

Abstract
Synovial sarcoma (SS) is an aggressive soft tissue sarcoma with a poor prognosis and, thus, novel therapeutic strategies for SS are urgently required. In the present study, we investigated the functional and therapeutic relevance of hepatocyte growth factor (HGF)/c‐MET signaling in SS. Both HGF and c‐MET were highly expressed in Yamato‐SS cells, resulting in activation of c‐MET and its downstream AKT and extracellular signal‐regulated kinase signaling pathways, whereas c‐MET was expressed but not activated in SYO‐1 or HS‐SY‐II cells. c‐MET‐activated Yamato‐SS cells showed higher anchorage‐independent growth ability and less sensitivity to chemotherapeutic agents than did c‐MET‐inactivated SYO‐1 or HS‐SY‐II cells. INC280, a selective c‐MET inhibitor, inhibited growth of Yamato‐SS cells both in vitro and in vivo but not that of SYO‐1 or HS‐SY‐II cells. INC280 induced cell cycle arrest and apoptosis, and blocked phosphorylation of c‐MET and its downstream effectors in Yamato‐SS cells. Co‐expression of HGF and c‐MET in SS clinical samples correlated with a poor prognosis in patients with SS. Taken together, activation of HGF/c‐MET signaling in an autocrine fashion leads to an aggressive phenotype in SS and targeting of this signaling exerts superior antitumor effects on c‐MET‐activated SS. HGF/c‐MET expression status is a potential biomarker for identification of SS patients with a worse prognosis who can benefit from c‐MET inhibitors.
Keywords: c‐MET, hepatocyte growth factor, INC280, synovial sarcoma, Yamato‐SS
Synovial sarcoma (SS) is a high‐grade malignant soft tissue sarcoma and accounts for 7–10% of all soft tissue sarcomas.1 SS most commonly arises in the extremities of young adults and is characterized by a specific translocation t(X;18)(p11.2;q11.2) that occurs in >95% of patients and leads to two main chimeric fusion genes, SS18‐SSX1 and SS18‐SSX2.1, 2, 3 Two histologically distinct subtypes of SS can be distinguished: biphasic tumors containing both epithelial‐like and spindle cells and monophasic fibrous tumors containing only spindle cells.4 Despite standardized treatment comprising surgical resection, chemotherapy and radiotherapy, the 5‐year overall survival rate of SS is only 30–70% and more than half of SS cases develop lung metastases, which worsens prognosis.1, 5, 6, 7, 8 Therefore, novel therapeutic approaches against SS are critically required.
c‐MET is a receptor tyrosine kinase (RTK) encoded by the proto‐oncogene MET and has a high affinity for hepatocyte growth factor (HGF). Cancer‐associated c‐MET activation triggers cell growth, survival, invasion, migration and angiogenesis.9, 10, 11 HGF stimulation induces c‐MET activation, which, in turn, activates multiple downstream signaling pathways, including the phosphatidylinositol 3‐kinase (PI3K)/AKT/mammalian target of rapamycin and MAPK/extracellular signal‐regulated kinase (ERK) pathways.12, 13 These pathways have crucial roles in regulating cell proliferation and survival.14, 15 Several c‐MET inhibitors are currently in clinical trials and show antitumor activities against non‐small cell lung cancer, papillary renal cell carcinoma and prostate cancer.16, 17, 18
Combined overexpression of HGF and c‐MET is observed in numerous soft tissue sarcomas such as epithelioid sarcomas, malignant peripheral nerve sheath tumors, and clear cell sarcomas, and HGF can activate c‐MET in an autocrine manner in these tumors.19, 20, 21, 22, 23 It has been reported that co‐expression of HGF and c‐MET was also frequently observed in SS clinical samples and was correlated with a poor prognosis.24, 25, 26, 27 However, little is known about the function of HGF/c‐MET signaling in SS and the antitumor effects of c‐MET inhibitors on SS.
In the present study, we first examined the mechanism of c‐MET activation and the functional role of c‐MET signaling in SS cell lines. Next, we assessed the antitumor effects of a selective c‐MET inhibitor, INC280, on SS cell lines both in vitro and in vivo, and sought a potential biomarker predictive of SS cell sensitivity to the c‐MET inhibitor. Finally, we investigated the expression status of HGF and c‐MET in SS clinical specimens and evaluated the relevance of HGF/c‐MET signaling and clinicopathological factors as well as patients’ survival.
Materials and Methods
Cell lines, reagents and antibodies
We used three human SS cell lines, Yamato‐SS, SYO‐1 and HS‐SY‐II. Yamato‐SS was established in our laboratory, as previously described.28 SYO‐1 was kindly provided by Dr Ozaki (Okayama University, Okayama, Japan).29 HS‐SY‐II was provided by the RIKEN BioResource Center through the National Bio‐Resource Project of the MEXT, Japan. Cells were grown in DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA). Cells were cultured in a humidified atmosphere at 37°C in 5% CO2. Doxorubicin was purchased by Wako Pure Chemical Industries (Osaka, Japan). Trabectedin was provided by Taiho Pharmaceutical (Tokyo, Japan). An ATP‐competitive selective c‐MET inhibitor, INC280, was provided by Novartis Pharma AG (Basel, Switzerland). The drugs were prepared in DMSO before being added to cell cultures for in vitro studies. According to the manufacturer's instructions, INC280 was diluted in 0.5% methylcellulose and 0.1% Tween 80 for in vivo experiments. Recombinant human HGF was purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against c‐MET, p‐MET (Tyr1234/1235), platelet‐derived growth factor receptor alpha (PDGFRα), p‐PDGFRα (Tyr849), AKT, p‐AKT (Ser473), ERK, p‐ERK (Thr202/Tyr204), cleaved caspase‐3 and beta‐actin were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against HGF and p‐PDGFRα (Tyr762) were purchased from R&D Systems. Antibodies against proliferating cell nuclear antigen (PCNA) and PDGFB were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HRP‐conjugated secondary antibodies were purchased from GE Healthcare Life Sciences (Piscataway, NJ, USA).
Patients
Forty‐two patients with SS treated in Osaka University Hospital or Osaka Medical Center for Cancer and Cardiovascular Diseases from 1986 to 2011 were enrolled in the present study. Clinical and survival data for these patients were collected from their medical records. All patients were histopathologically diagnosed as having SS. Tumor specimens were obtained with the patients’ informed consent and were used for additional immunohistochemical study. Follow‐up ranged from 3 to 314 months (mean, 83.0 months). To assess clinicopathological prognostic factors, SS18‐SSX fusion type, patient age at presentation, gender, primary tumor location, tumor size, histological subtype, and disease stage at presentation were analyzed. Extremity tumors were defined as tumors located in free extremities only but extremity girdles, including the shoulder, axilla, groin or buttock, were considered to be trunk locations. Tumor size was defined as the maximum dimension measured on a magnetic resonance imaging or computed tomographic scan. Disease stage was classified as localized or metastatic at initial diagnosis.
Western blot analysis
For the lysate preparation, cells were first washed with PBS and lysed in RIPA buffer (Thermo Scientific, Waltham, MA, USA). Tumor tissues were homogenized and lysed using a T‐PER tissue protein extraction buffer (Thermo Scientific). Protein concentrations were determined according to the bicinchoninic acid method (Thermo Scientific). Then, the cell lysates were separated on 4–12% Bis‐Tris gels (Life Technologies) and transferred to polyvinylidene difluoride membranes (Nippon Genetics, Tokyo, Japan). The membranes were incubated in 5% skim milk in TBS with Tween 20 (TBS‐T) at room temperature. Blocked membranes were incubated with primary antibodies at 4°C overnight, followed by incubation with secondary antibodies at room temperature for 1 h. After washing in TBS‐T, immunoreactive bands were visualized by enhanced chemiluminescence (GE Healthcare Life Sciences).
Elisa
Synovial sarcoma cells were cultured at a density of 1 × 105 cells/well in 6‐well plates. Cell culture supernatants were collected 48 h later. When the xenograft tumors reached 2 cm3, whole blood samples were collected by intracardiac puncture and sera were obtained. HGF concentrations in cell‐conditioned media or sera of xenografted mice were determined by ELISA using a Human HGF Quantikine ELISA Kit (R&D Systems), according to the manufacturer's instructions.
Immunohistochemistry
Specimens of tumors formed in nude mice and those of patients’ primary tumors were fixed in 10% neutral‐buffered formalin, embedded in paraffin, and sectioned at 4‐μm thickness. Paraffin‐embedded sections were deparaffinized and dehydrated. Antigens were retrieved at 95°C for 10 min in a 10‐mM citrate buffer. After blocking of endogenous peroxidase activity for 10 min with methanol containing 3% H2O2, the sections were reacted for 1 h with TBS containing 2% BSA at room temperature. The sections were incubated with primary antibodies at 4°C overnight. The next day, the sections were incubated for 1 h with secondary antibodies and stained with 3,3′‐diaminobenzidine tetrahydrochloride (Dako, Glostrup, Denmark). Finally, the sections were counterstained using hematoxylin. Immunohistochemical protein expression levels were determined using NIS‐Elements software (Nikon Corporation, Tokyo, Japan). Immunohistochemical results were interpreted as negative (<10% positive cells) or positive (>10% positive cells).
Soft agar colony formation assay
Five thousand SS cells were suspended in 1 mL of 0.5% SeaPlaque Agarose (Lonza, Basel, Switzerland) with normal growth medium and seeded over a basal layer of 0.6% agarose in 35‐mm culture dishes. The number of colonies (>100 μm in diameter) per well was counted under a light microscope 3 weeks later.
WST‐1 cell proliferation assay
Synovial sarcoma cells were seeded at a density of 1 × 103 cells/well in 96‐well plates for cell proliferation assays. The cells were treated with various concentrations of drugs or vehicle (DMSO) for drug experiments. Cell viability was assessed using the Premix WST‐1 cell proliferation assay system (Takara Bio, Otsu, Japan). By using a microplate reader, absorbance measurements read at 690 nm were subtracted from those read at 450 nm. Relative cell growth was expressed as: (absorbance of treated cells minus absorbance of cell‐free control)/(absorbance of untreated control − absorbance of cell‐free control).
RNA interference
Synovial sarcoma cells were seeded at a density of 3 × 105 cells/well in 6‐well plates and grown overnight. Cells were transfected with 20 nM small interfering RNA (siRNA) for 48 h using Lipofectamine 2000 (Life Technologies). Two kinds of siRNA targeting c‐MET (constructs I and II) and a non‐targeting siRNA were purchased from Cell Signaling Technology. siRNA against SS18‐SSX (siRNA‐A and siRNA‐B) and a control siRNA were designed as previously described.28
Cell cycle analysis
Synovial sarcoma cells were seeded at a density of 5 × 105 cells/dish in 10‐cm culture dishes and grown overnight, followed by treatment with INC280 or vehicle (DMSO). After 24‐h treatment, the cells were collected and stained with propidium iodide (PI) solution (25 μg/mL PI, 0.03% NP‐40, 0.02 mg/mL RNase A, 0.1% sodium citrate) for 30 min at room temperature. The cell cycle was analyzed using a BD FACSCanto II Flow Cytometer (BD Biosciences, San Jose, CA, USA).
In vivo animal xenograft model
Five‐week‐old athymic nude mice (BALB/c nu/nu; SLC, Shizuoka, Japan) were housed at the Institute of Experimental Animal Sciences, Osaka University Medical School, in accordance with guidelines approved by the Institutional Animal Care and Use Committee of the Osaka University Graduate School of Medicine. For the xenograft tumor growth assay, 1 × 107 SS cells were injected subcutaneously into the left side of the back. Therapy was initiated after tumor establishment (>5 mm in the longest diameter). INC280 was administered orally once a day. Xenograft tumor volume and mice body weight were measured twice a week. Tumor volume was measured using a caliper and was calculated according to the formula (A × B2)/2, with A being the longest diameter and B the shortest diameter of the tumor. Mice were killed when the total tumor burden reached 2 cm3 and the tumor weight was measured. The tumors were resected for immunohistochemical study.
Statistical analysis
Each experiment was performed in triplicate. All data are expressed as means ± SD. Student's t‐test for biological assays and Mann–Whitney's U‐test for animal experiments were used to evaluate the significance of differences. Values of P < 0.05 were considered to indicate statistical significance.
Results
c‐MET and its downstream signaling pathways are activated via autocrine hepatocyte growth factor stimulation in Yamato‐SS cells
To assess the role of c‐MET signaling in SS, we first examined the expression and phosphorylation of its related molecules in three human SS cell lines using western blot analysis. c‐MET expression was observed in all three SS cell lines (Fig. 1a). Among them, c‐MET was highly expressed and extensively phosphorylated in Yamato‐SS cells, whereas c‐MET was not phosphorylated in SYO‐1 or HS‐SY‐II cells (Fig. 1a), suggesting that Yamato‐SS was a c‐MET‐activated cell line and SYO‐1 and HS‐SY‐II were c‐MET‐inactivated cell lines. In addition, Yamato‐SS cells showed higher phosphorylation of c‐MET downstream AKT and ERK than did SYO‐1 or HS‐SY‐II cells (Fig. 1a). In contrast, exogenous recombinant HGF stimulation induced phosphorylation of c‐MET and its downstream effectors in SYO‐1 and HS‐SY‐II cells (Fig. 1b).
Figure 1.
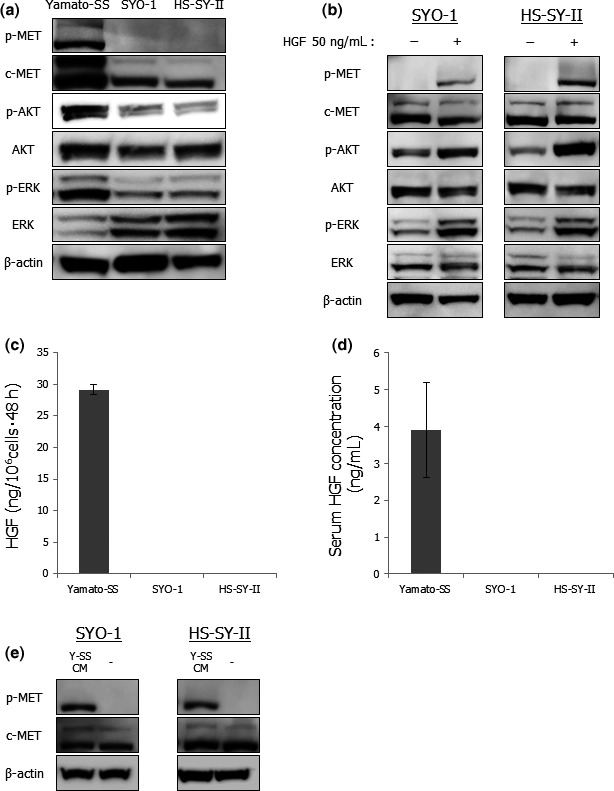
Autocrine hepatocyte growth factor (HGF) stimulation activates c‐MET and its downstream effectors in Yamato‐SS cells. (a) Expressions of proteins related to c‐MET signaling in three SS cell lines. The c‐MET protein presents a characteristic two band pattern, with the upper band corresponding to an unprocessed 170‐kDa precursor and the lower band corresponding to the 140‐kDa beta‐chain. (b) Effects of exogenous HGF stimulation on c‐MET signaling in SYO‐1 and HS‐SY‐II cells. The cells were stimulated by the addition of 50 ng/mL human recombinant HGF for 10 min and cell lysates were then prepared. (c) HGF levels in cell‐conditioned media of thre SS cell lines. Columns, mean; bars, SD. (d) HGF concentrations in sera of SS xenografted mice. Columns, mean; bars, SD. (e) Effects of cell‐conditioned media of Yamato‐SS cells on c‐MET in SYO‐1 and HS‐SY‐II cells. SYO‐1 and HS‐SY‐II cells were cultured overnight and the media were replaced with Yamato‐SS cell‐conditioned media 10 min before cell lysis. Y‐SS CM, Yamato‐SS cell‐conditioned media.
Second, to determine whether c‐MET phosphorylation was caused by the HGF autocrine loop in SS, we examined HGF secretion levels in SS cell lines by ELISA. Indeed, Yamato‐SS cells secreted high levels of HGF into culture media, whereas SYO‐1 or HS‐SY‐II cells did not produce HGF (Fig. 1c). Furthermore, we also detected high amounts of human HGF in the sera of mice bearing Yamato‐SS xenograft tumors but not in those of SYO‐1 or HS‐SY‐II xenografted mice (Fig. 1d). Conditioned media of Yamato‐SS cells induced c‐MET phosphorylation in SYO‐1 or HS‐SY‐II cells (Fig. 1e). These results implied that Yamato‐SS cells secreted high levels of functional HGF and that SYO‐1 and HS‐SY‐II cells conserved the c‐MET circuit. Co‐expression of HGF and c‐MET, and c‐MET phosphorylation were also observed in Yamato‐SS xenograft tumors, whereas only c‐MET was expressed in SYO‐1 or HS‐SY‐II tumors (Fig. S1). These data suggested that c‐MET was activated via autocrine HGF stimulation in a subset of SS both in vitro and in vivo and that autocrine HGF/c‐MET signaling maintained activation of c‐MET and its downstream signaling pathways.
c‐MET‐activated synovial sarcoma cells show higher anchorage‐independent growth ability and less sensitivity to chemotherapeutic agents than did c‐MET‐inactivated synovial sarcoma cells
It has been reported that c‐MET could serve as a marker for cancer stem cells of several malignancies and that c‐MET activation was associated with tumor sphere formation and resistance to chemotherapies.30, 31, 32, 33, 34, 35 We investigated anchorage‐independent growth abilities of three SS cell lines and their sensitivities to doxorubicin and trabectedin, which are used for treatment of soft tissue sarcomas.36, 37 c‐MET‐activated Yamato‐SS cells showed higher colony‐forming capacity than did c‐MET‐inactivated SYO‐1 or HS‐SY‐II cells (Fig. 2a,b). In addition, we calculated the 50% inhibitory concentration (IC50) of doxorubicin or trabectedin on three SS cell lines. The IC50 of doxorubicin or trabectedin on Yamato‐SS cells was higher than that on SYO‐1 or HS‐SY‐II cells (Fig. 2c). These results indicated that c‐MET‐activated SS cells showed higher anchorage‐independent growth ability and lesser sensitivity to chemotherapeutic agents than did c‐MET‐inactivated SS cells.
Figure 2.
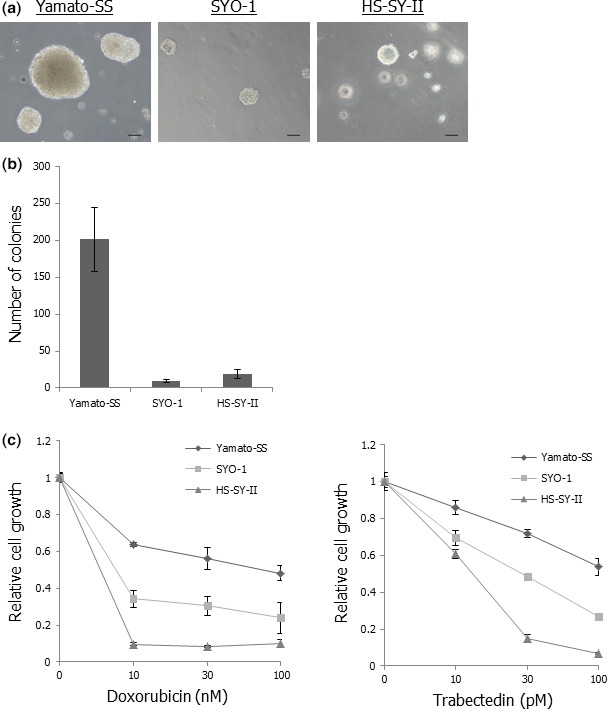
Yamato‐SS cells show higher anchorage‐independent growth ability and lesser sensitivity to chemotherapeutic agents than SYO‐1 or HS‐SY‐II cells. (a) Colony formation in three SS cell lines. Scale bars: 100 μm. (b) Numbers of colonies in three SS cell lines. Columns, mean; bars, SD. (c) Sensitivities of three SS cell lines to doxorubicin or trabectedin. The SS cells were exposed to different concentrations of doxorubicin or trabectedin for 72 h. Cell viability was determined by WST‐1 assay and relative cell growth was calculated. Points, mean; bars, SD.
Silencing of c‐MET expression suppresses proliferation and anchorage‐independent growth of c‐MET‐activated synovial sarcoma cells
To determine whether HGF/c‐MET signaling affected proliferation and anchorage‐independent growth of c‐MET‐activated SS cells, we used RNA interference technology in vitro. Two kinds of anti‐c‐MET‐specific siRNA were transfected into Yamato‐SS cells, resulting in significant inhibition of c‐MET and p‐MET expression (Fig. 3a). Silencing of c‐MET expression inhibited proliferation and colony formation of Yamato‐SS cells (Fig. 3b–d). These results suggested that autocrine activation of HGF/c‐MET signaling contributed to proliferation and anchorage‐independent growth of SS cells.
Figure 3.
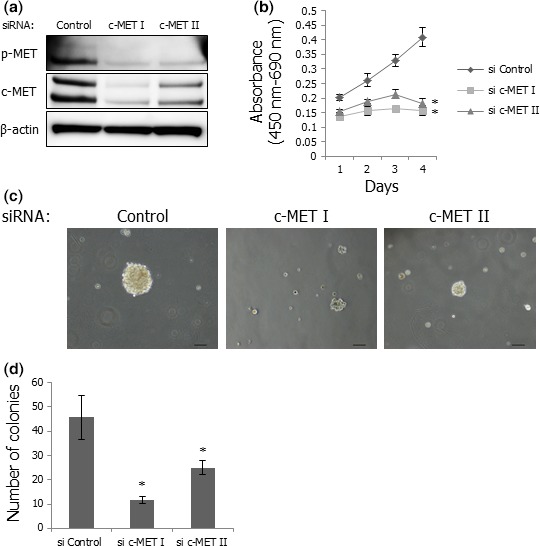
Silencing of c‐MET suppresses proliferation and colony formation of Yamato‐SS cells. (a) Expression of c‐MET and p‐MET in Yamato‐SS cells transfected with anti‐c‐MET siRNA or a control siRNA. (b) Proliferation of Yamato‐SS cells transfected with anti‐c‐MET siRNA or a control siRNA. Transfected cells were cultured for 96 h and cell viability was determined by WST‐1 assay every 24 h. Points, mean; bars, SD. *P < 0.05, compared with control. (c) Appearance of colonies of Yamato‐SS cells transfected with anti‐c‐MET siRNA or a control siRNA. Scale bars: 100 μm. (d) Numbers of colonies in Yamato‐SS cells transfected with anti‐c‐MET siRNA or a control siRNA. Columns, mean; bars, SD. *P < 0.05, compared with control.
INC280 inhibits the growth of c‐MET‐activated synovial sarcoma cells by blocking activation of c‐MET signaling pathways in vitro
A recently developed c‐MET inhibitor, INC280, has been shown to inhibit c‐MET‐dependent cell motility, proliferation and invasion in several cancer cell lines in vitro and in vivo.20, 38 First, we tested the antitumor effects of INC280 on proliferation and anchorage‐independent growth of SS cell lines in vitro. INC280 inhibited the proliferation of Yamato‐SS cells in a dose‐dependent manner but not that of SYO‐1 or HS‐SY‐II cells (Fig. 4a). These data implied that not c‐MET expression but c‐MET autocrine activation should be a potential biomarker to determine sensitivities of SS cells to the c‐MET inhibitor. In addition, colony formation of Yamato‐SS cells was blocked by treatment with INC280 (Figs 4b and S2). Flow cytometry analyses showed that INC280 induced a dose‐dependent increase in the G0/G1‐phase population and a decrease in the S‐phase population in Yamato‐SS cells (Fig. 4c). Furthermore, the expression of cleaved caspase‐3 was increased in a dose‐dependent fashion in Yamato‐SS cells (Fig. 4d). These results indicated that INC280 inhibited the growth of c‐MET‐activated SS cells by inducing G0/G1 cell cycle arrest and apoptosis in vitro.
Figure 4.
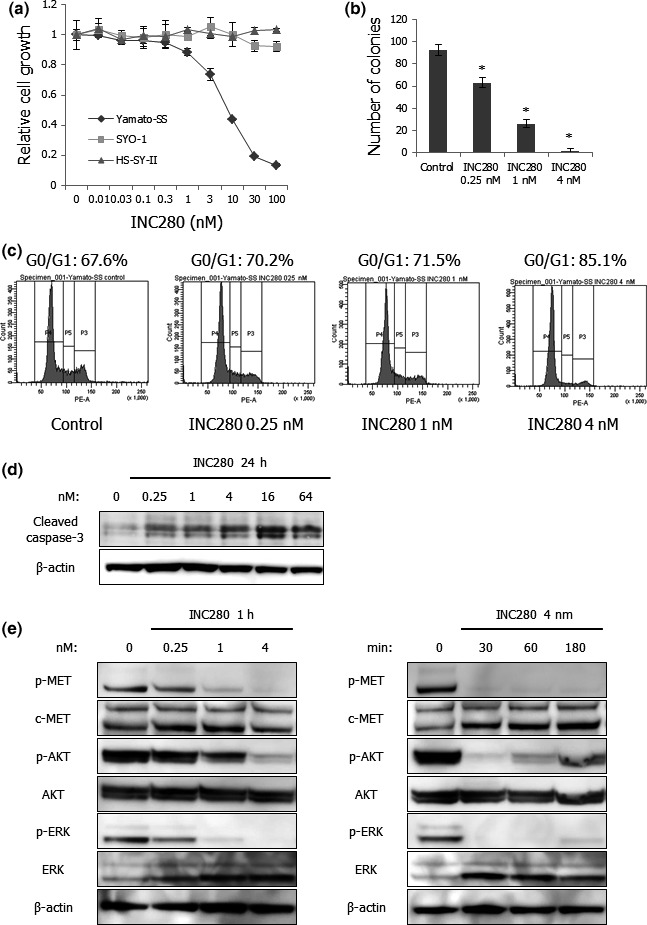
INC280 inhibits the growth of Yamato‐SS cells in vitro. (a) Sensitivities of three SS cell lines to INC280. SS cells were treated with various concentrations of INC280 for 72 h. Cell viability was determined by WST‐1 assay and relative cell growth was calculated. Points, mean; bars, SD. (b) Numbers of colonies in Yamato‐SS cells treated with 0.25–4 nM of INC280 or vehicle for 3 weeks. Columns, mean; bars, SD. *P < 0.05, compared with control. (c) PI staining and fluorescence‐activated cell sorting analyses of the DNA contents of Yamato‐SS cells in response to INC280. The cells were incubated with 0.25–4 nM of INC280 or vehicle for 24 h. (d) Effects of INC280 on caspase‐3 cleavage in Yamato‐SS cells. The cells were treated with 0.25–64 nM of INC280 or vehicle for 24 h. (e) Effects of INC280 on phosphorylation of c‐MET and its downstream effectors in Yamato‐SS cells. The cells were treated with 0.25–4 nM of INC280 or vehicle for 1 h (left) and with 4 nM INC280 for 30–180 min (right).
Second, we examined the effects of INC280 on c‐MET and its downstream effectors, including AKT and ERK, in Yamato‐SS cells by performing western blot analyses. INC280 strikingly blocked the phosphorylation of c‐MET, AKT and ERK in a dose‐dependent manner in Yamato‐SS cells (Fig. 4e). These results suggested that the PI3K/AKT and MAPK/ERK signaling pathways were highly dependent on autocrine HGF/c‐MET signaling in c‐MET‐activated SS cells and that the c‐MET inhibitor exerted significant antitumor effects on these cells by blocking c‐MET downstream signaling pathways.
INC280 abrogates the growth of c‐MET‐activated synovial sarcoma xenograft tumors
We next evaluated the antitumor effects of INC280 on growth of Yamato‐SS xenograft tumors. An INC280 dose of 3 or 10 mg/kg given orally once a day was selected on the basis of the previous observation that this therapeutic dose resulted in inhibition of c‐MET phosphorylation in vivo.20, 38 Administration of INC280 notably abrogated Yamato‐SS xenograft tumor growth relative to that of the vehicle control (Fig. 5a). No significant body weight loss was observed in INC280‐treated mice (data not shown). c‐MET phosphorylation was strongly suppressed in INC280‐treated Yamato‐SS xenograft tumors by immunohistochemical studies (Fig. 5b). In addition, a decrease in the rate of PCNA‐positive tumor cells and an increase in that of cleaved caspase‐3‐positive cells were immunohistochemically observed in INC280‐treated SS xenograft tumors (Fig. 5b–d). These results suggested that INC280 also exerted significant antitumor effects on the growth and survival of c‐MET‐activated SS xenograft tumors by blocking c‐MET signaling.
Figure 5.
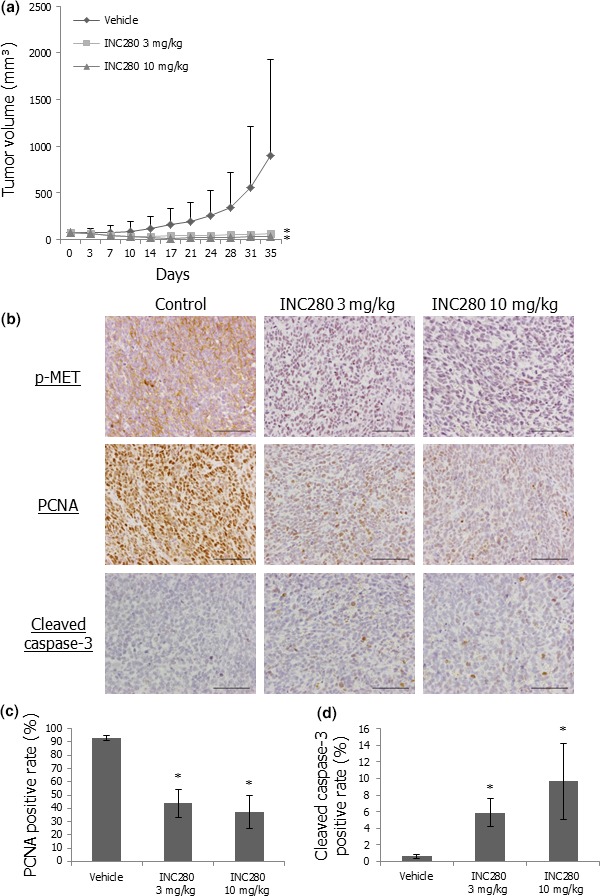
INC280 abrogates the growth of Yamato‐SS xenograft tumors by inactivating c‐MET signaling. (a) Effects of INC280 on Yamato‐SS xenograft tumor growth. Mice bearing Yamato‐SS (n = 6/group each) xenograft tumors were treated with 3 mg/kg INC280, 10 mg/kg INC280 or vehicle control. Points, mean; bars, SD. *P < 0.05, compared with control treatment. (b) Immunohistochemical staining of p‐MET, PCNA and cleaved caspase‐3 in Yamato‐SS xenograft tumors in the three groups. Scale bars: 100 μm. (c) PCNA‐positive rates of Yamato‐SS xenograft tumors in the three groups. Columns, mean; bars, SD. *P < 0.05, compared with control treatment. (d) Cleaved caspase‐3‐positive rates of Yamato‐SS xenograft tumors in the three groups. Columns, mean; bars, SD. *P < 0.05, compared with control treatment.
Co‐expression of hepatocyte growth factor and c‐MET is correlated with a worse prognosis in synovial sarcoma
To evaluate the clinical role of HGF/c‐MET signaling in patients with SS, we first examined the expression status of HGF and c‐MET in 42 SS clinical samples. HGF and c‐MET were expressed in 21 (50%) and 33 (78.6%) samples, respectively (Fig. 6a and Table S1). In addition, co‐expression of HGF and c‐MET was observed in 15 (35.7%) samples. These data indicated that the expression status of HGF and c‐MET varied considerably in SS clinical specimens.
Figure 6.
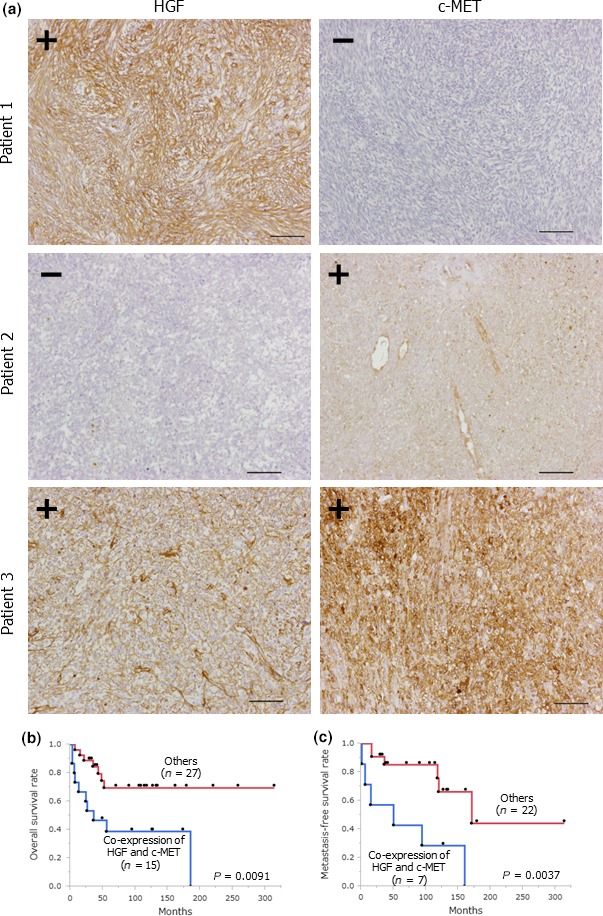
Co‐expression of hepatocyte growth factor (HGF) and c‐MET correlates with a poor prognosis in patients with synovial sarcoma (SS). (a) HGF and c‐MET expression in SS clinical samples. Scale bars: 100 μm. (b) Overall survival curve in SS patients whose tumors co‐expressed HGF and c‐MET (n = 15) and the other patients (n = 27). (c) Metastasis‐free survival curve in SS patients with localized diseases at initial diagnosis whose tumors were positive for both HGF and c‐MET (n = 7) and the other patients (n = 22).
Next, we compared clinicopathological characteristics and survival rates of the patients whose tumors co‐expressed HGF and c‐MET with those of the other patients. Co‐expression of HGF and c‐MET was associated with truncal tumor location (P = 0.0295), large tumor size (>5 cm) (P = 0.0134) and lung metastasis at initial diagnosis (P = 0.0189) (Table S2). Using the Kaplan–Meier method, overall survival of the patients whose tumors were positive for both HGF and c‐MET was significantly shorter than that of the other patients (Fig. 6b). Large tumor size (>5 cm) (P = 0.0377), lung metastasis at initial diagnosis (P < 0.001), and co‐expression of HGF and c‐MET (P = 0.0091) were significantly unfavorable predictors for overall survival in all SS patients according to the univariate analyses (Table S3). The results of the multivariable analyses showed that disease stage at initial diagnosis was the exclusively independent predictor for overall survival (P < 0.001) (Table S4). Among 29 patients with localized disease at initial diagnosis, co‐expression of HGF and c‐MET tended to be correlated with shorter overall survival than the other types of expression (P = 0.0659) (Fig. S3). In contrast, metastasis‐free survival of the patients with tumors positive for both HGF and c‐MET was significantly shorter than that of the other patients in the univariate analyses (P = 0.0037) (Fig. 6c). In addition, there was no significant correlation between the other factors and metastasis‐free survival (Table S5). These results suggested that the autocrine HGF/c‐MET signaling clinically played an important role in promoting the progression and metastasis of SS, resulting in a worse prognosis in patients with SS.
Discussion
Successful development of personalized molecular targeted therapy is dependent on identification of targets that are directly involved in tumorigenesis and tumor progression. HGF/c‐MET signaling reportedly has an important role in the development of several human cancers and in drug resistance in cancer cells.10, 11, 12, 13, 19, 20, 21, 22, 23, 30, 31, 32, 33, 34, 35 In the present study, we found that c‐MET‐activated Yamato‐SS cells were highly addicted to autocrine HGF/c‐MET signaling and exhibited higher anchorage‐independent growth ability and lesser sensitivity to chemotherapies than did c‐MET‐inactivated SYO‐1 or HS‐SY‐II cells. These results suggested that HGF/c‐MET signaling was one of the essential factors for tumorigenesis or tumor progression in SS and raised the possibility that a subset of SS driven by this signaling might be highly aggressive and intractable.
c‐MET inhibitors have shown antitumor efficacy in preclinical studies and are currently being evaluated in human cancer clinical trials.16, 17, 18, 20, 38 However, the antitumor effects of c‐MET‐targeted therapies on SS remain unresolved. c‐MET expression was frequently observed in SS clinical samples as well as in all three SS cell lines by western blotting analyses (Fig. S4). In this study, INC280, a selective c‐MET inhibitor, significantly inhibited the growth of c‐MET‐activated SS cells both in vitro and in vivo, whereas the proliferation of c‐MET‐inactivated SS cells was not suppressed. These data indicated that INC280 selectively blocked c‐MET activation and exerted notable antitumor effects on a subset of SS.
To identify patient subgroups most likely to benefit from c‐MET‐targeted therapeutics and enable more rational trial designs, a biomarker predicting response to c‐MET inhibitors is needed. It has been reported that HGF/c‐MET autocrine status correlated with c‐MET phosphorylation in glioblastoma cell lines and c‐MET activation predicted higher sensitivity to c‐MET inhibitors.39 Consistently, our results suggested that SS patients who were likely to benefit from treatment with c‐MET inhibitors could be selected on the basis of their c‐MET phosphorylation status. However, such an approach would be highly impractical because of the failure in retaining c‐MET phosphorylation in clinical samples.40 In fact, several studies have evaluated HGF/c‐MET expression in SS clinical specimens probably due to the difficulty of detecting c‐MET phosphorylation.24, 25, 26, 27 On the basis of our results, we proposed that co‐expression of HGF and c‐MET should result in c‐MET autocrine activation and be predictive of response to anti‐HGF/c‐MET therapies in SS. Therefore, pathological interrogation of both HGF and c‐MET expressions could permit selection of patients most likely to respond to c‐MET inhibition.
Previously, it has been demonstrated that the positive rate of both HGF and c‐MET in SS clinical samples was 14–68%, resulting in worse prognosis in SS.24, 26, 27 In the present study, we also showed that expression statuses of HGF and c‐MET differed among SS cell lines, suggesting that SS cell lines exhibited heterogeneity in dependency on HGF/c‐MET signaling. However, the mechanism underlying variations in expression statuses of HGF and c‐MET in SS remains unknown.
Prior publications have suggested that fusion genes and cellular origins were crucial for determining and maintaining the tumor phenotype in translocation‐related malignancies.28, 41, 42 c‐MET is also frequently activated in translocation‐related soft tissue sarcomas such as alveolar rhabdomyosarcoma and clear cell sarcoma.22, 23, 43 Alveolar rhabdomyosarcoma is characterized by a chromosomal translocation t(2,13)(q35;q14) generating the PAX3‐FOXO1 (also known as FKHR) fusion gene. It has been demonstrated that the expression of c‐MET correlated significantly with PAX3‐FOXO1 expression, indicating that c‐MET was a downstream target of PAX3‐FOXO1 in alveolar rhabdomyosarcoma.44 Furthermore, it has been shown that EWS‐ATF1, the product of the pathognomonic translocation associated with clear cell sarcoma, also induced c‐MET expression.22 In sharp contrast, silencing of SS18‐SSX in our parallel study did not decrease HGF/c‐MET expression or c‐MET phosphorylation in Yamato‐SS cells (Fig. S5a). We previously demonstrated that PDGFRα was activated in c‐MET‐inactivated SS cells,40 indicating that there was a diversity of activated RTK among SS cell lines. Similar to HGF/c‐MET signaling in Yamato‐SS cells, silencing of SS18‐SSX attenuated neither PDGFB/PDGFRα expression nor PDGFRα phosphorylation in PDGFRα‐activated HS‐SY‐II cells (Fig. S5b). These results suggested that activation of HGF/c‐MET or PDGFB/PDGFRα signaling in SS might be derived from not SS18‐SSX, the central genetic driver of SS, but the cell of origin for SS itself.
It has been reported that tumors with features of human SS developed when expression of the SS18‐SSX gene was induced in Myf5‐expressing murine myoblasts but not in the embryonic stage or in Myf6‐expressing myocytes.45 These findings suggested that the window of permissive cells was relatively narrow for SS18‐SSX.46 Previously, we demonstrated that one of the cellular origins for SS was a multipotent mesenchymal stem cell with potential to differentiate into mesenchymal and hematopoietic lineages by silencing of SS18‐SSX.28 Human mesenchymal stem cells express a large number of growth factors and growth factor receptors with different functions.47, 48 In mesenchymal stem cells, HGF/c‐MET signaling is important in the maintenance of stemness, whereas PDGF/PDGFR signaling is a key regulator of cell differentiation and mesenchymal tissue formation.49, 50 We propose that cellular origins of c‐MET‐activated SS might be more immature multipotent mesenchymal stem cells expressing higher levels of HGF/c‐MET, whereas those of PDGFR‐activated SS might be less immature mesenchymal stem cells dependent on PDGF/PDGFR signaling.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Immunohistochemical staining of hepatocyte growth factor (HGF), c‐MET and p‐MET in synovial sarcoma (SS) xenograft tumors. Scale bars: 100 μm.
Fig. S2. Appearance of colonies of Yamato‐SS cells treated with 0.25–4 nM of INC280 or vehicle for 3 weeks. Scale bars: 100 μm.
Fig. S3. Overall survival curve in synovial sarcoma (SS) patients with localized diseases at initial diagnosis whose tumors were positive for both hepatocyte growth factor (HGF) and c‐MET (n = 7), and the other patients (n = 22).
Fig. S4. c‐MET expression in synovial sarcoma (SS) clinical samples in western blotting analysis.
Fig. S5. (a) Expressions of c‐MET‐related proteins in Yamato‐SS cells transfected with siRNA targeting SS18‐SSX or a control siRNA. (b) Expressions of PDGFRα‐related proteins in HS‐SY‐II cells transfected with siRNA targeting SS18‐SSX or a control siRNA.
Table S1. Expression status of hepatocyte growth factor (HGF) and c‐MET in synovial sarcoma (SS) clinical samples.
Table S2. Association between hepatocyte growth factor (HGF)/c‐MET expression status and clinicopathologic factors in all synovial sarcoma (SS) patients.
Table S3. Association between 5‐year overall survival rate and clinicopathologic factors or hepatocyte growth factor (HGF)/c‐MET expression status in all synovial sarcoma (SS) patients.
Table S4. Multivariate overall survival analysis for clinicopathologic factors and hepatocyte growth factor (HGF)/c‐MET expression status.
Table S5. Association between 5‐year metastasis‐free survival rate and clinicopathologic factors or hepatocyte growth factor (HGF)/c‐MET expression status in synovial sarcoma (SS) patients with localized diseases at initial diagnosis.
Acknowledgments
We wish to thank Dr Toshifumi Ozaki for kindly providing the human SS cell line, SYO‐1. We also thank Mari Shinkawa and Asa Tada for their excellent technical assistance. This work was supported by grants from the Japan Society for the Promotion of Science, JSPS KAKENHI (Grant Numbers: 16H05448 and 26462264), the Osaka Medical Research Foundation for Intractable Diseases, and the Practical Research for Innovative Cancer Control from Japan Agency For Medical Research and development, AMED.
Cancer Sci 107 (2016) 1867–1876
Funding Information
The Practical Research for Innovative Cancer Control from Japan Agency For Medical Research and Development, AMED, the Osaka Medical Research Foundation for Intractable Diseases, the Japan Society for the Promotion of Science, JSPS KAKENHI, (Grant/Award Number: ‘16H05448’, ‘26462264’).
Contributor Information
Yoshinori Imura, Email: y.imura@hotmail.co.jp.
Norifumi Naka, Email: nnaka@ort.med.osaka-u.ac.jp.
References
- 1. Smith S, Reeves BR, Wong L et al A consistent chromosome translocation in synovial sarcoma. Cancer Genet Cytogenet 1987; 26: 179–80. [DOI] [PubMed] [Google Scholar]
- 2. Clark J, Rocques PJ, Crew AJ et al Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 1994; 7: 502–8. [DOI] [PubMed] [Google Scholar]
- 3. Crew AJ, Clark J, Fisher C et al Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel‐associated box in human synovial sarcoma. EMBO J 1995; 14: 2333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thway K. Pathology of soft tissue sarcomas. Clin Oncol 2009; 21: 695–705. [DOI] [PubMed] [Google Scholar]
- 5. Lewis JJ, Antonescu CR, Leung DH et al Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol 2000; 18: 2087–94. [DOI] [PubMed] [Google Scholar]
- 6. Spillane AJ, A'Hern R, Judson IR et al Synovial sarcoma: a clinicopathologic, staining, and prognostic assessment. J Clin Oncol 2000; 18: 3794–803. [DOI] [PubMed] [Google Scholar]
- 7. Takenaka S, Ueda T, Naka N et al Prognostic implication of SYT‐SSX fusion type in synovial sarcoma: a multi‐institutional retrospective analysis in Japan. Oncol Rep 2008; 19: 467–76. [PubMed] [Google Scholar]
- 8. Krieg AH, Hefti F, Speth BM et al Synovial sarcomas usually metastasize after >5 years: a multicenter retrospective analysis with minimum follow‐up of 10 years for survivors. Ann Oncol 2011; 22: 458–67. [DOI] [PubMed] [Google Scholar]
- 9. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets 2012; 16: 553–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawaguchi M, Kataoka H. Mechanisms of hepatocyte growth factor activation in cancer tissues. Cancers 2014; 6: 1890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakai K, Aoki S, Matsumoto K. Hepatocyte growth factor and Met in drug discovery. J Biochem 2015; 157: 915–25. [DOI] [PubMed] [Google Scholar]
- 12. Zeng Q, Chen S, You Z et al Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J Biol Chem 2002; 277: 25203–8. [DOI] [PubMed] [Google Scholar]
- 13. Dong G, Chen Z, Li ZY et al Hepatocyte growth factor/scatter factor‐induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin‐8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res 2001; 61: 5911–8. [PubMed] [Google Scholar]
- 14. Dudek H, Datta SR, Franke TF et al Regulation of neuronal survival by the serine‐threonine protein kinase Akt. Science 1997; 275: 661–5. [DOI] [PubMed] [Google Scholar]
- 15. Sontag E, Fedorov S, Kamibayashi C et al The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 1993; 75: 887–97. [DOI] [PubMed] [Google Scholar]
- 16. Sequist LV, von Pawel J, Garmey EG et al Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non‐small‐cell lung cancer. J Clin Oncol 2011; 29: 3307–15. [DOI] [PubMed] [Google Scholar]
- 17. Choueiri TK, Vaishampayan U, Rosenberg JE et al Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol 2013; 31: 181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith DC, Smith MR, Sweeney C et al Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol 2013; 31: 412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cortner J, Vande Woude GF, Rong S. The Met‐HGF/SF autocrine signaling mechanism is involved in sarcomagenesis. EXS 1995; 74: 89–121. [DOI] [PubMed] [Google Scholar]
- 20. Imura Y, Yasui H, Outani H et al Combined targeting of mTOR and c‐MET signaling pathways for effective management of epithelioid sarcoma. Mol Cancer 2014; 13: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Torres KE, Zhu QS, Bill K et al Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res 2011; 17: 3943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davis IJ, McFadden AW, Zhang Y et al Identification of the tyrosine kinase c‐Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma. Cancer Res 2010; 70: 639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Outani H, Tanaka T, Wakamatsu T et al Establishment of a novel clear cell sarcoma cell line (Hewga‐CCS), and investigation of the antitumor effects of pazopanib on Hewga‐CCS. BMC Cancer 2014; 14: 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuhnen C, Tolnay E, Steinau HU et al Expression of c‐Met receptor and hepatocyte growth factor/scatter factor in synovial sarcoma and epithelioid sarcoma. Virchows Arch 1998; 432: 337–42. [DOI] [PubMed] [Google Scholar]
- 25. Fukuda T, Ichimura E, Shinozaki T et al Coexpression of HGF and c‐Met/HGF receptor in human bone and soft tissue tumors. Pathol Int 1998; 48: 757–62. [DOI] [PubMed] [Google Scholar]
- 26. Motoi T, Ishida T, Kuroda M et al Coexpression of hepatocyte growth factor and c‐Met proto‐oncogene product in synovial sarcoma. Pathol Int 1998; 48: 769–75. [DOI] [PubMed] [Google Scholar]
- 27. Oda Y, Sakamoto A, Saito T et al Expression of hepatocyte growth factor (HGF)/scatter factor and its receptor c‐MET correlates with poor prognosis in synovial sarcoma. Hum Pathol 2000; 31: 185–92. [DOI] [PubMed] [Google Scholar]
- 28. Naka N, Takenaka S, Araki N et al Synovial sarcoma is a stem cell malignancy. Stem Cells 2010; 28: 1119–31. [DOI] [PubMed] [Google Scholar]
- 29. Kawai A, Naito N, Yoshida A et al Establishment and characterization of a biphasic synovial sarcoma cell line, SYO‐1. Cancer Lett 2004; 204: 105–13. [DOI] [PubMed] [Google Scholar]
- 30. Tamase A, Muraguchi T, Naka K et al Identification of tumor‐initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc Natl Acad Sci USA 2009; 106: 17163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 32. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 33. Straussman R, Morikawa T, Shee K et al Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487: 500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joo KM, Jin J, Kim E et al MET signaling regulates glioblastoma stem cells. Cancer Res 2012; 72: 3828–38. [DOI] [PubMed] [Google Scholar]
- 35. De Bacco F, Casanova E, Medico E et al The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res 2012; 72: 4537–50. [DOI] [PubMed] [Google Scholar]
- 36. Santoro A, Tursz T, Mouridsen H et al Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first‐line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995; 13: 1537–45. [DOI] [PubMed] [Google Scholar]
- 37. del Muro G, Solans X, Martin Broto J et al SEOM clinical guidelines for the management of adult soft tissue sarcomas. Clin Transl Oncol 2012; 14: 541–4. [DOI] [PubMed] [Google Scholar]
- 38. Liu X, Wang Q, Yang G et al A novel kinase inhibitor, INCB28060, blocks c‐MET‐dependent signaling, neoplastic activities, and cross‐talk with EGFR and HER‐3. Clin Cancer Res 2011; 17: 7127–38. [DOI] [PubMed] [Google Scholar]
- 39. Xie Q, Bradley R, Kang L et al Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci USA 2012; 109: 570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yasui H, Naka N, Imura Y et al Tailored therapeutic strategies for synovial sarcoma: receptor tyrosine kinase pathway analyses predict sensitivity to the mTOR inhibitor RAD001. Cancer Lett 2014; 347: 114–22. [DOI] [PubMed] [Google Scholar]
- 41. Riggi N, Suva ML, Suva D et al EWS‐FLI‐1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 2008; 68: 2176–85. [DOI] [PubMed] [Google Scholar]
- 42. Rodriguez R, Rubio R, Menendez P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res 2012; 22: 62–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taulli R, Scuoppo C, Bersani F et al Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res 2006; 66: 4742–9. [DOI] [PubMed] [Google Scholar]
- 44. Ginsberg JP, Davis RJ, Bennicelli JL et al Up‐regulation of MET but not neural cell adhesion molecule expression by the PAX3‐FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res 1998; 58: 3542–6. [PubMed] [Google Scholar]
- 45. Haldar M, Hancock JD, Coffin CM et al A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 2007; 11: 375–88. [DOI] [PubMed] [Google Scholar]
- 46. Toguchida J, Nakayama T. Molecular genetics of sarcomas: applications to diagnoses and therapy. Cancer Sci 2009; 100: 1573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol 2014; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baek SJ, Kang SK, Ra JC. In vitro migration capacity of human adipose tissue‐derived mesenchymal stem cells reflects their expression of receptors for chemokines and growth factors. Exp Mol Med 2011; 43: 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eom YW, Oh JE, Lee JI et al The role of growth factors in maintenance of stemness in bone marrow‐derived mesenchymal stem cells. Biochem Biophys Res Commun 2014; 445: 16–22. [DOI] [PubMed] [Google Scholar]
- 50. Ng F, Boucher S, Koh S et al PDGF, TGF‐beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008; 112: 295–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunohistochemical staining of hepatocyte growth factor (HGF), c‐MET and p‐MET in synovial sarcoma (SS) xenograft tumors. Scale bars: 100 μm.
Fig. S2. Appearance of colonies of Yamato‐SS cells treated with 0.25–4 nM of INC280 or vehicle for 3 weeks. Scale bars: 100 μm.
Fig. S3. Overall survival curve in synovial sarcoma (SS) patients with localized diseases at initial diagnosis whose tumors were positive for both hepatocyte growth factor (HGF) and c‐MET (n = 7), and the other patients (n = 22).
Fig. S4. c‐MET expression in synovial sarcoma (SS) clinical samples in western blotting analysis.
Fig. S5. (a) Expressions of c‐MET‐related proteins in Yamato‐SS cells transfected with siRNA targeting SS18‐SSX or a control siRNA. (b) Expressions of PDGFRα‐related proteins in HS‐SY‐II cells transfected with siRNA targeting SS18‐SSX or a control siRNA.
Table S1. Expression status of hepatocyte growth factor (HGF) and c‐MET in synovial sarcoma (SS) clinical samples.
Table S2. Association between hepatocyte growth factor (HGF)/c‐MET expression status and clinicopathologic factors in all synovial sarcoma (SS) patients.
Table S3. Association between 5‐year overall survival rate and clinicopathologic factors or hepatocyte growth factor (HGF)/c‐MET expression status in all synovial sarcoma (SS) patients.
Table S4. Multivariate overall survival analysis for clinicopathologic factors and hepatocyte growth factor (HGF)/c‐MET expression status.
Table S5. Association between 5‐year metastasis‐free survival rate and clinicopathologic factors or hepatocyte growth factor (HGF)/c‐MET expression status in synovial sarcoma (SS) patients with localized diseases at initial diagnosis.