 5G), compared to those of bleomycin-treated group, suggesting great clinical potential of this technology.
Fig. 5.

Intradermal injection of HSP47 siRNA (siHSP47) delivered with an antioxidant mesoporous silica-based nanoparticle in the bleomycin-induced skin fibrosis mouse model. (A) Dosing scheme of bleomycin induction and the siRNA-nanoparticle treatment. (B) Representative images of skin sections stained with hematoxylin and eosin (H&E), scale bar=200 µm. (C) Dermal thickness measured from skin sections in (B). Expression levels of (D) HSP47 and (E) NOX4 proteins as well as (F) α-SMA- and (G) COL I-positive area of skin sections harvested upon sacrifice. Immunofluorescence images can be found in ref [169]. Reproduced with modification (permission from Elsevier [169]).
5. Conclusions
Reactive oxygen species (ROS) have prominent roles in pathogenesis of fibrosis and cancer. Many researchers thus have studied antioxidant compounds, enzymes, and NOX inhibitors for treating such diseases, some of which have also been evaluated in clinical trials. However, the results to date have been suboptimal mainly due to their low systemic bioavailability and insufficient levels at the target sites. Antioxidant nanoparticles present a unique opportunity because they can be made larger than a kidney filtration cut-off size (~10 nm), hence prolonging circulation compared to small molecules. They can also be designed further to avoid rapid phagocytic clearance or to target specific sites and organs. Nanoparticles are also widely studied as delivery platforms for genes, oligonucleotides, drugs, and imaging agents, providing numerous opportunities in developing theranostic agents or co-delivering synergistic therapeutic agents within the same nanoparticle. For instance, we have recently reviewed different classes of nanoparticle candidates for siRNA delivery and the progress in clinical trials for systemic cancer treatment [179]. Nanoparticles are typically viewed as merely passive delivery carriers. Inherent antioxidant property of nanoparticles is thus one beneficial attribute that will set certain nanoparticle classes apart from others.
Conflict of interest disclosure
The authors declare the following competing financial interest(s): OHSU, J.M., W.N., and W.Y. have a significant financial interest in PDX Pharmaceuticals, LLC, a company that may have a commercial interest in the results of this research and technology. This potential personal and institutional conflict of interest has been reviewed and managed by OHSU.
Acknowledgements
We are grateful to Dr. Xiaolin Nan for independently reviewing this manuscript. This work was supported by National Cancer Institute of NIH under a contract HHSN261201300078C, the Prospect Creek Foundation, and OHSU’s Office of the Vice President for Research.
References
- 1.Brown D.I., Griendling K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015;116(3):531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liou G.-Y., Storz P. Reactive oxygen species in cancer. Free Radic. Res. 2010;44(5) doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Fiskum G., Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80(5):780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 4.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Fang F.C. Antimicrobial actions of reactive oxygen species. mBio. 2011;2(5) doi: 10.1128/mBio.00141-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowe L.A., Degtyareva N., Doetsch P.W., Damage-induced D.N.A. Reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008;45(8):1167–1177. doi: 10.1016/j.freeradbiomed.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.M.Schieber, N.Chandel, ROS function in redox signaling and oxidative stress, Curr. Biol., 24(10) , pp. R453–R462. [DOI] [PMC free article] [PubMed]
- 8.Waris G., Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J. Carcinog. 2006;5 doi: 10.1186/1477-3163-5-14. 14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babalola O., Mamalis A., Lev-Tov H., Jagdeo J. NADPH oxidase enzymes in skin fibrosis: molecular targets and therapeutic agents. Arch. Dermatol. Res. 2014;306(4):313–330. doi: 10.1007/s00403-013-1416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecker L., Cheng J., Thannickal V.J., Targeting NOX enzymes in pulmonary fibrosis. Cell Mol. Life Sci. 2012;69(14):2365–2371. doi: 10.1007/s00018-012-1012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Minicis S., Brenner D.A. NOX in liver fibrosis. Arch. Biochem. Biophys. 2007;462(2):266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holterman C.E., Read N.C., Kennedy C.R. Nox and renal disease. Clin. Sci. (Lond.) 2015;128(8):465–481. doi: 10.1042/CS20140361. [DOI] [PubMed] [Google Scholar]
- 13.Roy K., Wu Y., Meitzler J.L., Juhasz A., Liu H., Jiang G., Lu J., Antony S., Doroshow J.H. NADPH oxidases and cancer. Clinical Sci. (Lond.) 2015;128(12):863–875. doi: 10.1042/CS20140542. [DOI] [PubMed] [Google Scholar]
- 14.Quintavalle M., Elia L., Price J.H., Heynen-Genel S., Courtneidge S.A. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci. Signal. 2011;4(183):ra49. doi: 10.1126/scisignal.2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okon I.S., Zou M.-H., Mitochondrial ROS and cancer drug resistance: implications for therapy. Pharmacol. Res. 2015;100:170–174. doi: 10.1016/j.phrs.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008;266(1):53–59. doi: 10.1016/j.canlet.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 17.Olsson B., Johansson M., Gabrielsson J., Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988;34(1):77–82. doi: 10.1007/BF01061422. [DOI] [PubMed] [Google Scholar]
- 18.Souto E.B., Severino P., Basso R., Santana M.H. Encapsulation of antioxidants in gastrointestinal-resistant nanoparticulate carriers. Methods Mol. Biol. (Clifton, N.J.) 2013;1028:37–46. doi: 10.1007/978-1-62703-475-3_3. [DOI] [PubMed] [Google Scholar]
- 19.Sharpe E., Andreescu D., Andreescu S. American Chemical Society; Washington, DC: 2011. Artificial Nanoparticle Antioxidants, Oxidative Stress: Diagnostics, Prevention, and Therapy; pp. 235–253.http://pubs.acs.org/isbn/9780841226838 [Google Scholar]
- 20.Sandhir R., Yadav A., Sunkaria A., Singhal N. Nano-antioxidants: an emerging strategy for intervention against neurodegenerative conditions. Neurochem. Int. 2015;89:209–226. doi: 10.1016/j.neuint.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Sosa V., Moliné T., Somoza R., Paciucci R., Kondoh H., Lleonart M.E. Oxidative stress and cancer: an overview. Ageing Res. Rev. 2013;12(1):376–390. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Richter K., Konzack A., Pihlajaniemi T., Heljasvaara R., Kietzmann T. Redox-fibrosis: impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox Biol. 2015;6:344–352. doi: 10.1016/j.redox.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson J.M. Reactive oxygen species in phagocytic leukocytes. Histochem. Cell Biol. 2008;130(2):281–297. doi: 10.1007/s00418-008-0461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michiels C., Raes M., Toussaint O., Remacle J. Importance of SE-glutathione peroxidase, catalase, and CU/ZN-SOD for cell survival against oxidative stress. Free Radic. Biol. Med. 1994;17(3):235–248. doi: 10.1016/0891-5849(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 25.Wynn T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007;117(3):524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.J.Varga, D.Abraham, Systemic sclerosis: a prototypic multisystem fibrotic disorder, J. Clin. Investig., 117(3) , pp. 557–567. [DOI] [PMC free article] [PubMed]
- 27.Sebastiani G., Gkouvatsos K., Pantopoulos K. Chronic hepatitis C and liver fibrosis. World J. Gastroenterol. : WJG. 2014;20(32):11033–11053. doi: 10.3748/wjg.v20.i32.11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siegmund S.V., Brenner D.A. Molecular pathogenesis of alcohol-induced hepatic fibrosis. Alcohol. Clin. Exp. Res. 2005;29(11 Suppl):102s–109s. doi: 10.1097/01.alc.0000189275.97419.58. [DOI] [PubMed] [Google Scholar]
- 29.Pellicoro A., Ramachandran P., Iredale J.P., Fallowfield J.A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014;14(3):181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 30.P.W.Noble, C.E.Barkauskas, D.Jiang, Pulmonary fibrosis: patterns and perpetrators, J. Clin. Investig., 122(8) , pp. 2756–2762. [DOI] [PMC free article] [PubMed]
- 31.Bitterman P.B., Henke C.A. Fibroproliferative disorders. Chest. 1991;99(3 Suppl):81s–84s. doi: 10.1378/chest.99.3_supplement.81s. [DOI] [PubMed] [Google Scholar]
- 32.Li C., Kuemmerle J.F. The mechanisms that mediate the development of fibrosis in patients with Crohn's disease. Inflamm. Bowel Dis. 2014;20(7):1250–1258. doi: 10.1097/MIB.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kendall R.T., Feghali-Bostwick C.A. Fibroblasts in fibrosis: novel roles and mediators. Front. Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pohlers D., Brenmoehl J., Löffler I., Müller C.K., Leipner C., Schultze-Mosgau S., Stallmach A., Kinne R.W., Wolf G. TGF-β and fibrosis in different organs — molecular pathway imprints. Biochim. Biophys. Acta (BBA) – Mol. Basis Dis. 2009;1792(8):746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Lepparanta O., Sens C., Salmenkivi K., Kinnula V.L., Keski-Oja J., Myllarniemi M., Koli K. Regulation of TGF-beta storage and activation in the human idiopathic pulmonary fibrosis lung. Cell Tissue Res. 2012;348(3):491–503. doi: 10.1007/s00441-012-1385-9. [DOI] [PubMed] [Google Scholar]
- 36.Dooley S., ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347(1):245–256. doi: 10.1007/s00441-011-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanzler S., Lohse A.W., Keil A., Henninger J., Dienes H.P., Schirmacher P., Rose-John S., zum Buschenfelde K.H., Blessing M. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am. J. Physiol. 1999;276(4 Pt 1):G1059–G1068. doi: 10.1152/ajpgi.1999.276.4.G1059. [DOI] [PubMed] [Google Scholar]
- 38.Corbel M., Caulet-Maugendre S., Germain N., Molet S., Lagente V., Boichot E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. J. Pathol. 2001;193(4):538–545. doi: 10.1002/path.826. [DOI] [PubMed] [Google Scholar]
- 39.Lakos G., Takagawa S., Chen S.J., Ferreira A.M., Han G., Masuda K., Wang X.J., DiPietro L.A., Varga J. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am. J. Pathol. 2004;165(1):203–217. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu R.-M., Desai L.P. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan E.C., Peshavariya H.M., Liu G.S., Jiang F., Lim S.Y., Dusting G.J. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochem. Biophys. Res. Commun. 2013;430(3):918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 42.Nakano H., Nakajima A., Sakon-Komazawa S., Piao J.H., Xue X., Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13(5):730–737. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 43.Trachootham D., Lu W., Ogasawara M.A., Valle N.R.-D., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.U.S. Cancer Statistics Working Group, United States Cancer Statistics: 1999–2013 Incidence and Mortality Web-based Report, 〈www.cdc.gov/uscs〉, 2016.
- 45.B.W. Stewart, C.P. Wild, World Cancer Report 2014, 2014.
- 46.Szatrowski T.P., Nathan C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]
- 47.Panieri E., Santoro M.M. ROS homeostasis and metabolism: a dangerous liaison in cancer cells. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peiris-Pagès M., Martinez-Outschoorn Ubaldo E., Sotgia F., Lisanti Michael P. Metastasis and oxidative stress: are antioxidants a metabolic driver of progression? Cell Metab. 2015;22(6):956–958. doi: 10.1016/j.cmet.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 49.P.Schumacker, Reactive oxygen species in cancer: a dance with the devil, Cancer Cell, 27(2) , pp. 156–157. [DOI] [PubMed]
- 50.Grimsrud P.A., Xie H., Griffin T.J., Bernlohr D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008;283(32):21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B., Chen Y., Clair D.K. St. ROS and p53: versatile partnership. Free Radic. Biol. Med. 2008;44(8):1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim Y.P., Lim T.T., Chan Y.L., Song A.C., Yeo B.H., Vojtesek B., Coomber D., Rajagopal G., Lane D. The p53 knowledgebase: an integrated information resource for p53 research. Oncogene. 2007;26(11):1517–1521. doi: 10.1038/sj.onc.1209952. [DOI] [PubMed] [Google Scholar]
- 53.Rivlin N., Brosh R., Oren M., Rotter V. Mutations in the p53 Tumor Suppressor Gene: important Milestones at the Various Steps of Tumorigenesis. Genes Cancer. 2011;2(4):466–474. doi: 10.1177/1947601911408889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 55.Graeber T.G., Osmanian C., Jacks T., Housman D.E., Koch C.J., Lowe S.W., Giaccia A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 56.Semenza G.L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012;33(4):207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsieh C.-H., Shyu W.-C., Chiang C.-Y., Kuo J.-W., Shen W.-C., Liu R.-S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS One. 2011;6(9):e23945. doi: 10.1371/journal.pone.0023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu W., Shen S.-M., Zhao X.-Y., Chen G.-Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012;3(2):165–178. [PMC free article] [PubMed] [Google Scholar]
- 59.Huang L.E., Arany Z., Livingston D.M., Bunn H.F. Activation of Hypoxia-inducible Transcription Factor Depends Primarily upon Redox-sensitive Stabilization of Its α Subunit. J. Biol. Chem. 1996;271(50):32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 60.Chang T.-C., Huang C.-J., Tam K., Chen S.-F., Tan K.T., Tsai M.-S., Lin T.-N., Shyue S.-K. Stabilization of hypoxia-inducible factor-1α by prostacyclin under prolonged hypoxia via reducing reactive oxygen species level in endothelial cells. J. Biol. Chem. 2005;280(44):36567–36574. doi: 10.1074/jbc.M504280200. [DOI] [PubMed] [Google Scholar]
- 61.Srinivas V., Leshchinsky I., Sang N., King M.P., Minchenko A., Caro J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J. Biol. Chem. 2001;276(25):21995–21998. doi: 10.1074/jbc.C100177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hiraga R., Kato M., Miyagawa S., Kamata T., Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013;33(10):4431–4438. [PubMed] [Google Scholar]
- 63.Krstic J., Trivanovic D., Mojsilovic S., Santibanez J.F. Transforming growth factor-beta and oxidative stress interplay: implications in tumorigenesis and cancer progression. Oxid. Med. Cell Longev. 2015;2015:654594. doi: 10.1155/2015/654594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Massague J. TGFbeta in cancer. Cell. 2008;134(2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lebrun J.-J. The dual role of TGF in human cancer: from tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012;2012:28. doi: 10.5402/2012/381428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gorelik L., Flavell R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-[beta] signaling in T cells. Nat. Med. 2001;7(10):1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 67.Sakurai T., Kudo M. Signaling pathways governing tumor angiogenesis. Oncology. 2011;81(Suppl 1):24–29. doi: 10.1159/000333256. [DOI] [PubMed] [Google Scholar]
- 68.Heldin C.-H., Vanlandewijck M., Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012;586(14):1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 69.Nakajima S., Doi R., Toyoda E., Tsuji S., Wada M., Koizumi M., Tulachan S.S., Ito D., Kami K., Mori T., Kawaguchi Y., Fujimoto K., Hosotani R., Imamura M. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2004;10(12 Pt 1):4125–4133. doi: 10.1158/1078-0432.CCR-0578-03. [DOI] [PubMed] [Google Scholar]
- 70.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bukholm I.K., Nesland J.M., Borresen-Dale A.L. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients [see comments] J. Pathol. 2000;190(1):15–19. doi: 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 72.Kase S., Sugio K., Yamazaki K., Okamoto T., Yano T., Sugimachi K. Expression of E-cadherin and beta-catenin in human non-small cell lung cancer and the clinical significance. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 2000;6(12):4789–4796. [PubMed] [Google Scholar]
- 73.Hermiston M.L., Wong M.H., Gordon J.I. Forced expression of E-cadherin in the mouse intestinal epithelium slows cell migration and provides evidence for nonautonomous regulation of cell fate in a self-renewing system. Genes Dev. 1996;10(8):985–996. doi: 10.1101/gad.10.8.985. [DOI] [PubMed] [Google Scholar]
- 74.Hazan R.B., Phillips G.R., Qiao R.F., Norton L., Aaronson S.A. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000;148(4):779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimada K., Fujii T., Anai S., Fujimoto K., Konishi N. ROS generation via NOX4 and its utility in the cytological diagnosis of urothelial carcinoma of the urinary bladder. BMC Urol. 2011;11(1):1–12. doi: 10.1186/1471-2490-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tobar N., Guerrero J., Smith P.C., Martinez J. NOX4-dependent ROS production by stromal mammary cells modulates epithelial MCF-7 cell migration. Br. J. Cancer. 2010;103(7) doi: 10.1038/sj.bjc.6605847. 1040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seo J.M., Park S., Kim J.H. Leukotriene B4 receptor-2 promotes invasiveness and metastasis of ovarian cancer cells through signal transducer and activator of transcription 3 (STAT3)-dependent up-regulation of matrix metalloproteinase 2. J. Biol. Chem. 2012;287(17):13840–13849. doi: 10.1074/jbc.M111.317131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.G.P.Gupta, J.Massagué, Cancer Metastasis: Building a Framework, Cell, 127(4) , pp. 679–695. [DOI] [PubMed]
- 79.Nelson K.K., Melendez J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004;37(6):768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 80.Diaz B., Shani G., Pass I., Anderson D., Quintavalle M., Courtneidge S.A. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci. Signal. 2009;2(88):ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vaquero E.C., Edderkaoui M., Pandol S.J., Gukovsky I., Gukovskaya A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004;279(33):34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 82.Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc. Res. 2006;71(2):289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 83.Diaz B., Courtneidge S.A. Redox signaling at invasive microdomains in cancer cells. Free Radic. Biol. Med. 2012;52(2):247–256. doi: 10.1016/j.freeradbiomed.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Block K., Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer. 2012;12(9):627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lambeth J.D., Kawahara T., Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007;43(3):319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amara N., Goven D., Prost F., Muloway R., Crestani B., Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65(8):733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spadoni T., Svegliati Baroni S., Amico D., Albani L., Moroncini G., Avvedimento E.V., Gabrielli A. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. 2015;67(6):1611–1622. doi: 10.1002/art.39084. [DOI] [PubMed] [Google Scholar]
- 88.Aoyama T., Paik Y.H., Watanabe S., Laleu B., Gaggini F., Fioraso-Cartier L., Molango S., Heitz F., Merlot C., Szyndralewiez C., Page P., Brenner D.A. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology (Baltimore Md. ) 2012;56(6):2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jarman E.R., Khambata V.S., Cope C., Jones P., Roger J., Ye L.Y., Duggan N., Head D., Pearce A., Press N.J., Bellenie B., Sohal B., Jarai G. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014;50(1):158–169. doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 90.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C., Pennathur S., Martinez F.J., Thannickal V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15(9):1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sedeek M., Callera G., Montezano A., Gutsol A., Heitz F., Szyndralewiez C., Page P., Kennedy C.R.J., Burns K.D., Touyz R.M., Hébert R.L. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. – Renal Physiol. 2010;299(6):F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 92.Bondi C.D., Manickam N., Lee D.Y., Block K., Gorin Y., Abboud H.E., Barnes J.L. NAD(P)H oxidase mediates TGF-β1–induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. : JASN. 2010;21(1):93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ismail S., Sturrock A., Wu P., Cahill B., Norman K., Huecksteadt T., Sanders K., Kennedy T., Hoidal J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296(3):L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Michaeloudes C., Sukkar M.B., Khorasani N.M., Bhavsar P.K., Chung K.F. TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. – Lung Cell. Mol. Physiol. 2011;300(2):L295–L304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boudreau H.E., Casterline B.W., Rada B., Korzeniowska A., Leto T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012;53(7):1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shono T., Yokoyama N., Uesaka T., Kuroda J., Takeya R., Yamasaki T., Amano T., Mizoguchi M., Suzuki S.O., Niiro H., Miyamoto K., Akashi K., Iwaki T., Sumimoto H., Sasaki T. Enhanced expression of NADPH oxidase Nox4 in human gliomas and its roles in cell proliferation and survival. Int. J. Cancer. 2008;123(4):787–792. doi: 10.1002/ijc.23569. [DOI] [PubMed] [Google Scholar]
- 97.Yamaura M., Mitsushita J., Furuta S., Kiniwa Y., Ashida A., Goto Y., Shang W.H., Kubodera M., Kato M., Takata M., Saida T., Kamata T. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009;69(6):2647–2654. doi: 10.1158/0008-5472.CAN-08-3745. [DOI] [PubMed] [Google Scholar]
- 98.Graham K.A., Kulawiec M., Owens K.M., Li X., Desouki M.M., Chandra D., Singh K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010;10(3):223–231. doi: 10.4161/cbt.10.3.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang C., Lan T., Hou J., Li J., Fang R., Yang Z., Zhang M., Liu J., Liu B. NOX4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget. 2014;5(12):4392–4405. doi: 10.18632/oncotarget.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gregg J.L., Turner R.M., Chang G., Joshi D., Zhan Y., Chen L., Maranchie J.K. NADPH oxidase NOX4 supports renal tumorigenesis by promoting the expression and nuclear accumulation of HIF2α. Cancer Res. 2014;74(13):3501–3511. doi: 10.1158/0008-5472.CAN-13-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Halliwell B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007;35(Pt 5):1147–1150. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 102.Fuchs-Tarlovsky V. Role of antioxidants in cancer therapy. Nutrition. 2013;29(1):15–21. doi: 10.1016/j.nut.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 103.Packer L., Weber S.U., Rimbach G. Molecular aspects of α-tocotrienol antioxidant action and cell signalling. J. Nutr. 2001;131(2):369S–373S. doi: 10.1093/jn/131.2.369S. [DOI] [PubMed] [Google Scholar]
- 104.Thakur V., Morley S., Manor D. The hepatic alpha tocopherol transfer protein (TTP): ligand-induced protection from proteasomal degradation. Biochemistry. 2010;49(43):9339–9344. doi: 10.1021/bi100960b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fiedor J., Burda K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients. 2014;6(2):466–488. doi: 10.3390/nu6020466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rahmanto A.S., Davies M.J. Selenium-containing amino acids as direct and indirect antioxidants. IUBMB Life. 2012;64(11):863–871. doi: 10.1002/iub.1084. [DOI] [PubMed] [Google Scholar]
- 107.Blot W.J., Li J.Y., Taylor P.R., Guo W., Dawsey S., Wang G.Q., Yang C.S., Zheng S.F., Gail M., Li G.Y. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 1993;85(18):1483–1492. doi: 10.1093/jnci/85.18.1483. [DOI] [PubMed] [Google Scholar]
- 108.Klein E.A., Thompson I.M., Jr., Tangen C.M., Crowley J.J., Lucia M.S., Goodman P.J., Minasian L.M., Ford L.G., Parnes H.L., Gaziano J.M., Karp D.D., Lieber M.M., Walther P.J., Klotz L., Parsons J.K., Chin J.L., Darke A.K., Lippman S.M., Goodman G.E., Meyskens F.L., Jr., Baker L.H. Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT) JAMA. 2011;306(14):1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lippman S.M., Klein E.A., Goodman P.J., Lucia M.S., Thompson I.M., Ford L.G., Parnes H.L., Minasian L.M., Gaziano J.M., Hartline J.A., Parsons J.K., Bearden J.D., 3rd, Crawford E.D., Goodman G.E., Claudio J., Winquist E., Cook E.D., Karp D.D., Walther P., Lieber M.M., Kristal A.R., Darke A.K., Arnold K.B., Ganz P.A., Santella R.M., Albanes D., Taylor P.R., Probstfield J.L., Jagpal T.J., Crowley J.J., Meyskens F.L., Jr., Baker L.H., Coltman C.A., Jr. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de la Maza M.P., Petermann M., Bunout D., Hirsch S. Effects of long-term vitamin E supplementation in alcoholic cirrhotics. J. Am. Coll. Nutr. 1995;14(2):192–196. doi: 10.1080/07315724.1995.10718493. [DOI] [PubMed] [Google Scholar]
- 111.van Zandwijk N., Dalesio O., Pastorino U., de Vries N., van Tinteren H. EUROSCAN, a randomized trial of vitamin A and N-acetylcysteine in patients with head and neck cancer or lung cancer. For the European organization for research and treatment of cancer head and neck and lung cancer cooperative groups. J. Natl. Cancer Inst. 2000;92(12):977–986. doi: 10.1093/jnci/92.12.977. [DOI] [PubMed] [Google Scholar]
- 112.Heinonen O., Huttunen J., Albanes D. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N. Engl. J. Med. 1994;330(15):1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 113.Goodman G.E., Thornquist M.D., Balmes J., Cullen M.R., Meyskens F.L., Jr., Omenn G.S., Valanis B., Williams J.H., Jr. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J. Natl. Cancer Inst. 2004;96(23):1743–1750. doi: 10.1093/jnci/djh320. [DOI] [PubMed] [Google Scholar]
- 114.Hennekens C.H., Buring J.E., Manson J.E., Stampfer M., Rosner B., Cook N.R., Belanger C., LaMotte F., Gaziano J.M., Ridker P.M., Willett W., Peto R. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N. Engl. J. Med. 1996;334(18):1145–1149. doi: 10.1056/NEJM199605023341801. [DOI] [PubMed] [Google Scholar]
- 115.Lee I.M., Cook N.R., Manson J.E., Buring J.E., Hennekens C.H. Beta-carotene supplementation and incidence of cancer and cardiovascular disease: the Women's Health Study. J. Natl. Cancer Inst. 1999;91(24):2102–2106. doi: 10.1093/jnci/91.24.2102. [DOI] [PubMed] [Google Scholar]
- 116.M.L. Neuhouser, M.J. Barnett, A.R. Kristal, C.B. Ambrosone, I.B. King, M. Thornquist, G.G. Goodman, Dietary supplement use and prostate cancer risk in the Carotene and Retinol Efficacy Trial, Cancer epidemiology, biomarkers and prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive, Oncology 18(8), 2009, 2202-6. [DOI] [PMC free article] [PubMed]
- 117.Heinonen O., Huttunen J., Albanes D. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994;330(15):1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 118.Hajhashemi V., Vaseghi G., Pourfarzam M., Abdollahi A. Are antioxidants helpful for disease prevention? Res. Pharm. Sci. 2010;5(1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 119.Chakravarthi S., Jessop C.E., Bulleid N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006;7(3):271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meister A. Glutathione, ascorbate, and cellular protection. Cancer Res. 1994;54(7 Suppl):1969s–1975s. [PubMed] [Google Scholar]
- 121.Schmitt B., Vicenzi M., Garrel C., Denis F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: a comparative crossover study. Redox Biol. 2015;6:198–205. doi: 10.1016/j.redox.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cook G.C., Sherlock S. Results of a controlled clinical trial of glutathione in cases of hepatic cirrhosis. Gut. 1965;6(5):472–476. doi: 10.1136/gut.6.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khoshbaten M., Aliasgarzadeh A., Masnadi K., Tarzamani M.K., Farhang S., Babaei H., Kiani J., Zaare M., Najafipoor F. N-acetylcysteine improves liver function in patients with non-alcoholic fatty liver disease. Hepat. Mon. 2010;10(1):12–16. [PMC free article] [PubMed] [Google Scholar]
- 124.T.I.P.F.C.R. Network Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370(22):2093–2101. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Behr J., Demedts M., Buhl R., Costabel U., Dekhuijzen R.P., Jansen H.M., MacNee W., Thomeer M., Wallaert B., Laurent F., Nicholson A.G., Verbeken E.K., Verschakelen J., Flower C., Petruzzelli S., De Vuyst P., Bosch vdJ., Rodriguez-Becerra E., Lankhorst I., Sardina M., Boissard G. Lung function in idiopathic pulmonary fibrosis – extended analyses of the IFIGENIA trial. Respir. Res. 2009;10(1):1–9. doi: 10.1186/1465-9921-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang H., Forman H.J., Choi J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005;401:468–483. doi: 10.1016/S0076-6879(05)01028-1. [DOI] [PubMed] [Google Scholar]
- 127.Hong S.-Y., Gil H.-W., Yang J.-O., Lee E.-Y., Kim H.-K., Kim S.-H., Chung Y.-H., Lee E.-M., Hwang S.-K. Effect of high-dose intravenous N-acetylcysteine on the concentration of plasma sulfur-containing amino acids. Korean J. Intern. Med. 2005;20(3):217–223. doi: 10.3904/kjim.2005.20.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez F.J., de Andrade J.A., Anstrom K.J., King T.E., Jr., Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014;370(22):2093–2101. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang H.J., Yan T., Oberley T.D., Oberley L.W. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res. 1999;59(24):6276–6283. [PubMed] [Google Scholar]
- 130.Weydert C.J., Waugh T.A., Ritchie J.M., Iyer K.S., Smith J.L., Li L., Spitz D.R., Oberley L.W. Overexpression of manganese or copper–zinc superoxide dismutase inhibits breast cancer growth. Free Radic. Biol. Med. 2006;41(2):226–237. doi: 10.1016/j.freeradbiomed.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 131.Zhong W., Oberley L.W., Oberley T.D., St Clair D.K. Suppression of the malignant phenotype of human glioma cells by overexpression of manganese superoxide dismutase. Oncogene. 1997;14(4):481–490. doi: 10.1038/sj.onc.1200852. [DOI] [PubMed] [Google Scholar]
- 132.Church S.L., Grant J.W., Ridnour L.A., Oberley L.W., Swanson P.E., Meltzer P.S., Trent J.M. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc. Natl. Acad. Sci. 1993;90(7):3113–3117. doi: 10.1073/pnas.90.7.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hu Y., Rosen D.G., Zhou Y., Feng L., Yang G., Liu J., Huang P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005;280(47):39485–39492. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 134.Zhang Y., Zhao W., Zhang H.J., Domann F.E., Oberley L.W. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002;62(4):1205–1212. [PubMed] [Google Scholar]
- 135.Epperly M.W., Carpenter M., Agarwal A., Mitra P., Nie S., Greenberger J.S. Intraoral manganese superoxide dismutase-plasmid/liposome (MnSOD-PL) radioprotective gene therapy decreases ionizing irradiation-induced murine mucosal cell cycling and apoptosis. In vivo (Athens, Greece) 2004;18(4):401–410. [PubMed] [Google Scholar]
- 136.Niu Y., Epperly M.W., Shen H., Smith T., Wang H., Greenberger J.S. Intraesophageal MnSOD-plasmid liposome enhances engraftment and self-renewal of bone marrow derived progenitors of esophageal squamous epithelium. Gene Ther. 2007;15(5):347–356. doi: 10.1038/sj.gt.3303089. [DOI] [PubMed] [Google Scholar]
- 137.Tarhini A.A., Belani C.P., Luketich J.D., Argiris A., Ramalingam S.S., Gooding W., Pennathur A., Petro D., Kane K., Liggitt D., ChampionSmith T., Zhang X., Epperly M.W., Greenberger J.S. A phase I study of concurrent chemotherapy (paclitaxel and carboplatin) and thoracic radiotherapy with swallowed manganese superoxide dismutase plasmid liposome protection in patients with locally advanced stage III non-small-cell lung cancer. Hum. Gene Ther. 2011;22(3):336–342. doi: 10.1089/hum.2010.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Campana F., Zervoudis S., Perdereau B., Gez E., Fourquet A., Badiu C., Tsakiris G., Koulaloglou S. Topical superoxide dismutase reduces post-irradiation breast cancer fibrosis. J. Cell. Mol. Med. 2004;8(1):109–116. doi: 10.1111/j.1582-4934.2004.tb00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Apeiron, Press Release: Positive Outcome in Clinical Study to Prevent Radiation-induced Dermatitis in Breast Cancer Patients, Vienna, Austria, 2013.
- 140.Thomas R., Sharifi N. SOD mimetics: a novel class of androgen receptor inhibitors that suppresses castration-resistant growth of prostate cancer. Mol. Cancer Ther. 2012;11(1):87–97. doi: 10.1158/1535-7163.MCT-11-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shah M.H., Liu G.-S., Thompson E.W., Dusting G.J., Peshavariya H.M. Differential effects of superoxide dismutase and superoxide dismutase/catalase mimetics on human breast cancer cells. Breast Cancer Res. Treat. 2015;150(3):523–534. doi: 10.1007/s10549-015-3329-z. [DOI] [PubMed] [Google Scholar]
- 142.Yan S., Liu G., Pei C., Chen W., Li P., Wang Q., Jin X., Zhu J., Wang M., Liu X. Inhibition of NADPH oxidase protects against metastasis of human lung cancer by decreasing microRNA-21. AntiCancer Drugs. 2015;26(4):388–398. doi: 10.1097/CAD.0000000000000198. [DOI] [PubMed] [Google Scholar]
- 143.Dosoki H., Stegemann A., Taha M., Schnittler H., Luger T.A., Schroder K., Distler J.H., Kerkhoff C., Bohm M. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp. Dermatol. 2016 doi: 10.1111/exd.13180. [DOI] [PubMed] [Google Scholar]
- 144.S.S.Bhandarkar, M.Jaconi, L.E.Fried, M.Y.Bonner, B.Lefkove, B.Govindarajan, B.N.Perry, R.Parhar, J.Mackelfresh, A.Sohn, M.Stouffs, U.Knaus, G.Yancopoulos, Y.Reiss, A.V.Benest, H.G.Augustin, J.L.Arbiser, Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice, J. Clin. Investig., 119(8), pp. 2359–2365. [DOI] [PMC free article] [PubMed]
- 145.Jaquet V., Marcoux J., Forest E., Leidal K.G., McCormick S., Westermaier Y., Perozzo R., Plastre O., Fioraso-Cartier L., Diebold B., Scapozza L., Nauseef W.M., Fieschi F., Krause K.H., Bedard K. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011;164(2b):507–520. doi: 10.1111/j.1476-5381.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhu H., Liu X.-W., Cai T.-Y., Cao J., Tu C.-X., Lu W., He Q.-J., Yang B. Celastrol acts as a potent antimetastatic agent targeting β1 integrin and inhibiting cell-extracellular matrix adhesion, in Part via the p38 mitogen-activated protein kinase pathway. J. Pharmacol. Exp. Ther. 2010;334(2):489–499. doi: 10.1124/jpet.110.165654. [DOI] [PubMed] [Google Scholar]
- 147.Jiang J.X., Chen X., Serizawa N., Szyndralewiez C., Page P., Schroder K., Brandes R.P., Devaraj S., Torok N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012;53(2):289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cifuentes-Pagano E., Meijles D.N., Pagano P.J. The quest for selective nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid. Redox Signal. 2014;20(17):2741–2754. doi: 10.1089/ars.2013.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Crosas-Molist E., Bertran E., Sancho P., López-Luque J., Fernando J., Sánchez A., Fernández M., Navarro E., Fabregat I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014;69:338–347. doi: 10.1016/j.freeradbiomed.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 150.Wind S., Beuerlein K., Eucker T., Müller H., Scheurer P., Armitage M.E., Ho H., Schmidt H., Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010;161(4):885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dong K.R., Zheng S., Xiao X. Conservative management of neonatal hepatic hemangioma: a report from one institute. Pediatr. Surg. Int. 2009;25(6):493–498. doi: 10.1007/s00383-009-2373-3. [DOI] [PubMed] [Google Scholar]
- 152.Munson J.M., Fried L., Rowson S.A., Bonner M.Y., Karumbaiah L., Diaz B., Courtneidge S.A., Knaus U.G., Brat D.J., Arbiser J.L., Bellamkonda R.V. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 2012;4(127) doi: 10.1126/scitranslmed.3003016. 127ra36-127ra36. [DOI] [PubMed] [Google Scholar]
- 153.Yang W.H., Su Y.H., Hsu W.H., Wang C.C., Arbiser J.L., Yang M.H. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Oncogene. 2016;35(18):2287–2298. doi: 10.1038/onc.2015.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Garrido-Urbani S., Jemelin S., Deffert C., Carnesecchi S., Basset O., Szyndralewiez C., Heitz F., Page P., Montet X., Michalik L., Arbiser J., Rüegg C., Krause K.H., Imhof B. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One. 2011;6(2):e14665. doi: 10.1371/journal.pone.0014665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Das S., Dowding J.M., Klump K.E., McGinnis J.F., Self W., Seal S. Cerium oxide nanoparticles: applications and prospects in nanomedicine. Nanomedicine (Lond. Engl.) 2013;8(9):1483–1508. doi: 10.2217/nnm.13.133. [DOI] [PubMed] [Google Scholar]
- 156.Giri S., Karakoti A., Graham R.P., Maguire J.L., Reilly C.M., Seal S., Rattan R., Shridhar V. Nanoceria: a rare-earth nanoparticle as a novel anti-angiogenic therapeutic agent in ovarian. Cancer PLoS One. 2013;8(1):e54578. doi: 10.1371/journal.pone.0054578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hijaz M., Das S., Mert I., Gupta A., Al-Wahab Z., Tebbe C., Dar S., Chhina J., Giri S., Munkarah A., Seal S., Rattan R. Folic acid tagged nanoceria as a novel therapeutic agent in ovarian cancer. BMC Cancer. 2016;16:220. doi: 10.1186/s12885-016-2206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Siu K.S., Chen D., Zheng X., Zhang X., Johnston N., Liu Y., Yuan K., Koropatnick J., Gillies E.R., Min W.P. Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials. 2014;35(10):3435–3442. doi: 10.1016/j.biomaterials.2013.12.079. [DOI] [PubMed] [Google Scholar]
- 159.Hirst S.M., Karakoti A., Singh S., Self W., Tyler R., Seal S., Reilly C.M. Bio-distribution and in vivo antioxidant effects of cerium oxide nanoparticles in mice. Environ. Toxicol. 2013;28(2):107–118. doi: 10.1002/tox.20704. [DOI] [PubMed] [Google Scholar]
- 160.Grebowski J., Krokosz A. Fullerenes in radiobiology. Post. Biochem. 2010;56(4):456–462. [PubMed] [Google Scholar]
- 161.Andrievsky G.V., Bruskov V.I., Tykhomyrov A.A., Gudkov S.V. Peculiarities of the antioxidant and radioprotective effects of hydrated C60 fullerene nanostuctures in vitro and in vivo. Free Radic. Biol. Med. 2009;47(6):786–793. doi: 10.1016/j.freeradbiomed.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 162.Xu J.-Y., Su Y.-Y., Cheng J.-S., Li S.-X., Liu R., Li W.-X., Xu G.-T., Li Q.-N. Protective effects of fullerenol on carbon tetrachloride-induced acute hepatotoxicity and nephrotoxicity in rats. Carbon. 2010;48(5):1388–1396. [Google Scholar]
- 163.Prylutska S., Grynyuk I., Matyshevska O., Prylutskyy Y., Evstigneev M., Scharff P., Ritter U. C(60) fullerene as synergistic agent in tumor-inhibitory doxorubicin treatment. Drugs R&D. 2014;14(4):333–340. doi: 10.1007/s40268-014-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Katsumi H., Fukui K., Sato K., Maruyama S., Yamashita S., Mizumoto E., Kusamori K., Oyama M., Sano M., Sakane T., Yamamoto A. Pharmacokinetics and preventive effects of platinum nanoparticles as reactive oxygen species scavengers on hepatic ischemia/reperfusion injury in mice. Metallomics. 2014;6(5):1050–1056. doi: 10.1039/c4mt00018h. [DOI] [PubMed] [Google Scholar]
- 165.Shibuya S., Ozawa Y., Watanabe K., Izuo N., Toda T., Yokote K., Shimizu T. Palladium and platinum nanoparticles attenuate aging-like skin atrophy via antioxidant activity in mice. PLoS One. 2014;9(10):e109288. doi: 10.1371/journal.pone.0109288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yoshihisa Y., Honda A., Zhao Q.L., Makino T., Abe R., Matsui K., Shimizu H., Miyamoto Y., Kondo T., Shimizu T. Protective effects of platinum nanoparticles against UV-light-induced epidermal inflammation. Exp. Dermatol. 2010;19(11):1000–1006. doi: 10.1111/j.1600-0625.2010.01128.x. [DOI] [PubMed] [Google Scholar]
- 167.Hao N., Yang H., Li L., Li L., Tang F. The shape effect of mesoporous silica nanoparticles on intracellular reactive oxygen species in A375 cells. N. J. Chem. 2014;38(9):4258–4266. [Google Scholar]
- 168.Huang X., Zhuang J., Teng X., Li L., Chen D., Yan X., Tang F. The promotion of human malignant melanoma growth by mesoporous silica nanoparticles through decreased reactive oxygen species. Biomaterials. 2010;31(24):6142–6153. doi: 10.1016/j.biomaterials.2010.04.055. [DOI] [PubMed] [Google Scholar]
- 169.Morry J., Ngamcherdtrakul W., Gu S., Goodyear S.M., Castro D.J., Reda M.M., Sangvanich T., Yantasee W. Dermal delivery of HSP47 siRNA with NOX4-modulating mesoporous silica-based nanoparticles for treating fibrosis. Biomaterials. 2015;66:41–52. doi: 10.1016/j.biomaterials.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Slowing I.I., Trewyn B.G., Giri S., Lin V.S.Y. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv. Funct. Mater. 2007;17(8):1225–1236. [Google Scholar]
- 171.Tarn D., Ashley C.E., Xue M., Carnes E.C., Zink J.I., Brinker C.J. Mesoporous silica nanoparticle nanocarriers: biofunctionality and biocompatibility. Acc. Chem. Res. 2013;46(3):792–801. doi: 10.1021/ar3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Trewyn B.G., Nieweg J.A., Zhao Y., Lin V.S.Y. Biocompatible mesoporous silica nanoparticles with different morphologies for animal cell membrane penetration. Chem. Eng. J. 2008;137(1):23–29. [Google Scholar]
- 173.Ngamcherdtrakul W., Morry J., Gu S., Castro D.J., Goodyear S.M., Sangvanich T., Reda M.M., Lee R., Mihelic S.A., Beckman B.L., Hu Z., Gray J.W., Yantasee W. Cationic polymer modified mesoporous silica nanoparticles for targeted siRNA delivery to HER2+ breast cancer. Adv. Funct. Mater. 2015;25(18):2646–2659. doi: 10.1002/adfm.201404629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.He Q., Zhang Z., Gao F., Li Y., Shi J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: effects of particle size and PEGylation. Small. 2011;7(2):271–280. doi: 10.1002/smll.201001459. [DOI] [PubMed] [Google Scholar]
- 175.Kumar R., Roy I., Ohulchanskky T.Y., Vathy L.A., Bergey E.J., Sajjad M., Prasad P.N. In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles. ACS Nano. 2010;4(2):699–708. doi: 10.1021/nn901146y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee J.-A., Kim M.-K., Paek H.-J., Kim Y.-R., Kim M.-K., Lee J.-K., Jeong J., Choi S.-J. Tissue distribution and excretion kinetics of orally administered silica nanoparticles in rats. Int. J. Nanomed. 2014;9(Suppl. 2):251–260. doi: 10.2147/IJN.S57939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Phillips E., Penate-Medina O., Zanzonico P.B., Carvajal R.D., Mohan P., Ye Y., Humm J., Gönen M., Kalaigian H., Schöder H., Strauss H.W., Larson S.M., Wiesner U., Bradbury M.S. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci. Transl. Med. 2014;6(260):260ra149. doi: 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ishida Y., Nagata K. Hsp47 as a collagen-specific molecular chaperone. Methods Enzymol. 2011;499:167–182. doi: 10.1016/B978-0-12-386471-0.00009-2. [DOI] [PubMed] [Google Scholar]
- 179.Ngamcherdtrakul W., Castro D.J., Gu S., Morry J., Reda M., Gray J.W., Yantasee W. Current development of targeted oligonucleotide-based cancer therapies: perspective on HER2-positive breast cancer treatment. Cancer Treat. Rev. 2016;45:19–29. doi: 10.1016/j.ctrv.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]