

Abstract
Coactivator-associated arginine methyltransferase 1 (CARM1) is a type I protein arginine methyltransferase (PRMT) that catalyzes the conversion of arginine into monomethylarginine (MMA) and further into asymmetric dimethylarginine (ADMA). CARM1 methylates histone 3 arginines 17 and 26, as well as numerous non-histone proteins including CBP/p300, SRC-3, NCOA2, PABP1, and SAP49, while also functioning as a coactivator for various proteins that have been linked to cancer such as p53, NF-κβ, β-catenin, E2F1 and steroid hormone receptor ERα. As a result, CARM1 is involved in transcriptional activation, cellular differentiation, cell cycle progression, RNA splicing and DNA damage response. It has been associated with several human cancers including breast, colon, prostate and lung cancers and thus, is a potential oncological target. Herein, we present the design and synthesis of a series of CARM1 inhibitors. Based on a fragment hit, we discovered compound 9 as a potent inhibitor that displayed selectivity for CARM1 over other PRMTs.
Graphical Abstract
Structure-activity relationship studies, starting from a fragment hit, resulted in discovery of the compound 9, an inhibitor of CARM1 with high potency and selectivity.
Introduction
Coactivator-associated arginine methyltransferase 1 (CARM1, also known as PRMT4) is a type I protein arginine methyltransferase (PRMT) along with PRMT1, PRMT3, PRMT6 and PRMT8, which are responsible for converting arginine into monomethylarginine (MMA) and further into asymmetric dimethylarginine (ADMA).1 CARM1 methylates histone 3 arginines 17 and 26,2, 3 as well as numerous non-histone proteins including CBP/p300, SRC-3, NCOA2, PABP1, SmB, HuR, HuD, CA150, SAP49, and U1C.1 Therefore, it plays a role in cellular processes such as transcriptional activation,4 RNA splicing,4, 5 cellular differentiation,6 cell cycle progression7 and DNA repair.8 It has been shown that the enzymatic activity of CARM1 is essential for most of its in vivo functions.9 CARM1 functions as a coactivator for various proteins that have been linked to cancer including p53, NF-κβ, β-catenin, E2F1 and steroid hormone receptor ERα.1, 10–12 Overexpression of CARM1 in several human cancers including breast,7 colon,13 prostate14 and lung15 cancers has been reported. Thus, CARM1 is a potentially attractive therapeutical target for cancer and as a result there have been numerous studies directed toward the discovery of small molecule CARM1 inhibitors.16, 17
Several high throughput screening (HTS) campaigns resulted in the identification of pyrrazole-amide as well as benzo[d]imidazole CARM1 inhibitors.18, 19 These initial reports were followed by structure-activity relationship (SAR) studies, which led to the discovery of small molecule inhibitors 1 and 2 with IC50 values around 30 nM. These inhibitors displayed selectivity for CARM1 over PRMT1 and PRMT3 while the selectivity over other PRMTs was not reported (Figure 1).20–23 We have recently reported a type I PRMT chemical probe, MS023 (3), which is potent against all type I PRMTs, but inactive against type II and type III PRMTs as well as other methyltransferases.24 A series of adenosine-based CARM1 inhibitors with high potency and selectivity over PRMT1 and PRMT6 was also published.25 Herein we report the discovery of selective CARM1 inhibitors and describe their design, synthesis and biological evaluation.
Figure 1.

Structures of known CARM1 inhibitors 1, 2 and the type I PRMT chemical probe 3.
Results and Discussion
The alkyl-diamino tail, shown to be bound in the substrate-binding site of CARM1,21 is the shared feature of inhibitors 1-3 (Figure 1) as well as of a recently published PRMT6 inhibitor.26 In a recent study, a commercially available, diverse fragment library of compounds mimicking this alkyl-diamino tail were tested against PRMT6 and compound 4 was reported as a PRMT6 inhibitor with IC50 of 300 ± 40 nM (Figure 2).27 Compound 4 was 3- and 7-fold less potent for CARM1 (IC50 = 1,000 ± 40 nM) and PRMT8 (IC50 = 2100 ± 200 nM), respectively. It was not very potent against PRMT1 and PRMT3 (>40-fold less potent) and did not inhibit other methyltransferases. We sought to discover potent CARM1 selective inhibitors by using the fragment hit 4 as a starting point. Our initial SAR studies resulted in compounds 5 and 6 as promising CARM1 inhibitors (Figure 2). Compound 5 shares the (piperidinyl)ethan-1-amine core of 4, but it is connected to the phenyl group via a methylene-amine (-CH2NH-) linker instead of methylene (-CH2-) linker. It was found to be 10-fold more potent for CARM1 (IC50 = 471 ± 36 nM) over PRMT6 (IC50 = 4,564 ± 1210 nM), exhibiting some selectivity (Figure 2). In addition, it had no appreciable inhibitory activity against PRMT1, PRMT3 and PRMT8 as well as PRMT5 and PRMT7 (IC50 > 50,000 nM for all). On the other hand, compound 6 contains an (azetidinyl)ethan-1-amine core and –CH2O- connection to the aryl ring (Figure 2). This inhibitor displayed good potency for CARM1 (IC50 = 144 ± 37 nM) and was less than 10-fold selective over PRMT6 and PRMT8 (IC50 = 1,079 ± 75 and 1,200 ± 70 nM, respectively) while it was inactive against other PRMTs.
Figure 2.

Structures of the fragment hit 4, and initial hits 5 and 6.
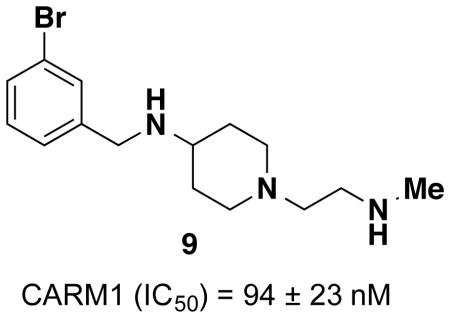
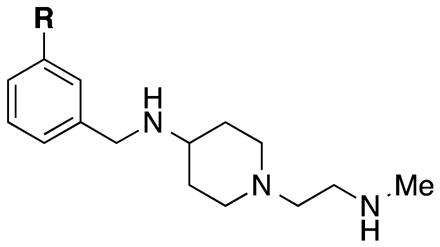
Since inserting a nitrogen between the piperidine and benzyl groups of compound 4 (compound 5, Figure 2) resulted in some selectivity for CARM1 over PRMT6, we further explored this scaffold and synthesized derivatives of 5 by adding substituents on the phenyl ring (Table 1). We mainly investigated meta-substitution based on the encouraging potency of compound 6 for CARM1. Among the meta-fluoro (7), meta-chloro (8) and meta-bromo (9) derivatives, 9 displayed the best potency for CARM1 with IC50 of 94 ± 23 nM as well as selectivity over PRMT6 (around 20–fold, IC50 = 2,160 ± 68 nM). The electron-withdrawing trifluoromethyl (10), or electron-donating triflourometoxy (11) substitution did not result in a more potent or selective inhibitor than 9. The phenyl-substituted derivative (12) reversed the selectivity in favour of PRMT6 (IC50 = 120 ± 13 nM) over CARM1 (IC50 = 330 ± 43 nM).
Table 1.
IC50 values of compounds 5, 7-12.
| |||
|---|---|---|---|
| Cmpd | R | CARM1 IC50 (nM) |
PRMT6 IC50 (nM) |
| 5 | H | 471 ± 36 | 4,560 ± 1210 |
| 7 | F | 223 ± 57 | 1,890 ± 456 |
| 8 | Cl | 157 ± 54 | 2,920 ± 303 |
| 9 | Br | 94 ± 23 | 2,160 ± 68 |
| 10 | CF3 | 302 ± 41 | 2,980 ± 298 |
| 11 | OCF3 | 342 ± 48 | 4,730 ± 174 |
| 12 | Ph | 330 ± 43 | 120 ± 13 |
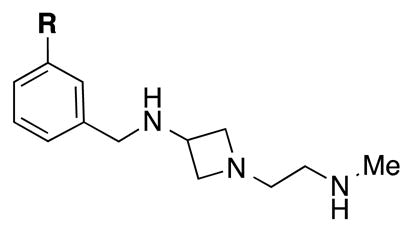
We then focused our attention on the (azetidinyl)ethan-1-amine sca1fold represented by compound 6 (Figure 2). Since this scaffold with an oxygen linker did not show good selectivity over either PRMT6 or PRMT8, we utilized the methylene-amine (-CH2NH-) linker as in the piperidinyl series and synthesized substituted phenyl derivatives 13-18 (Table 2). While there was an overall increase in potency of these inhibitors as compared to compounds 7-12 for CARM1, the potency for PRMT6 was even greater in comparison and thus, the selectivity over PRMT6 suffered significantly. For example the meta-chloro (14) and meta-bromo (15) derivatives were potent for CARM1 with IC50 of 50 ± 14 and 67 ± 9 nM, respectively. However, 14 and 15 displayed high potency for PRMT6 with IC50 of 133 ± 8 and 249 ± 22 nM respectively, resulting in only 3 to 4-fold selectivity for CARM1 over PRMT6.
Table 2.
IC50 values of compounds 13-18.
| |||
|---|---|---|---|
| Cmpd | R | CARM1 IC50 (nM) |
PRMT6 IC50 (nM) |
| 13 | F | 130 ± 29 | 101 ± 7 |
| 14 | Cl | 50 ± 14 | 133 ± 8 |
| 15 | Br | 67 ± 9 | 249 ± 22 |
| 16 | CF3 | 94 ± 17 | 469 ± 64 |
| 17 | OCF3 | 90 ± 25 | 434 ±27 |
| 18 | Ph | 69 ± 10 | 67 ± 13 |
The synthetic route for the preparation of compounds 7-12 is shown in Scheme 1. The synthesis started with a nucleophilic displacement reaction between 4-amino-1-Boc-piperidine (19) and meta-substituted benzyl bromides (20). The resulting diamines were then treated with methanolic HCl to remove the tert-butoxy carbamate group. The reductive amination of amines 21 with N-Boc-(methylamino)acetaldehyde (22), followed by a deprotection reaction, afforded the desired compounds (5 and 7-12). A similar synthetic route was employed for the synthesis of compounds 13-18 starting with 3-amino-1-Boc-azetidine (see supporting information for detailed procedures).
Scheme 1.

General scheme for the synthesis of compounds 5, 7-12. Conditions: a. Et3N, DCM b. Methanolic HCl (3N) c. Et3N, NaBH(OAC)3 d. Methanolic HCl (3N).
Among all the derivatives synthesized, compound 9 emerged as the best inhibitor with good potency and around 20-fold selectivity toward CARM1 over PRMT6. In addition, we have tested Inhibitor 9 against other PRMTs in biochemical assays and found that it showed excellent selectivity for CARM1 over other type I PRMTs, PRMT1, PRMT3 (IC50 > 50,000 nM) and PRMT8 (IC50 = 9,200 ± 500 nM) as well as the type II and type III PRMTs, PRMT 5 and 7 (IC50 > 50,000 nM).
To assess the mechanism of action (MOA) of inhibitor 9, we evaluated the effect of the peptide substrate and cofactor S-adenosyl-L-methionine (SAM) concentrations on IC50 values of 9 against CARM1. As shown in Figure 3, increasing the peptide substrate or SAM concentrations had no effect on the IC50 values of 9 against CARM1, suggesting that this inhibitor is noncompetitive with both the cofactor SAM and peptide substrate. It has been previously reported that active site binding inhibitors can display noncompetitive behavior in MOA studies.28, 29 We and others have observed this phenomenon with PRMT inhibitors.24, 26 For example, MOA studies of MS023 (3) suggested that the inhibitor was noncompetitive with both SAM and peptide substrate while the X-ray crystal structure of MS023 in complex with PRMT6 clearly showed that it occupies the substrate binding pocket. Therefore, based on the literature precedents, it is very likely that inhibitor 9 interacts with the substrate binding site of CARM1 to assert its inhibitory effect.
Figure 3.

MOA study of compound 9 against CARM1 with changing concentrations of the peptide substrate (top) and the cofactor SAM (bottom).
Conclusions
Starting from a reported fragment hit (4), a PRMT6 inhibitor, we discovered compound 9, a potent and selective inhibitor of CARM1, through a SAR study. In biochemical assays, 9 displayed high potency (IC50 = 94 ± 23 nM) for CARM1 and was around 20-fold selective for CARM1 over PRMT6 and highly selective over PRMT1, PRMT3, PRMT8 as well as PRMT5 and PRMT7. We believe that inhibitor 9 could be a useful tool for studying the role of CARM1 in health and disease. We anticipate that the work described here will facilitate further development of CARM1 selective inhibitors and chemical tools.
Supplementary Material
Acknowledgments
The research described here was supported by the grant R01GM103893 (to J. J.) from the U.S. National Institutes of Health. The Structural Genomics Consortium (SGC) is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Sao Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust.
Footnotes
The authors declare no competing interests.
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x.
Notes and references
- 1.Bedford MT, Clarke SG. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW. Biochemistry. 2001;40:5747–5756. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- 3.Jacques SL, Aquino KP, Gureasko J, Boriack-Sjodin PA, Porter Scott M, Copeland RA, Riera TV. Biochemistry. 2016;55:1635–1644. doi: 10.1021/acs.biochem.5b01071. [DOI] [PubMed] [Google Scholar]
- 4.Cheng D, Cote J, Shaaban S, Bedford MT. Mol Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 5.Ohkura N, Takahashi M, Yaguchi H, Nagamura Y, Tsukada T. J Biol Chem. 2005;280:28927–28935. doi: 10.1074/jbc.M502173200. [DOI] [PubMed] [Google Scholar]
- 6.Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GE. J Biol Chem. 2002;277:4324–4333. doi: 10.1074/jbc.M109835200. [DOI] [PubMed] [Google Scholar]
- 7.El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng DH, Theillet C, Vandel L, Bedford MT, Sardet C. P Natl Acad Sci USA. 2006;103:13351–13356. doi: 10.1073/pnas.0605692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee YH, Stallcup MR. Cell cycle. 2011;10:1343–1344. doi: 10.4161/cc.10.9.15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D, Lee J, Cheng D, Li J, Carter C, Richie E, Bedford MT. J Biol Chem. 2010;285:1147–1152. doi: 10.1074/jbc.M109.035865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh SS, Li H, Lee YH, Widelitz RB, Chuong CM, Stallcup MR. J Biol Chem. 2002;277:26031–26035. doi: 10.1074/jbc.M110865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, Hottiger MO. EMBO J. 2005;24:85–96. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Dhaheri M, Wu J, Skliris GP, Li J, Higashimato K, Wang Y, White KP, Lambert P, Zhu Y, Murphy L, Xu W. Cancer Res. 2011;71:2118–2128. doi: 10.1158/0008-5472.CAN-10-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YR, Lee BK, Park RY, Nguyen NT, Bae JA, Kwon DD, Jung C. BMC Cancer. 2010;10:197. doi: 10.1186/1471-2407-10-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong H, Kao C, Jeng MH, Eble JN, Koch MO, Gardner TA, Zhang S, Li L, Pan CX, Hu Z, MacLennan GT, Cheng L. Cancer. 2004;101:83–89. doi: 10.1002/cncr.20327. [DOI] [PubMed] [Google Scholar]
- 15.Elakoum R, Gauchotte G, Oussalah A, Wissler MP, Clement-Duchene C, Vignaud JM, Gueant JL, Namour F. Biochimie. 2014;97:210–218. doi: 10.1016/j.biochi.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 16.Kaniskan HU, Konze KD, Jin J. J Med Chem. 2015;58:1596–1629. doi: 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaniskan HU, Jin J. ACS Chem Biol. 2015;10:40–50. doi: 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purandare AV, Chen Z, Huynh T, Pang S, Geng J, Vaccaro W, Poss MA, Oconnell J, Nowak K, Jayaraman L. Bioorganic & Medicinal Chemistry Letters. 2008;18:4438–4441. doi: 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 19.Wan HH, Huynh T, Pang SH, Geng JP, Vaccaro W, Poss MA, Trainor GL, Lorenzi MV, Gottardis M, Jayaraman L, Purandare AV. Bioorganic & Medicinal Chemistry Letters. 2009;19:5063–5066. doi: 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- 20.Huynh T, Chen Z, Pang SH, Geng JP, Bandiera T, Bindi S, Vianello P, Roletto F, Thieffine S, Galvani A, Vaccaro W, Poss MA, Trainor GL, Lorenzi MV, Gottardis M, Jayaraman L, Purandare AV. Bioorganic & Medicinal Chemistry Letters. 2009;19:2924–2927. doi: 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- 21.Sack JS, Thieffine S, Bandiera T, Fasolini M, Duke GJ, Jayaraman L, Kish KF, Klei HE, Purandare AV, Rosettani P, Troiani S, Xie D, Bertrand JA. Biochem J. 2011;436:331–339. doi: 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- 22.Allan M, Manku S, Therrien E, Nguyen N, Styhler S, Robert MF, Goulet AC, Petschner AJ, Rahil G, MacLeod AR, Deziel R, Besterman JM, Nguyen H, Wahhab A. Bioorganic & Medicinal Chemistry Letters. 2009;19:1218–1223. doi: 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- 23.Therrien E, Larouche G, Manku S, Allan M, Nguyen N, Styhler S, Robert MF, Goulet AC, Besterman JM, Nguyen H, Wahhab A. Bioorganic & Medicinal Chemistry Letters. 2009;19:6725–6732. doi: 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- 24.Eram MS, Shen Y, Szewczyk MM, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HU, Speed BA, Dela Sena C, Dong A, Zeng H, Schapira M, Brown PJ, Arrowsmith CH, Barsyte-Lovejoy D, Liu J, Vedadi M, Jin J. ACS Chem Biol. 2016;11:772–781. doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Haren M, van Ufford LQ, Moret EE, Martin NI. Organic & biomolecular chemistry. 2015;13:549–560. doi: 10.1039/c4ob01734j. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell LH, Drew AE, Ribich SA, Rioux N, Swinger KK, Jacques SL, Lingaraj T, Boriack-Sjodin PA, Waters NJ, Wigle TJ, Moradei O, Jin L, Riera T, Porter-Scott M, Moyer MP, Smith JJ, Chesworth R, Copeland RA. ACS Med Chem Lett. 2015;6:655–659. doi: 10.1021/acsmedchemlett.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira de Freitas R, Eram MS, Szewczyk MM, Steuber H, Smil D, Wu H, Li F, Senisterra G, Dong A, Brown PJ, Hitchcock M, Moosmayer D, Stegmann CM, Egner U, Arrowsmith C, Barsyte-Lovejoy D, Vedadi M, Schapira M. J Med Chem. 2016;59:1176–1183. doi: 10.1021/acs.jmedchem.5b01772. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell LH, Boriack-Sjodin PA, Smith S, Thomenius M, Rioux N, Munchhof M, Mills JE, Klaus C, Totman J, Riera TV, Raimondi A, Jacques SL, West K, Foley M, Waters NJ, Kuntz KW, Wigle TJ, Scott MP, Copeland RA, Smith JJ, Chesworth R. ACS Med Chem Lett. 2016;7:134–138. doi: 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blat Y. Chem Biol Drug Des. 2010;75:535–540. doi: 10.1111/j.1747-0285.2010.00972.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.