

Editor’s Note:
Did you know that glial cells are more numerous than neurons in the brain? Scientists have found that one type of glial cell that is prevalent in the cortex—the astrocyte—communicates with its brethren, sends information to neurons, and controls blood flow to regions of brain activity. Because of all these properties, and since the cortex is believed responsible for cognition, the role of astrocytes in sleep, learning, and memory is being determined.
For much of the 20th century, our ability to decode brain signals has been limited to recording and stimulating the electrical activity of neurons. But in the 1990s, more powerful microscopes allowed neuroscientists to observe dynamic, second-to-second chemical changes in the brain. As a consequence, they observed alterations in the levels of chemical signals within certain non-neuronal cells—astrocytes—that fueled a revolution in the understanding of brain function.1
In addition to the electrically active neurons, the brain contains several non-neuronal glial cells (glia is Greek for “glue”) as depicted in Figure 1. Astrocytes are the most numerous glial cell type and comprise over half of our brain volume. Although they were once thought to be merely the material that supports brain structure, like the mortar that holds a house together, we now know that they are quite active and intimately involved in signal transmission in the nervous system, responding to and instructing neuronal activity.2 Because of work performed in the last 25 years, we know that by modulating neural excitation, inhibition, and synaptic transmission, astrocytes influence sleep, learning, and memory 3–6 and that dysfunction in these cells can lead to debilitating disorders.
Figure 1:
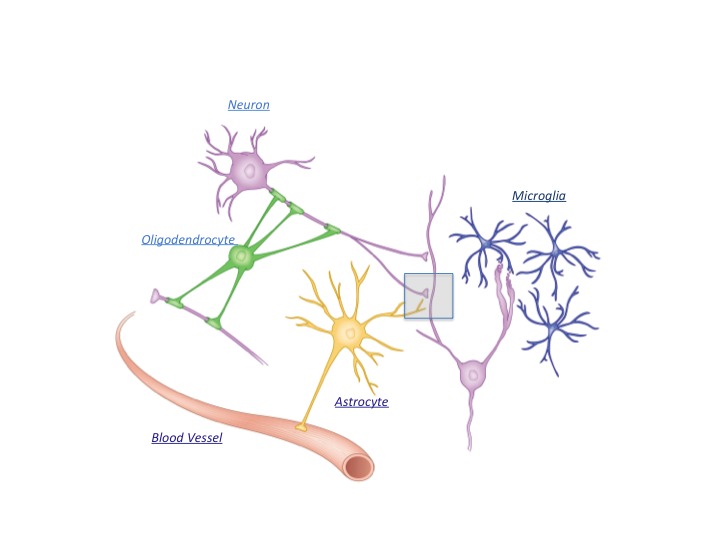
In addition to the electrically active neurons, the brain contains numerous glial cell types, including astrocytes, oligodendrocytes, and microglia. Astrocytes are the most plentiful of these glial cells and have unique physical attributes: They contract blood vessels as well as neuronal synapses at a structure called the tripartite synapse (rectangular box and see figure 2). Consequently, they play important roles in synaptic development and modulation/homeostasis as well as in the delivery of nutrients from the circulation to neurons and act as an intermediate to relay neuronal activity to the vasculature to control blood flow.
Astrocytes’ Role in Brain Function
It was soon realized that their physical linkages to both the vasculature and synapses give astrocytes the potential for significant roles in brain function, such as regulating blood vessel dilatation, and modulating synaptic function at a structure known as the tripartite synapse (Figure 2).3
Figure 2:
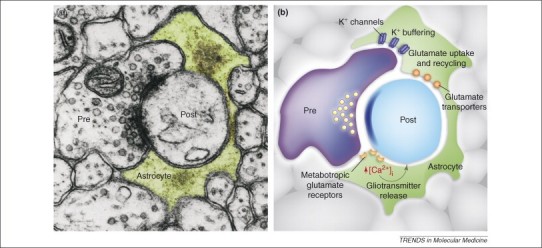
The astrocytic process is the third active element forming the tripartite synapse. (a) Electron micrograph showing a presynaptic (Pre) and postsynaptic (Post) terminal enwrapped by the astrocytic process (green) forming the tripartite synapse. (b) The close association of the astrocytic process with the presynaptic and postsynaptic terminals exerts crucial roles in clearing K+ ions that accumulate following neuronal activity, and in the uptake of the synaptic transmitter glutamate by the activity of plasma-membrane glutamate transporters. Additionally, neurotransmitter release from presynaptic terminals can activate astrocytic receptors that induce Ca2+ elevations which in turn triggers the release of gliotransmitters from these cells.7
Courtesy of Cell Press
With the development of methods that allowed intracellular imaging, several research groups observed that astrocytes exhibited calcium encoding, in which rapid shifts in calcium ion concentration transmit information from one part of the cell to another; they noted how application of the synaptic transmitter glutamate induces calcium encoding signals in astrocytes2 that stimulate such processes as the release of neurotransmitters from nerve terminals, muscle contraction, and hormone secretion. Changes in calcium ion levels in astrocytes thus gave scientists their first indication that glial cells were active participants in brain function, paving the way to new insights.
Researchers reached another major milestone when they determined that astrocytic calcium encoding signals could induce neuronal activation mediated by glutamate receptors.8 Within a few short years, we had advanced from knowing little about astrocytes to identifying bidirectional signaling between neurons and these non-neuronal cells, at least in cell culture.9–11 Subsequent studies examined freshly isolated slices of living brain tissue,12–14 but the relative imprecision of these techniques limited researchers’ ability to draw conclusions about the operation of these signaling pathways. If new insights about the role of astrocytes in neuronal networks were to emerge, new methods needed to be developed.
Modulating Nerve Terminals
The first of these came in 2005, when a cell-specific molecular genetic method enabled researchers to probe the functional role of the astrocyte within the context of neural networks.15 In this study, a promoter (the DNA region that initiates the synthesis of gene products) that is turned on selectively within astrocytes was used to provoke the expression of proteins that inhibit the release of chemical transmitters from astrocytes. With this approach, my laboratory was able to show that the astrocyte continuously modulates nerve terminals through a chemical signal called adenosine.
Adenosine is known to promote drowsiness (we drink beverages containing caffeine, an antagonist of adenosine receptors, to feel more awake). Hence we used a mouse model, genetically modified to inhibit the release of adenosine from astrocytes to determine whether sleep homeostasis (the balance between sleep and wakefulness) was consequently perturbed. As expected, the drive to fall asleep was impaired when we thus prevented adenosine-mediated gliotransmission,16 an effect that is replicated by pharmacological inhibition of adenosine receptors in normal mice. Over a 15-year time span, we were able to make the transition from identifying bidirectional signaling between astrocytes and neurons to demonstrating roles for these glia cells in the modulation of synaptic activity and a specific behavior. In support of this exciting observation, others have replicated the contribution of astrocytes to sleep by stimulating the astrocyte by optogenetics, rather than inhibiting it. When activated by light, astrocytes provoked slow oscillations in neurons,17 a result that is compatible with the consequences of genetic inhibition of gliotransmission cited above. Together, these studies have led to the predominant view that astrocytes are generally integrated within neural circuits, where they modify brain function and influence behavior.
Synapse Development
One of the many mysteries of the brain concerns the cues that guide and regulate its development. Of particular interest is this question: which signals determine whether two neurons are able to functionally connect with one another at a synapse? Some early clues to the answer came from cell-culture studies where scientists plate young neurons into a plastic culture dish and observe whether synaptic connections are able to form. It was found that in the absence of astrocytes, synapse formation was hindered, while in mixed cultures containing both neurons and astrocytes, exuberant synapses formed between neurons. This led to the concept that astrocytes regulate synapse formation. Elegant studies have since shown that proteins called thrombospondins (TSPs) are released from astrocytes and regulate specific aspects of synapse development. The addition of pure TSPs to neuronal cultures obviates the need for other astrocyte factors, and depletion of TSP from astrocytes impairs the ability of this medium to stimulate synapse formation.18,19
Interestingly, gabapentin, a therapeutic agent used in the treatment of neuropathic pain and epilepsy, inhibits the receptor for TSPs, suggesting that it may exert its action by preventing new synapse formation in these pathological conditions. In support of this idea is the observation that injury to the cortex, which normally leads to the development of hyperexcitability, is attenuated by treatment with gabapentin.20
Astrocytic Metabolic Support
The primary source of energy in the brain is glucose. Astrocytes project a process (called an end foot) that wraps around the blood vessels in the brain to take up glucose from the circulation. But neurons do not make this kind of physical contacts with the vasculature; how then do they obtain energy? One prominent theory holds that astrocytes metabolize the glucose they take up from the circulation to lactate, then transfer it to neighboring neurons that use it as an energy source. This lactate shuttle hypothesis 21 has generated significant debate, but several intriguing observations support it. In particular, astrocytes and neurons express transporters (proteins in a cell membrane that bind to a molecule and transfer it across the membrane) that promote the transfer of lactate from astrocyte to neuron. Additionally, in glucose-depleted conditions, lactate has been shown capable of supporting the energy demands of the neuron.
Cerebral Blood Flow Control
As discussed above, astrocytes, not neurons, predominantly contact the vasculature. Yet neuronal electrical activity induces changes in blood flow. How is this achieved in the absence of direct interactions with the vasculature? Since astrocytes make physical contact with both neurons and the vasculature, they are physically positioned to act as a signal conduit from the first to the second. Since neuronal activity can induce calcium changes in astrocytes, scientists asked whether direct stimulations of calcium changes in astrocytes would lead to changes in blood vessels. To answer this question,22 initial studies were performed in isolated slices of brain tissue and an optical technique was used to elevate calcium. A chemically synthesized calcium “cage” was introduced into the astrocyte. When light was turned on, the cage opened, liberating calcium in the astrocyte to stimulate calcium dependent biological events. When this optical technique was used to stimulate calcium dependent events in the astrocyte, the adjacent blood vessels dilated.
Mechanistically, it has been demonstrated that this neuron-astrocyte-vasculature interaction derives from calcium-dependent stimulation of an enzyme called phospholipase A2 (PLA2), which leads to the production of arachidonic acid and its downstream signaling cascades. After initial pharmacological studies supported this idea, when the gene for PLA2 was deleted, astrocytic calcium signals no longer evoked changes in vessel diameter.
Alexander’s Disease
Alexander’s disease is often referred to as a disease of the astrocyte because it is known to follow from mutations in an astrocytic protein, glial fibrillary acidic protein (GFAP).23 Alexander’s disease is lethal, generally during childhood. Mutations in the GFAP gene are correlated with more than 90 percent of the cases of this disorder, and nearly a third of these cases result from mutations at one of two amino acid sites within the GFAP protein.
A consequence of these mutations is that GFAP, which normally forms fine filaments, instead causes the formation of thick bundles of what are called Rosenthal fibers. The current idea is that mutations in the GFAP protein endow it with new functions such as, according to one prevailing view, binding or sequestering proteins that normally protect the cell. Once sequestered, these proteins cease to be protective, and cellular damage and toxicity result. With these insights, researchers are trying to determine whether they can reduce the amount of the GFAP protein and thus reduce the sequestration of the protective proteins, or alternatively increase the amount of these proteins so they can regain their normal role.
Glutamate Homeostasis and Neuroprotection
A critical function of the astrocyte is helping control the level of extracellular neurotransmitters. The excitatory transmitter glutamate plays pivotal roles in signal transmission in the brain, but its accumulation in the extracellular space can lead to neurodegeneration/toxicity mediated by the persistent activity of the NMDA receptor. These glutamate receptors allow calcium influx into neurons, an important stimulus for learning and memory. But their persistent activity leads to continuous accumulation of calcium, with neurotoxic consequences. Indeed, widespread neuronal death following stroke is, in part, due to this process during and following injury.
The clearance of glutamate from the synapse was one of the first prominent functional roles identified for astrocytes: they abundantly express glutamate transporters, particularly GLT-1, which removes the neurotransmitter from the extracellular space. Deleting this transporter from astrocytes leads to epilepsy and reduced lifespan, reflecting the importance of astrocyte-mediated glutamate control.24
Glutamine and Epilepsy
Once glutamate is taken up into astrocytes by glutamate transporters, it is converted to glutamine by an enzyme called glutamine synthetase (GS). Glutamine is an important precursor of both glutamate and the inhibitory neurotransmitter GABA. In certain types of epilepsy, the amount of GS in the brain declines significantly.25 The consequences of GS loss for brain function were elucidated when, through a technical accident, researchers found that the amount of GS was reduced in normal mice when a specific virus infected astrocytes. This accidental observation provided an opportunity to evaluate the consequence of reduced astrocytic GS on brain function.
Through recordings of synaptic connections between neurons, researchers found an impairment of GABA-mediated inhibitory synaptic transmission when astrocytes did not synthesize GS, which disrupted the balance between excitation and inhibition. [An analogy might be cars driving in a city with brake pedals (inhibition) disabled and gas pedals (excitation) intact.] The brain circuits involved became hyperexcitable and discharged epileptic-like electrical events. Importantly, the addition of glutamine, which is normally synthesized by GS, restored the normal level of inhibition and effectively put a brake on the mass excitation and epileptic-like discharges.26 Thus it appears possible that a primary deficit in astrocytic GS leads to a loss of inhibition that results in epileptic seizures.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive disorder in which the motor neurons that cause muscle contractions degenerate, ultimately leading to paralysis and an inability to breathe. The disorder is also sometimes called Lou Gehrig’s disease, after the baseball player Lou Gehrig, who suffered from ALS. There are two forms of the disease—familial (5–10 percent of cases) and sporadic (90–95 percent). The familial form is caused by mutations in the genome and thus can be inherited from affected individuals. In fact, the mutations in the genetic code for certain proteins causing ALS have been identified, allowing researchers to replicate miscoded genes in mice to further our understanding of the disorder.
For many years, the primary cause of ALS was thought to be within the neuron that degenerates. Cell-culture studies have allowed this idea to be tested since it is possible to introduce neurons and their glial partners, alone or in combination, into the culture dish. When neurons from normal mice were plated into cultures together with astrocytes from mice that carried a familial mutation, the neuron was found to degenerate. Thus, the genetic mutation causes changes to the astrocyte that promote the demise of the neuron, potentially by modulating inflammatory pathways. Further work will allow us to identify potential ways to treat such disorders of the nervous system.
APOE4 and Alzheimer’s Disease (AD)
There are two general forms of Alzheimer’s disease—early onset (1–5 percent of cases) and late onset (95–99 percent of cases). Apolipoprotein 4 (APOE4) is the largest known genetic risk factor for late onset AD. Apolipoproteins are expressed by astrocytes and can come in three different forms, in which slight changes in the genetic code have led to small changes in amino acid sequences. These different apolipoproteins are called APOE2, 3, or 4. APOE2 is protective in regard to AD; APOE3 is a mild risk factor for late onset AD, and APOE4 increases AD risk substantially. For example, individuals carrying two copies of APOE4 are 15 times more likely to get late onset AD than APOE3 carriers; subjects with two copies of the APOE2 gene are 40 percent less likely to develop the disease than APOE3 carriers. About 40 percent of patients with late onset AD carry the APOE4 gene.
Mice have been developed in which human APOE genes replaced the mouse APOE genes. This has allowed investigation into the mechanism by which these mutations change the risk of developing AD. One study showed that APOE2 increases while APOE4 reduces the ability of the astrocyte to clear cellular debris, leading the authors to speculate that reduction in this function underlies APOE4-linked AD risk, via an increase in synaptic vulnerability and neurodegeneration.27 Because the APOE4 allelic variant is the greatest known risk factor for late onset AD and, because astrocytes express the product of this gene, there is intense scientific focus on how astrocytes and APOE4 contribute to this form of dementia.
Over the past 25 years, our understanding of the biology of astrocytes has increased tremendously. Where we initially thought astrocytes to be simple static cells that support neurons, we now know that they listen to and detect the activity of neurons, signal the vasculature to increase local blood flow to provide new nutrients as needed, and act as a conduit to provide energy-rich chemicals to active neurons. We’ve learned that they also provide feedback signals and regulate processes such as sleep. We are realizing that defects in astrocytes may be the primary signal that causes neuronal degeneration in such diverse disorders as ALS, epilepsy, and Alzheimer’s disease. In the near future, it would not be surprising to see the development of new drugs for neurological and psychiatric disorders that target known astrocytic pathways, to protect neurons from neurodegeneration and, ultimately, increase quality of life.
Footnotes
Philip G. Haydon, Ph.D., is the Annetta and Gustav Grisard Professor and chair of the neuroscience department at Tufts University School of Medicine. He received his Ph.D. from the University of Leeds, UK. While a faculty member at Iowa State University in 1994, he performed a landmark study by demonstrating that astrocytes release chemical transmitters. In 2001 he joined the faculty of the University of Pennsylvania School of Medicine and, in 2008, moved to Tufts. For 25 years his research has addressed the roles played by glial cells. His recent focus is on the use of glial targets as therapeutic interventions for brain disorders. He has received the Alfred P. Sloan Scholarship, the McKnight Innovator Award, and the Jacob Javits Award from the National Institute of Neurological Disorders and Stroke.
References
- 1.Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. The Journal of biological chemistry. 1989;264:8171–8178. [PubMed] [Google Scholar]
- 2.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 3.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends in neurosciences. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 4.Haydon PG. GLIA: listening and talking to the synapse. Nature reviews Neuroscience. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 5.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annual review of physiology. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Araque A, et al. Gliotransmitters travel in time and space. Neuron. 2014;81:728–739. doi: 10.1016/j.neuron.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends in molecular medicine. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Parpura V, et al. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 9.Araque A, Li N, Doyle RT, Haydon PG. SNARE protein-dependent glutamate release from astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:666–673. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. The European journal of neuroscience. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- 11.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fellin T, et al. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Bezzi P, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 15.Pascual O, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- 16.Halassa MM, et al. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poskanzer KE, Yuste R. Astrocytes regulate cortical state switching in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1520759113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanco-Suarez E, Caldwell AL, Allen NJ. Role of astrocyte-synapse interactions in CNS disorders. The Journal of physiology. 2016 doi: 10.1113/JP270988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung WS, Allen NJ, Eroglu C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harbor perspectives in biology. 2015;7:a020370. doi: 10.1101/cshperspect.a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andresen L, et al. Gabapentin attenuates hyperexcitability in the freeze-lesion model of developmental cortical malformation. Neurobiology of disease. 2014;71:305–316. doi: 10.1016/j.nbd.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:1152–1166. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harbor perspectives in biology. 2015;7 doi: 10.1101/cshperspect.a020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messing A, LaPash Daniels CM, Hagemann TL. Strategies for treatment in Alexander disease. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:507–515. doi: 10.1016/j.nurt.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rimmele TS, Rosenberg PA. GLT-1: The elusive presynaptic glutamate transporter. Neurochemistry international. 2016;98:19–28. doi: 10.1016/j.neuint.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eid T, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- 26.Ortinski PI, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nature neuroscience. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung WS, et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:10186–10191. doi: 10.1073/pnas.1609896113. [DOI] [PMC free article] [PubMed] [Google Scholar]