

Abstract
Cancer/testis antigen MAGEC2, a member of the type I melanoma‐associated antigen family, is expressed in a wide variety of cancer types but not in normal somatic cells. MAGEC2 has long been recognized as a tumor‐specific target, however, its functions remain largely unknown. In this study, we established MAGEC2‐knockout A375 melanoma cell lines using the CRISPR/Cas9 system. Seven clonal cell lines were generated by using four single guide RNAs targeting the coding region of the MAGEC2 gene, which produced indels that abolished MAGEC2 protein expression. To identify the differentially expressed protein profiles associated with MAGEC2 loss, isobaric tag for relative quantitation‐based comparative proteomics experiments were carried out on the MAGEC2‐knockcout and control A375 cells. Mining of the proteomics data identified a total 224 (61.6% upregulated and 38.4% downregulated) proteins to be significantly altered in expression level in MAGEC2‐knockcout cells. Ingenuity Pathway Analysis indicated that the significantly altered proteins were involved in critical neoplasia‐related biological functions such as cell death, proliferation, and movement. Gene ontology analysis identified “apoptosis signaling” as the top‐most upregulated pathway associated with MAGEC2 loss. We showed that knockout or knockdown of the MAGEC2 gene sensitized melanoma cells to tumor necrosis factor‐α‐induced apoptosis. Interestingly, actin‐based motility by Rho and RhoA signaling, known to promote cell migration, were also identified as the top downregulated pathways in MAGEC2‐knockout A375 cells. In short, our study provides a suitable cell model for exploring the biological functions of MAGEC2 in malignant cells, and sheds light on the molecular pathway by which MAGEC2 promotes tumor development.
Keywords: Bioinformatics, CRISPR‐Cas systems, gene knockout techniques, MAGEC2, proteomics
The type I melanoma‐associated antigens (MAGE‐I) are a group of well‐characterized members of the cancer‐testis antigen family, which are aberrantly expressed in a wide variety of tumors but absent from normal adult tissues other than immunoprivileged germ‐line tissues.1 Although the cellular and physiological functions of MAGE‐I proteins in cancer have not been fully elucidated, recent studies have indicated that they are associated with hallmarks of aggressive cancers. Cancer/testis antigen MAGEC2, a member of the MAGE‐I protein family, is absent in normal adult somatic tissues but expressed in many types of tumors.2, 3, 4, 5, 6 Clinical studies indicate that expression of MAGEC2 is a potent predictor associated with sentinel lymph node metastasis in melanoma,7 and nuclear MAGEC2 expression in prostate cancer indicates a higher risk for biochemical recurrence after radical prostatectomy.8 The function of MAGEC2 in tumor cells was first reported by Yang et al. in 2007.9, 10 They found that MAGEC2 promotes cell viability in both mast and melanoma cell lines. Subsequent studies showed that MAGEC2 can form complexes with RING domain protein TRIM28 and promotes the degradation of p53 in a proteasome‐dependent manner by TRIM28.11 MAGEC2 was also reported to increase phosphorylation of TRIM28 and improve DNA repair after double‐stranded breaks.12 Recently, we reported that MAGEC2 binds directly to the RING domain protein Rbx1, decreases the degradation of cyclin E, and promotes cell cycle progression in tumor cells.13 The common strategies used in all these studies were to knock down endogenous MAGEC2 in cell lines by RNAi. A shortcoming of this approach, however, is that neither siRNA nor shRNA is 100% effective at eliminating the expression of a target gene product. To comprehensively understand the biological functions of MAGEC2 in tumor cells, establishing cell lines that completely lack the expression of MAGEC2 are necessary.
Recently, the bacterial clustered regularly interspaced short palindromic repeats (CRISPR)‐associated endonuclease 9 (CRISPR/Cas9) system has been successfully used as an easy‐to‐handle, highly specific, and efficient approach for genome editing in mammalian cells.14, 15 In this study, we applied the CRISPR/Cas9 system to human melanoma cell line A375 which endogenously expresses MAGEC2, and successfully established MAGEC2‐knockout A375 clones. To identify the differentially expressed protein profiles associated with loss of MAGEC2 expression, isobaric tag for relative quantitation (iTRAQ)16 based comparative proteomics experiments were carried out on the MAGEC2‐knockout and control A375 cells. Analysis with bioinformatics and experimental validation showed that MAGEC2 was involved in tumorigenesis and cancer development. This is the first study to establish MAGEC2‐knockout cells and thoroughly screen the altered proteins related with MAGEC2 loss, which will open a new window for investigating the contributions of cancer/testis antigen MAGEC2 in tumorigenesis and cancer development.
Materials and Methods
Cells and reagents
Human melanoma cell line A375 were purchased from ATCC (Rockville, MA, USA) and maintained in DMEM (ATCC) supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA), and human melanoma cell line Hs 695T cells were purchased from Cobioer Biosciences Company (Nanjing, China) and maintained in MEM (Gibco, Gaithersburg, MD, USA) supplemented with non‐essential amino acids, 1 mM sodium pyruvate, and 10% FBS (Invitrogen). MAGEC2‐knockdown Hs 695T cells were established by transfecting pGPU6/Neo‐shMAGEC2 or pGPU6/Neo‐shNC vectors and selected using 1.5 mg/mL G418 for 2 weeks. Target sequences are as follows: MAGEC2 shRNA, 5′‐CAATTGATACCGCAGATGA‐3′; and control shRNA, 5′‐TTCTCCGAACGTGTCACGT‐3′. Recombinant human tumor necrosis factor‐α (TNF‐α) was purchased from PeproTech (Rocky Hill, NJ, USA) and used at 20 ng/mL, and cycloheximide (CHX) was from Sigma‐Aldrich (St. Louis, MO, USA) and used at 10 μg/mL.
MAGEC2 single guide RNA expression vector construction
The MAGEC2 guide RNAs (gRNAs) were designed using the online CRISPR design tool (http://crispr.mit.edu). For effective gene repression, the first 200 bp of the gene‐coding region of MAGEC2 were selected to be targeted and submitted to the website, which provided a ranked list of all possible guides in the query sequence ordered by faithfulness of on‐target activity computed as 100% minus a weighted sum of off‐target hit‐scores in the target genome. The top six individual gRNAs, which had scores >80, were selected and six pairs of complementary oligos containing these MAGEC2 guide sequences and BbsI ligation adapters were synthesized (Table S1). Each pair of oligos for different targeting sites was annealed and ligated to the BbsI‐digested pX458 vector (provided by Professor Fengming Lu, Department of Microbiology, Peking University Health Science Center, Beijing, China), a bicistronic vector containing cDNAs encoding human codon‐optimized Streptococcus pyogenes Cas9 and an adaptable CRISPR RNA/trans‐activating CRISPR RNA chimera containing adjacent BbsI cloning sites for protospacer guide sequence insertion. The constructed plasmids were verified by sequencing analysis.
Transfection and clone validation
The pX458 plasmid containing each target gRNA sequence or pX458 empty vector was transfected into A375 cells with Lipofectamine 2000 (Invitrogen). To obtain single clones of MAGEC2‐knockout cells, serial dilution was carried out to achieve a cell density of 8 cells/mL medium, and a 100‐μL aliquot was seeded into each well of a 96‐well plate. Single colonies were selected and MAGEC2 protein expression levels in each clone were detected using Western blot. Genomic DNA was isolated from edited clones and non‐edited A375 control cells, followed by PCR amplification of the region surrounding the CRISPR/Cas9 target site using primers annealed to the flanking sequences of the single guide RNAs (sgRNAs) (forward primer, TTCCCAGCAGACAAACTCCC; reverse primer, GGACCCTGTGGAGGACTACT). The PCR products were cloned into a TA cloning vector and sequenced. The indels of the MAGEC2 gene were identified by comparing with the wild‐type MAGEC2 sequence.
Analysis of off‐target cleavage
Potential off‐target sites of the sgRNAs were predicted using the online off‐target searching tool, Cas‐OFFinder (http://www.rgenome.net/cas-offinder).17 As all the gRNAs we designed have at least a three‐nucleotide mismatch with the other sequence, the regions with a three‐nucleotide mismatch with the MAGEC2 gRNA that were most likely to produce off‐target mutations were selected and subjected to PCR and sequence analysis. The off‐target locations for each gRNA and primers to amplify these regions are listed in Table S2.
Proteomics sample preparation
The harvested cells were lysed in 25 mM triethylammonium bicarbonate, 8 M urea, 2% Triton X‐100, 0.1% SDS, 50 μg/mL DNase I, 50 μg/mL RNase A, and 1× protease inhibitor cocktail (Pierce, Rockford, IL, USA). Unsolubilized material was removed by centrifugation at 18 800g for 1 h at 15°C and the supernatants were collected. Protein concentrations were determined using 2‐D Quan kit (GE Healthcare, Piscataway, NJ, USA).
Isobaric tag for relative quantitation labeling
The iTRAQ labeling was carried out using an iTRAQ Reagent 8‐Plex kit (AB Sciex, Foster City, CA, USA) based on the manufacturer's protocol with minor modifications. Cells lysates from three pairs of MAGEC2‐WT (WT1, WT2, and WT3) and MAGEC2‐Knockout (KO1, KO2, and KO3) A375 cells were labeled with iTRAQ labeling reagent 113, 114, 115, 116, 117, and 118, respectively. Briefly, 100 μg protein from each of the six cell lines was reduced with 2 μL of 50 mM tris‐(2‐carboxyethyl) phosphine at 37°C for 1 h, cysteine blocked with 1 μL of 200 mM methyl methanethiosulfonate at 37°C for 30 min, and digested with 3 μg trypsin (Roche) at 37°C for 15 h. The peptides were then dried and labeled with isobaric tags, and incubated at room temperature for 2 h before being combined.
High‐pH reversed‐phase HPLC fractionation
The combined iTRAQ‐labeled peptides were subjected to high‐pH reversed‐phase separation using a Gemini C18 (4.6 × 250 mm, 5‐μm particles) column (Phenomenex, Torrance, CA, USA) on a Shimadzu LC‐20AT HPLC system (Kyoto, Japan), using mobile phase A (20 mM ammonium formate, pH 10) and mobile phase B (20 mM ammonium formate, 80% acetonitrile, pH 10) at a flow rate of 500 μL/min with the following gradient: 3% mobile B (40 min), 3–8% mobile B (1 min), 8–30% mobile B (55 min), 30–50% mobile B (20 min), 50–100% mobile B (1 min), 100% mobile B (9 min), 100‐2% mobile B (2 min), and 2% mobile B (9 min). Beginning at 40 min of peptide elution, fractions were collected every 1 min. A total of 90 fractions were collected and subsequently combined to 22 fractions. All the fractions were dried with a centrifugal vacuum concentrator.
Analysis with HPLC‐MS/MS
All spectra were acquired on an Orbitrap Fusion (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Easy‐nLC 1000 (Thermo Scientific) ultra‐high pressure liquid chromatography pump. Each fraction was resuspended in 0.1% formic acid and separated on a 75 μm × 15 cm Easy‐spray column (C18, 5 μm; Thermo Scientific) using mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) at a flow rate of 400 nL/min with the following gradient: 3–6% mobile B (3 min), 6–23% mobile B (72 min), 23–35% mobile B (15 min), 35–90% mobile B (5 min), and 90% mobile B (5 min). The mass spectrometry (MS) analysis was carried out on Orbitrap Fusion Mass Spectrometer in a data‐dependent mode. The MS1 scans were acquired over a range of 400–1600 m/z at a resolution of 120 000 at m/z 200, with an automated gain control target of 200 000, and a maximum ion injection time of 100 ms. The MS2 precursors were isolated in the quadrupole with an isolation window of 1.6 m/z. Precursors were fragmented by high‐energy collision dissociation at a collision energy of 38%.
Database searching
Tandem mass spectra were extracted by Mascot Distiller version 2.5.1.0 (Matrix Science, London, UK) for peak list generation. All MS/MS samples were analyzed using Mascot version 2.5.1. Mascot was set up to search the SwissProt_2015_04 database (selected for Homo sapiens, 2015.04, 20 205 entries) assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.020 Da and a parent ion tolerance of 10.0 p.p.m. Methylthio of cysteine and iTRAQ 8 plex of lysine and the N‐terminus were specified in Mascot as fixed modifications. Oxidation of methionine and iTRAQ 8 plex of tyrosine were specified in Mascot as variable modifications.
Criteria for protein identification and quantification
Scaffold (version Scaffold_4.4.3; Proteome Software Inc., Portland, OR, USA) was used to validate the MS/MS based peptide and protein identifications and quantification. Peptide identifications were accepted if they could be established at >70.0% probability to achieve a false discovery rate (FDR) <1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at >99.0% probability to achieve an FDR <5.0% and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.18 Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Channels were corrected in all samples according to the algorithm described in i‐Tracker.19 Acquired intensities in the experiment were globally normalized across all acquisition runs. Individual quantitative samples were normalized within each acquisition run. Intensities for each peptide identification were normalized within the assigned protein. The reference channels were normalized to produce a 1:1 fold change. All normalization calculations were carried out using medians to multiplicatively normalize data.
Statistical analysis of differentially expressed proteins between the WT group (tags 113, 114, and 115) and KO group (tags 116, 117, and 118) was carried out using the Mann–Whitney test provided by the Scaffold software and proteins with P < 0.05 were selected. The log2‐transformed fold change was provided by the Scaffold software and subsequently, the meaningful cut‐off for fold change (>1.2 or <0.833) was finalized by the use of biological replicate method proposed by Gan et al.20, 21
Ingenuity pathway analysis
The differentially expressed proteome was evaluated using Ingenuity Pathway Analysis (IPA) software (Ingenuity H Systems, Redwood City, CA, USA; http://www.ingenuity.com) for associated biological functions and predominant canonical pathways. Briefly, a dataset containing the differential proteome and corresponding extremum of expression values was uploaded into “Dataset Files” of the IPA, and core analysis module was selected to rank the proteins into the top canonical pathways involved, significantly changed upstream regulators, biological functions, as well as top networks. The reference set and parameters for IPA on a significantly altered protein list were as follows: (i) reference set, ingenuity knowledge base (genes + endogenous chemicals); (ii) relationship to include, direct and indirect; (iii) filter summary, consider only molecules and/or relationships where (species = human OR rat OR mouse AND (confidence = experimentally observed); and (iv) cut‐off, −1.2‐ to 1.2‐fold change. Protein–protein interaction maps were built using the STRING database version 10 (http://www.string-db.org/), which assembles functional protein networks on the basis of compiled evidence.22
Western blot validation of iTRAQ data
Western blot experiments were carried out as previously described.13 The blots were visualized by enhanced chemiluminescence kit (Biodragon Immunotechnologies, Beijing, China). Commercial antibodies used in this study were as follows: anti‐GAPDH (Bioworld, St Louis, MO, USA), anti‐NES (Novus, littleton, CO, USA), anti‐ALDH1A3 (Origene, Rockville, MD, USA), anti‐SERPINB2 (Bioworld), anti‐HTRA2 (Proteintech, Rosemont, IL, USA), anti‐BID (Origene), anti‐cleaved poly(ADP‐ribose) polymerase (PARP; Cell Signaling Technology, Danvers, MA, USA), anti‐cleaved caspase‐3 (Cell Signaling Technology). Anti‐MAGEC2 polyclonal and monoclonal antibodies have been described previously.23, 24 Briefly, three New Zealand rabbits were immunized s.c. with purified MAGEC2 protein (100 μg) admixed with complete Freund's adjuvant/incomplete Freund's adjuvant, four injections at 3‐week intervals. The resultant polyclonal antibody was purified by immunoaffinity chromatography.
Cell apoptosis assay–annexin V/PI
MAGEC2‐KO A375 or MAGEC2‐knockdown Hs 695T cells stimulated with TNF‐α (20 ng/mL) plus CHX (10 μg/mL) for 12–18 h were harvested, washed, and then incubated with annexin V–FITC for 30 min and 7‐aminoactinomycin D for 5 min before analyzing with a flow cytometer. Percentages of annexin V‐positive cells were compared.
Animal study
Animal experiments were undertaken at Peking University Health Science Center, and were approved by the Institutional Animal Care and Use Committee of Peking University Health Science Center. Mice were housed under pathogen‐free conditions in an animal center. NOD/SCID mice were purchased from Vital River Laboratory Animal Technology (Beijing, China). For the xenograft study, 1 × 105 MAGEC2‐WT or MAGEC2‐KO A375 cells were injected s.c. into 6‐week‐old NOD/SCID mice. The long (L) and short (S) axes of the tumor were measured by caliper every 2 or 3 days and tumor volume was calculated using the formula L × S2 × 0.5. The xenograft experiment was terminated when tumors were no more than 1400 mm3 in volume.
Results
Generation of MAGEC2‐knockout cell lines
To delete the MAGEC2 gene, we designed six gRNAs immediately downstream of the MAGEC2 start codon. Targeting at this site will create double‐stranded breaks repaired by non‐homologous end joining that will result in frameshift insertions or deletions (indels) and premature termination, and thus “knock out” functional MAGEC2 protein (Fig. 1a). A375 cells were transfected with the pX458‐MAGEC2 sgRNA plasmids, and individual clones of transfected cells were expanded. All the clones were probed for MAGEC2 protein expression by Western blot, and seven clones from four sgRNAs (#2, #3, #4, and #6) showed a complete absence of MAGEC2 expression compared to the unedited control, when probed with both monoclonal and polyclonal anti‐MAGEC2 antibodies (Fig. 1b). To exclude the possibilities of off‐target depletion of other members of MAGE family, we detected the expression of MAGEC3, MAGEA1, and MAGEA6 in four MAGEC2‐knockout clones; however, there were no significant changes for these proteins compared with control clones (Fig. S1a). MAGEC1 was not detected as it was not expressed in A375 cells (Fig. S1b).
Figure 1.
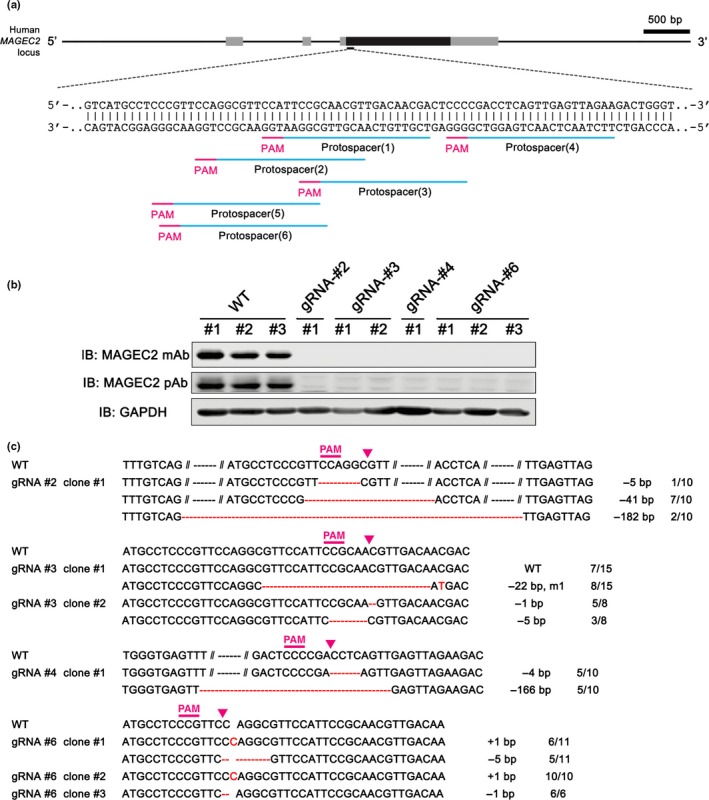
Generation of MAGEC2‐knockout A375 cell lines using the bacterial clustered regularly interspaced short palindromic repeats‐associated endonuclease 9 (CRISPR/Cas9) system. (a) Schematic representation of the MAGEC2‐targeting guide RNA (gRNA) sequences. Single gRNA target sites and protospacer adjacent motif (PAM) are indicated by blue and magenta bars, respectively. (b) MAGEC2 protein levels in MAGEC2‐WT and MAGEC2‐knockout (KO) A375 cell clones. MAGEC2 protein expression was analyzed by Western blot analysis with monoclonal (mAb) and polyclonal (pAb) anti‐MAGEC2 antibodies. GAPDH was used as the loading control. IB, immunoblot. (c) The sequences of 11 mutant alleles in the seven MAGEC2‐KO cell clones. TA clones from the PCR products were analyzed by DNA sequencing. PAM sequence and the Cas9 cleavage site are labeled by magenta bars and arrowheads. The number of sequences analyzed for each mutant allele is indicated behind the sequences. Red dashes, deleted bases (−); red bases, insertions (+) or mutations (m).
To characterize the nature of gene editing in MAGEC2‐knockout cell lines, genomic DNA PCR products from the seven individual clonal populations were cloned into a TA vector and 6–15 amplicons from each knockout cell lines were sequenced. A total of nine different indels were detected in MAGEC2 sequences isolated from seven MAGEC2 knockout cell lines. Most of the indels contained deletions of −1 to −182 nucleotides, one had an insertion of +1 nucleotide and one had a deletion/mutation (−22/m1) (Fig. 1c).
We next examined the off‐target activity of the four sgRNAs by amplifying the potential off‐target sequences with PCR and undertaking sequencing analyses. It revealed that there were no genome mutations at the potential off‐target sites (not shown). Three MAGEC2‐WT clones labeled as WT1, WT2, and WT3 as well as four MAGEC2‐KO clones labeled as KO1 (gRNA #2 clone #1), KO2 (gRNA #3 clone #1), KO3 (gRNA #6 clone #1), and KO4 (gRNA #6 clone #2) were used for the following experiments.
Workflow of global proteome comparison of MAGEC2‐KO versus MAGEC2‐WT A375 cells
To understand how the loss of MAGEC2 expression may alter cellular protein homeostasis, global proteome analyses were carried out with three pairs of biological replicates from the aforementioned CRISPR‐Cas9‐generated A375 cell clones (WT1–3 vs KO1–3). Cells were grown in standard growth medium, then harvested, lysed, and subjected to iTRAQ labeling. Thereafter, relative protein expression differences between MAGEC2‐WT and MAGEC2‐KO A375 cells were measured by mass spectrometry (Fig. 2). Protein identification and quantitation were carried out using the Scaffold software. A total of 4424 proteins were identified based on a threshold of at least 99.0% probability to achieve an FDR <5.0% and contained at least two identified peptides. Details of all identified proteins are provided in Table S3.
Figure 2.
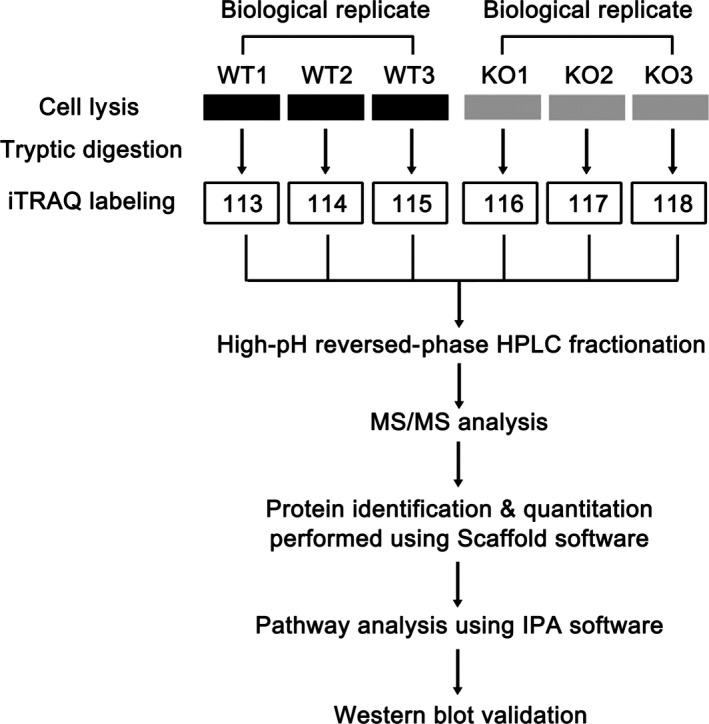
Schematic representation of the experimental design for isobaric tag for relative quantitation (iTRAQ) labeling showing biological replicates. Three biological replicates from MAGEC2‐WT A375 cells were labeled with 113, 114, and 115, respectively; replicates from MAGEC2‐knockout (KO) A375 cells were labeled with 116, 117, and 118, respectively. IPA, Ingenuity Pathway Analysis; MS/MS, tandem mass spectrometry.
To determine the cut‐off iTRAQ ratios for the differentially expressed proteins, the biological replicate method reported by Gan et al. was applied to our dataset, and approximately 15% variation corresponding to 88% coverage of data was observed (Fig. S2). Thus, 1.2‐fold (20% variation) was used as the cut‐off value corresponding to the iTRAQ ratio of >1.2 for upregulation and <0.83 for downregulation. The Mann–Whitney U‐test was applied to assess significant differences between the KO and WT groups with a P‐value <0.05 considered significant. Finally, proteins that qualified the cut‐off value as well as P‐value were chosen as significantly altered ones. A total of 229 proteins were finally screened as differentially expressed, with 141 (61.6%) upregulated and 88 (38.4%) downregulated (Table S4).
Ingenuity pathway analysis of differentially expressed proteins in MAGEC2‐KO A375 cells
To identify the biological functions and pathways associated with MAGEC2 deletion, the differentially expressed proteome was evaluated using the core analysis module of the IPA software.25 The significantly affected canonical pathways after MAGEC2 deletion are shown in Figure 3. After MAGEC2 deletion, the most significantly upregulated pathway is “apoptosis signaling” and the most significantly downregulated pathway is “regulation of actin‐based motility by RhoA”. The analysis of “top upstream regulators”, “top diseases and bio functions”, and “top networks” are summarized in Table 1. Chief of all, “cell death and survival” was identified among the top list of “molecular and cellular functions” and “top networks”, and TNF signaling was predicted to be activated as the top upstream regulator after MAGEC2 deletion. Network analysis was also carried out to provide a graphical representation with differentially expressed proteins associated with “cell death and survival” using the STRING protein–protein interaction database. As shown in Figure 4, 64 proteins were involved in “cell death and survival”‐related protein–protein interaction, of which 40 were upregulated and 24 were downregulated.
Figure 3.
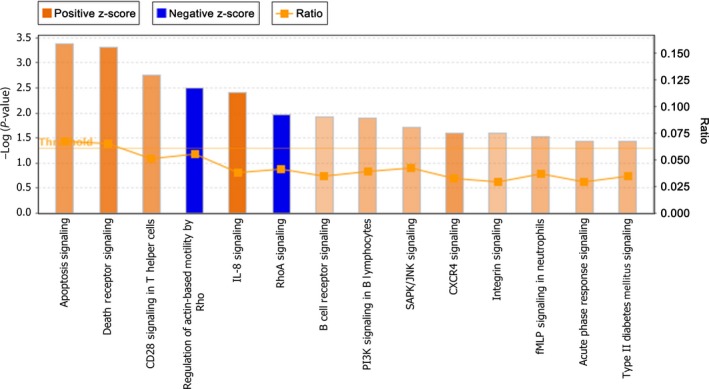
Significantly changed canonical pathways after MAGEC2 deletion analyzed by Ingenuity Pathway Analysis software. The top canonical pathways analysis was filtered by P‐value < 0.05 and z‐score > 0.5. The primary Y‐axis displays the −log of P‐value, which is calculated by Fisher's exact test right‐tailed. The orange and blue colored bars indicate predicted pathway activation and inhibition, respectively (z‐score). The orange points connected by a thin line represent the ratio which is calculated as the number of genes differentially expressed divided by the total number of genes for each pathway. CXCR4, chemokine (C‐X‐C motif) receptor 4; fMLP, N‐formyl‐methionyl‐leucyl‐phenylalanine; IL‐8, interleukin‐8; PI3K, phosphatidylinositol 3‐kinase; SAPK, stress‐activated protein kinase.
Table 1.
Summary of Ingenuity Pathway Analysis of proteins with significantly altered expression level in MAGEC2‐knockout cells
| Top upstream regulators | P‐value† | Z‐score |
|---|---|---|
| TNF | 5.71E‐05 | 1.801 |
| miR‐183‐5p | 9.72E‐05 | |
| TP63 | 1.14E‐04 | 1.257 |
| EPAS1 | 1.74E‐04 | 1.264 |
| HIF1A | 3.22E‐04 | 1.145 |
| Top diseases and bio functions | P‐value | Molecules‡ |
| Diseases and disorders | ||
| Cancer | 1.08E‐02–1.30E‐06 | 200 |
| Organismal injury and abnormalities | 1.08E‐02–1.30E‐06 | 204 |
| Reproductive system disease | 1.08E‐02–1.30E‐06 | 112 |
| Dermatological diseases and conditions | 1.08E‐02–4.95E‐06 | 43 |
| Developmental disorder | 1.08E‐02–4.95E‐06 | 36 |
| Molecular and cellular functions | ||
| Cell death and survival | 1.08E‐02–1.45E‐09 | 94 |
| Cellular assembly and organization | 1.08E‐02–3.31E‐09 | 79 |
| Cellular function and maintenance | 1.05E‐02–1.07E‐06 | 82 |
| Cellular growth and proliferation | 9.72E‐03–2.54E‐06 | 87 |
| Cellular movement | 9.72E‐03–6.80E‐06 | 53 |
| Physiological system development and function | ||
| Tissue development | 9.72E‐03–3.31E‐09 | 71 |
| Organismal survival | 1.12E‐03–4.24E‐05 | 67 |
| Organismal development | 8.86E‐03–3.47E‐05 | 45 |
| Nervous system development and function | 1.08E‐02–5.62E‐05 | 40 |
| Organ morphology | 1.05E‐02–5.62E‐05 | 31 |
| Associated network functions | IPA score§ | |
| Cell death and survival, cellular movement, hematological disease | 55 | |
| Cell death and survival, dermatological diseases and conditions, developmental disorder | 55 | |
| Cardiovascular system development and function, organismal development, organismal injury and abnormalities | 39 | |
| Cell death and survival, hematological disease, hematological system development and function | 35 | |
| Embryonic development, organismal development, cell cycle | 32 | |
†P‐values are displayed in E notation: aEb indicates a value of a × 10b. ‡number of molecules involved. §Ingenuity Pathway Analysis (IPA) scores were generated using a right‐tailed Fisher's exact test and represent the absolute value of the exponent to which base 10 is raised in the resulting P‐value, for example, an IPA score of 55 indicates a P‐value of 10E‐55. EPAS1, endothelial PAS domain protein 1; HIF1A, hypoxia‐inducible factor 1α; miR, microRNA; TNF, tumor necrosis factor; TP63, tumor protein 63.
Figure 4.
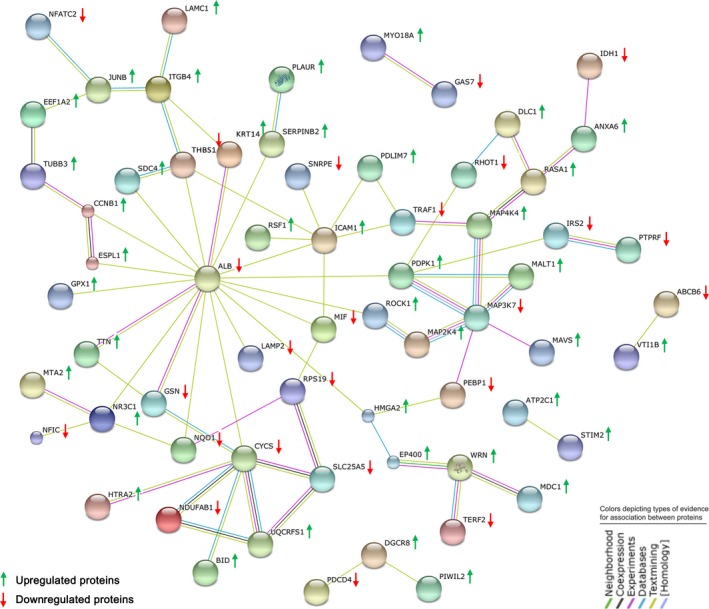
Network analysis of differentially expressed proteins associated with “cell death and survival” after MAGEC2 deletion using the String 9.1 program. Red arrows represent downregulated proteins, and green arrows represent upregulated proteins in MAGEC2‐knockout A375 cells.
Validation of iTRAQ dataset proteins
Based on the IPA analysis of functional pathways and networks, three proteins (HTRA2, BID, and SERPINB2) related to “cell death and survival”26, 27, 28 were selected for validating iTRAQ results using Western blot in the biological replicates used for iTRAQ labeling (WT1, WT3, KO1, KO2, and KO3) and additional MAGEC2‐KO clone (KO4). We also detected two cancer stem cell markers, ALDH1A329 and NES,30 which were found to be downregulated after MAGEC2 deletion in the iTRAQ results. Results from Western blot of the selected five proteins were consistent with our iTRAQ results (Fig. 5).
Figure 5.
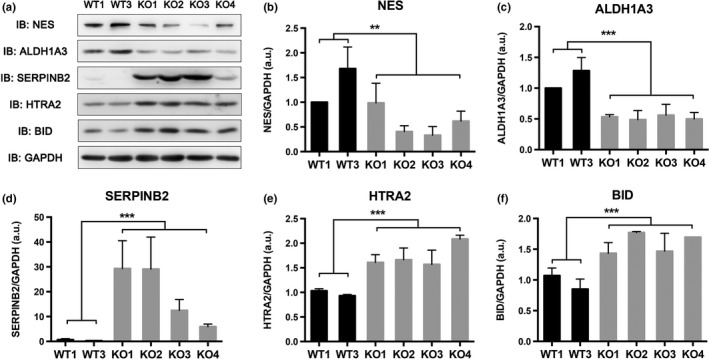
Validation of isobaric tag for relative quantitation (iTRAQ) results. (a) Western blot analysis to validate differential expression pattern of five candidate proteins from the iTRAQ dataset. NES and ALDH1A3 were downregulated, whereas SERPINB2, HTRA2, and BID were upregulated in MAGEC2‐knockout (KO) cells compared with the MAGEC2‐WT cells, which was in conformity with the iTRAQ results. GAPDH was used as a loading control. IB, immunoblot. (b–f) Relative protein level of pY‐STAT3/GAPDH was quantified by ImageJ software and the average results from more than two samples are shown for each protein. Data are represented as mean ± SD. **P < 0.01; ***P < 0.001.
Effect of MAGEC2 deletion on TNF‐α/CHX‐evoked apoptosis
As “cell death and survival” as well as “TNF signaling” were identified as the major changes after MAGEC2 deletion, we investigated whether MAGEC2 could influence TNF‐α‐induced apoptosis. Because TNF‐α is insufficient to trigger apoptosis in A375 cells by the expression of protective factors, CHX, a translational inhibitor, was used to block survival signals and enhance responsiveness to death. Two pairs of MAGEC2‐WT (WT1 and WT3) and MAGEC2‐KO (KO1 and KO4) A375 cells were treated with TNF‐α/CHX for 12 h to induce apoptosis and were measured by flow cytometry. The number of apoptotic cells was assessed by annexin V staining. MAGEC2 deletion resulted in a significant increase in the number of apoptotic cells (Fig. 6a). In addition, the activity of caspase‐3 and PARP, which are cleaved and activated in the final stage of apoptosis, was also measured in these MAGEC2‐WT or KO A375 cells. Western blot analysis showed that both cleaved caspase‐3 and cleaved PARP were significantly increased in the TNF‐α/CHX‐treated MAGEC2‐KO group, compared with the MAGEC2‐WT group (Fig. 6b). We also found that MAGEC2‐knockdown Hs 695T cells were more sensitive to apoptosis induced by TNF‐α/CHX (Fig. 6c,d). Taken together, these data suggest that MAGEC2‐silencing could promote TNF‐α/CHX‐evoked apoptosis in cancer cells.
Figure 6.
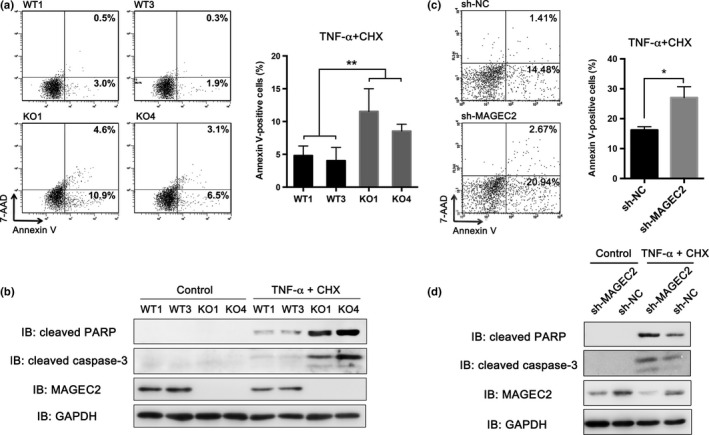
MAGEC2 deletion promotes tumor necrosis factor‐α/cycloheximide (TNF‐α/CHX)‐evoked apoptosis in melanoma cells. (a,c) Control and MAGEC2‐knockout (KO) A375 cells or MAGEC2‐knockdown Hs 695T cells treated with the TNF‐α (20 ng/mL) plus CHX (10 μg/mL) for 12–18 h were stained with annexin V/7‐aminoactinomycin D (7‐AAD) and subjected to FACS analysis (left panel). Apoptosis rates (right panel) are presented as mean ± SD of three separate experiments and expressed as the percentage of annexin V‐positive cells. *P < 0.05; **P < 0.01. (b,d) Western blot analysis of cleaved poly(ADP‐ribose) polymerase (PARP) and caspase‐3 in control and MAGEC2‐KO A375 cells or MAGEC2‐knockdown Hs 695T cells treated with TNF‐α (20 ng/mL) plus CHX (10 μg/mL) for 12 h. IB, immunoblot; NC, negative control.
Effect of MAGEC2 deletion on tumor growth in vivo
A tumor xenograft experiment was used to determine whether the observed effect of MAGEC2 on tumor apoptosis in vitro might be translated into the regulation of tumor growth in vivo model. We implanted MAGEC2‐WT (WT1 and WT3) and KO (KO1, KO2, and KO4) A375 cells s.c. into NOD/SCID mice, and the subsequent analysis of tumor volume was carried out with tumor‐bearing mice in each group. As shown in Figure 7, tumor growth was significantly retarded in the MAGEC2‐KO group compared with the MAGEC2‐WT group, and the tumor volume in the MAGEC2‐KO group was decreased by 64% compared with the control group at the termination of the experiment. These results corroborated our in vitro results and suggested that MAGEC2 promotes survival of melanoma cells both in vitro and in vivo.
Figure 7.
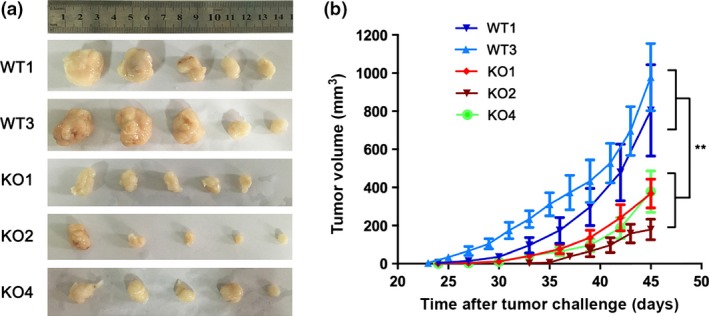
MAGEC2 deletion represses tumor growth in vivo. MAGEC2‐WT or MAGEC2‐knockout (KO) A375 cells (1 × 105) were injected s.c. into 6‐week‐old NOD/SCID mice, five mice per group. The sizes of xenograft tumors at the termination of the experiments (a) and tumor growth curves (b) are shown. Data are represented as mean ± SD. **P < 0.01.
Discussion
Cancer/testis antigen MAGEC2, overexpressed in various types of human cancer, has been extensively studied as a candidate cancer vaccine in previous reports.23, 31 Recent accumulating evidence has indicated that MAGEC2 may play an active role in tumorigenesis and cancer development.11, 12, 13 In this study, we used the CRISPR/Cas9 system to establish MAGEC2‐knockout melanoma cell lines, and identified the differentially expressed protein profiles related to the loss of expression of MAGEC2 with a high resolution tandem mass spectrometry proteomics approach.
To our knowledge, this is the first study on depletion of MAGEC2 using genomic‐editing technology, namely the CRISPR/Cas9 system, which is a novel approach that enables researchers to manipulate target genes in various cell types for gene function studies.14, 15 We designed six sgRNAs targeting coding regions in the MAGEC2 genome, and successfully obtained seven MAGEC2‐knockout clones from four sgRNA. It is not surprising that one clone showed three indels, which is consistent with the chromosome abnormalities in A375 melanoma cells. A major concern for CRISPR/Cas9‐mediated genome editing is off‐target cleavage (non‐specifically cleaved genomic DNA sequences containing base‐pair mismatches).32 To reduce potential off‐target cleavage in the genome, we used bioinformatic tools based on base mismatches for predicting the most likely potential off‐target sites when designing the sgRNA. We avoided target sequences with fewer than three mismatches, and guaranteed at least one base mismatch was within the “seed” sequence of the sgRNA.32, 33 In addition, we carried out sequence analysis on the genome sequences that contained three base mismatches to the sgRNA, but no mutagenesis was observed at any of these sites.
To explore the biological functions and molecular pathways associated with MAGEC2 expression, we analyzed protein profiles in MAGEC2‐knockout and control A375 cells using the iTRAQ proteomics approach. We identified 4424 proteins, of which 229 were significantly differentially regulated. Functional analysis of the differentially expressed proteins indicated that they were involved in critical neoplasia‐related biological functions: cell death and survival, cellular assembly and organization, cellular function and maintenance, cellular growth and proliferation, and cellular movement. Molecular pathway analysis identified “apoptosis signaling” and “death receptor signaling” as the top pathways associated with MAGEC2 loss. We tested the effect of suppression of MAGEC2 on apoptosis, and found that transfection of MAGEC2 siRNA in 375 cells was insufficient to trigger cell apoptosis. Our result is inconsistent with a previous report.12 However, when we treated cells with TNF and CHX, a high percentage of apoptotic cells was found in MAGEC2‐knockout A375 cells compared with control groups. Thus, we propose that MAGEC2 might play a role in resistance to TNF‐α‐induced apoptosis. In addition, we also found that loss of MAGEC2 in A375 cells inhibited tumor growth in NOD/SCID mice, which complies with results from suppression of MAGEC2 in MRA‐13 cells.12
Interestingly, “regulation of actin‐based motility by Rho” and “RhoA signaling” were identified as the most downregulated pathways associated with MAGEC2 depletion in our pathway analysis (IPA), suggesting that MAGEC2 may be involved in cell motility and metastasis. Previous studies on clinical tumor tissues indicated that MAGEC2 expression is correlated with metastasis in breast cancer and melanoma patients.7, 34 It is worth investigating the biological functions of MAGEC2 in promoting tumor metastasis as well as the association of MAGEC2 with RhoA signaling. Moreover, hypoxia‐inducible factors HIF1 and HIF2 were also identified among the top hit list of upstream regulators associated with MAGEC2. Oxygen deprivation is a common feature of solid tumors.35 MAGE‐11, a member of MAGE family, has been reported to interact with the major HIF‐α hydroxylating enzyme PHD2 and activate hypoxic response.36 Whether MAGEC2 plays a role in activation of HIFs needs further investigation.
In summary, we successfully established MAGEC2‐knockout A375 cell lines using the CRISPR/Cas9 system, providing an ideal cell model for exploring the roles of MAGEC2 in tumor cells. Quantitative proteomics and bioinformatics analyses on MAGEC2 knockout cells provide important implications for describing the widest landscape of MAGEC2's functions and revealing the molecular pathways that MAGEC2 may be involved in cancer development.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Expression of type I melanoma‐associated antigens (MAGE‐I) family members in MAGEC2‐knockout (KO) A375 cell clones.
Fig. S2. Determination of experimental variation using all 4424 proteins identified in both biological replicates.
Table S1. Primer sequences for single guide RNA cloning.
Table S2. Off‐target locations for each guide RNA and primers to amplify these regions.
Table S3. All identified proteins in isobaric tag for relative quantitation (iTRAQ).
Table S4. Differentially expressed proteins in isobaric tag for relative quantitation (iTRAQ).
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81472645) and the Natural Science Foundation of Beijing Municipality (Grant No. 7142087).
Cancer Sci 107 (2016) 1888–1897
References
- 1. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci 2009; 100: 2014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brasseur F, Rimoldi D, Lienard D et al Expression of MAGE genes in primary and metastatic cutaneous melanoma. Int J Cancer 1995; 63: 375–80. [DOI] [PubMed] [Google Scholar]
- 3. Riener MO, Wild PJ, Soll C et al Frequent expression of the novel cancer testis antigen MAGE‐C2/CT‐10 in hepatocellular carcinoma. Int J Cancer 2009; 124: 352–7. [DOI] [PubMed] [Google Scholar]
- 4. Jang SJ, Soria JC, Wang L et al Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis. Cancer Res 2001; 61: 7959–63. [PubMed] [Google Scholar]
- 5. Gure AO, Chua R, Williamson B et al Cancer‐testis genes are coordinately expressed and are markers of poor outcome in non‐small cell lung cancer. Clin Cancer Res 2005; 11: 8055–62. [DOI] [PubMed] [Google Scholar]
- 6. Otte M, Zafrakas M, Riethdorf L et al MAGE‐A gene expression pattern in primary breast cancer. Cancer Res 2001; 61: 6682–7. [PubMed] [Google Scholar]
- 7. Curioni‐Fontecedro A, Nuber N, Mihic‐Probst D et al Expression of MAGE‐C1/CT7 and MAGE‐C2/CT10 predicts lymph node metastasis in melanoma patients. PLoS ONE 2011; 6: e21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. von Boehmer L, Keller L, Mortezavi A et al MAGE‐C2/CT10 protein expression is an independent predictor of recurrence in prostate cancer. PLoS ONE 2011; 6: e21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang B, O'Herrin S, Wu J et al Select cancer testes antigens of the MAGE‐A, ‐B, and ‐C families are expressed in mast cell lines and promote cell viability in vitro and in vivo . J Invest Dermatol 2007; 127: 267–75. [DOI] [PubMed] [Google Scholar]
- 10. Yang B, O'Herrin SM, Wu J et al MAGE‐A, mMage‐b, and MAGE‐C proteins form complexes with KAP1 and suppress p53‐dependent apoptosis in MAGE‐positive cell lines. Cancer Res 2007; 67: 9954–62. [DOI] [PubMed] [Google Scholar]
- 11. Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE‐RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell 2010; 39: 963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhatia N, Xiao TZ, Rosenthal KA et al MAGE‐C2 promotes growth and tumorigenicity of melanoma cells, phosphorylation of KAP1, and DNA damage repair. J Invest Dermatol 2013; 133: 759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hao J, Song X, Wang J et al Cancer‐testis antigen MAGE‐C2 binds Rbx1 and inhibits ubiquitin ligase‐mediated turnover of cyclin E. Oncotarget 2015; 6: 42028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilbert LA, Horlbeck MA, Adamson B et al Genome‐scale CRISPR‐mediated control of gene repression and activation. Cell 2014; 159: 647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shalem O, Sanjana NE, Hartenian E et al Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 2014; 343: 84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Unwin RD, Griffiths JR, Whetton AD. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC‐MS/MS. Nat Protoc 2010; 5: 1574–82. [DOI] [PubMed] [Google Scholar]
- 17. Bae S, Park J, Kim JS. Cas‐OFFinder: a fast and versatile algorithm that searches for potential off‐target sites of Cas9 RNA‐guided endonucleases. Bioinformatics 2014; 30: 1473–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 2003; 75: 4646–58. [DOI] [PubMed] [Google Scholar]
- 19. Shadforth IP, Dunkley TP, Lilley KS, Bessant C. i‐Tracker: for quantitative proteomics using iTRAQ. BMC Genom 2005; 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J Proteome Res 2007; 6: 821–7. [DOI] [PubMed] [Google Scholar]
- 21. Ghosh D, Li Z, Tan XF, Lim TK, Mao Y, Lin Q. iTRAQ based quantitative proteomics approach validated the role of calcyclin binding protein (CacyBP) in promoting colorectal cancer metastasis. Mol Cell Proteomics 2013; 12: 1865–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szklarczyk D, Franceschini A, Kuhn M et al The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011; 39: D561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen J, Zhang L, Wen W et al Induction of HCA587‐specific antitumor immunity with HCA587 protein formulated with CpG and ISCOM in mice. PLoS ONE 2012; 7: e47219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li B, Qian XP, Pang XW et al HCA587 antigen expression in normal tissues and cancers: correlation with tumor differentiation in hepatocellular carcinoma. Lab Invest 2003; 83: 1185–92. [DOI] [PubMed] [Google Scholar]
- 25. Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014; 30: 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN. Hax1‐mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 2008; 452: 98–102. [DOI] [PubMed] [Google Scholar]
- 27. Zinkel SS, Hurov KE, Gross A. Bid plays a role in the DNA damage response. Cell 2007; 130: 9–10; author reply ‐1. [DOI] [PubMed] [Google Scholar]
- 28. Tonnetti L, Netzel‐Arnett S, Darnell GA et al SerpinB2 protection of retinoblastoma protein from calpain enhances tumor cell survival. Cancer Res 2008; 68: 5648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu X, Chai S, Wang P, Zhang C, Yang Y, Wang K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett 2015; 369: 50–7. [DOI] [PubMed] [Google Scholar]
- 30. Neradil J, Veselska R. Nestin as a marker of cancer stem cells. Cancer Sci 2015; 106: 803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol 2015; 37: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin Y, Cradick TJ, Brown MT et al CRISPR/Cas9 systems have off‐target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 2014; 42: 7473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cong L, Ran FA, Cox D et al Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339: 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang F, Zhou X, Miao X et al MAGEC2, an epithelial‐mesenchymal transition inducer, is associated with breast cancer metastasis. Breast Cancer Res Treat 2014; 145: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004; 4: 437–47. [DOI] [PubMed] [Google Scholar]
- 36. Aprelikova O, Pandolfi S, Tackett S et al Melanoma antigen‐11 inhibits the hypoxia‐inducible factor prolyl hydroxylase 2 and activates hypoxic response. Cancer Res 2009; 69: 616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression of type I melanoma‐associated antigens (MAGE‐I) family members in MAGEC2‐knockout (KO) A375 cell clones.
Fig. S2. Determination of experimental variation using all 4424 proteins identified in both biological replicates.
Table S1. Primer sequences for single guide RNA cloning.
Table S2. Off‐target locations for each guide RNA and primers to amplify these regions.
Table S3. All identified proteins in isobaric tag for relative quantitation (iTRAQ).
Table S4. Differentially expressed proteins in isobaric tag for relative quantitation (iTRAQ).